
Genomic Analysis of Reproductive Trait Divergence in Duroc and Yorkshire Pigs: A Comparison of Mixed Models and Selective Sweep Detection

Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Populations and Genotypes
2.2. Dissection of Additive and Dominant Genetic Effects
2.3. Selection Sweep Detection
2.4. Functional Annotation of Significant Loci
3. Results
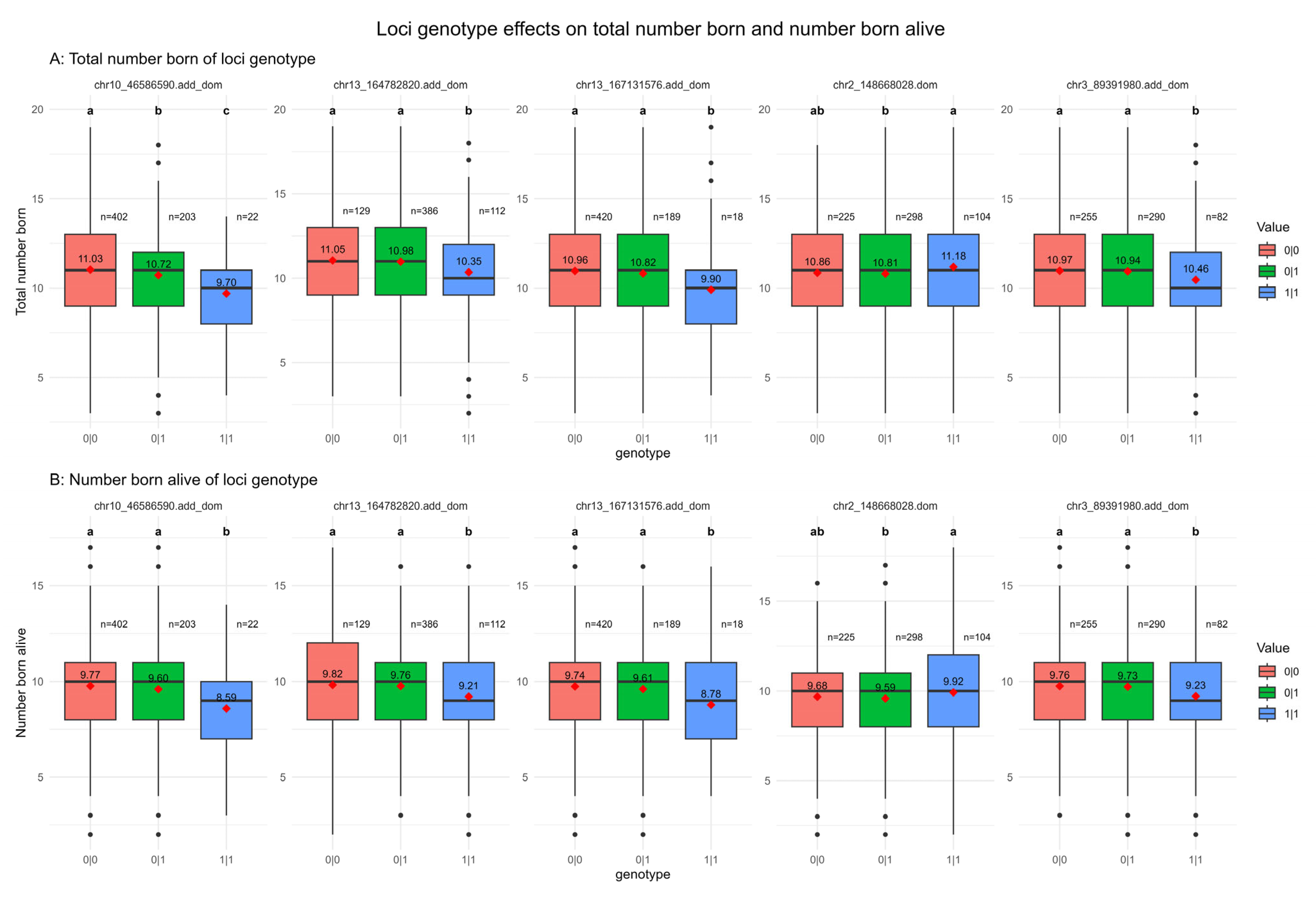
3.1. Additive and Dominant Effect Detection
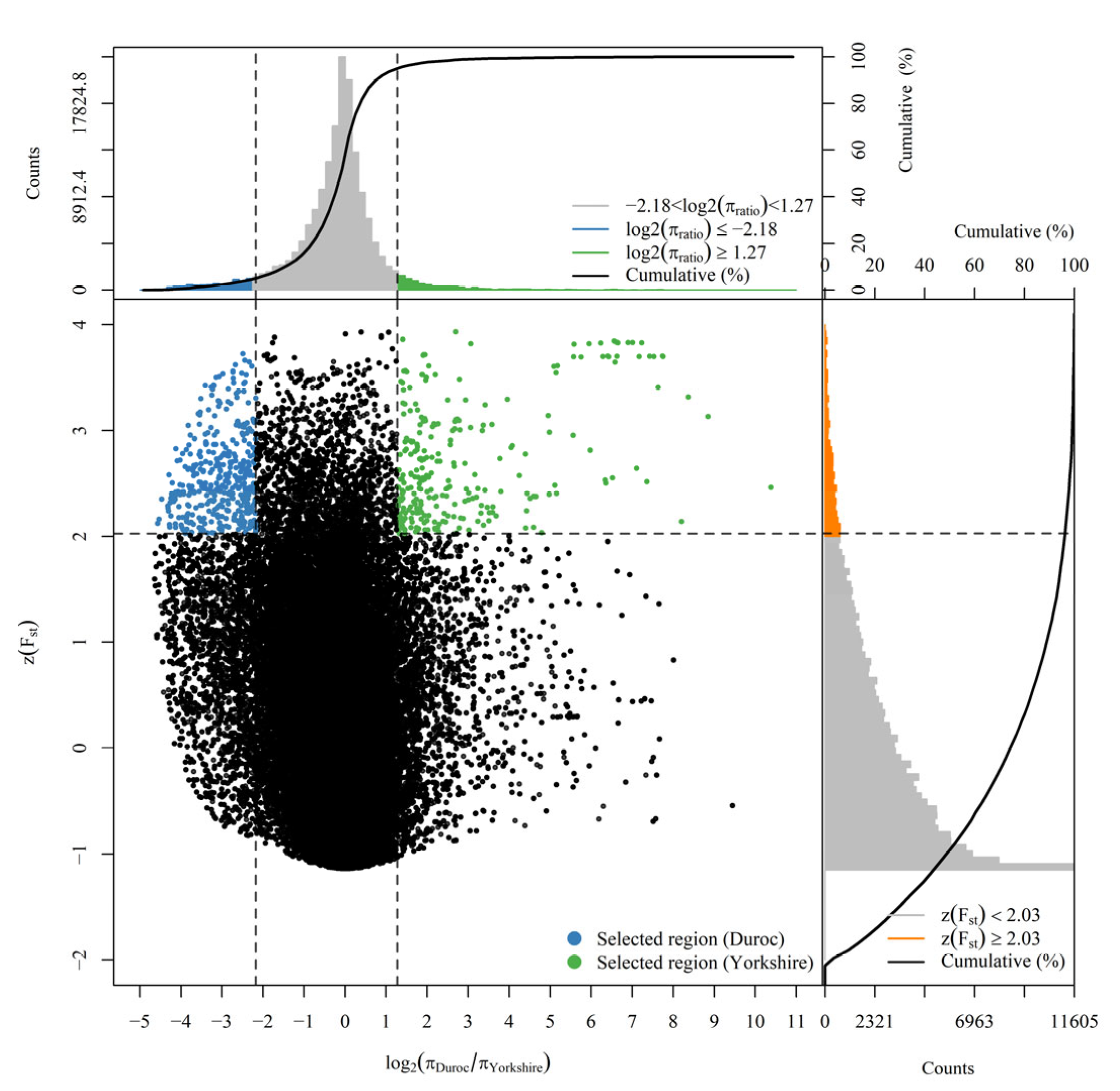
3.2. The Result of Selection Sweep Detection
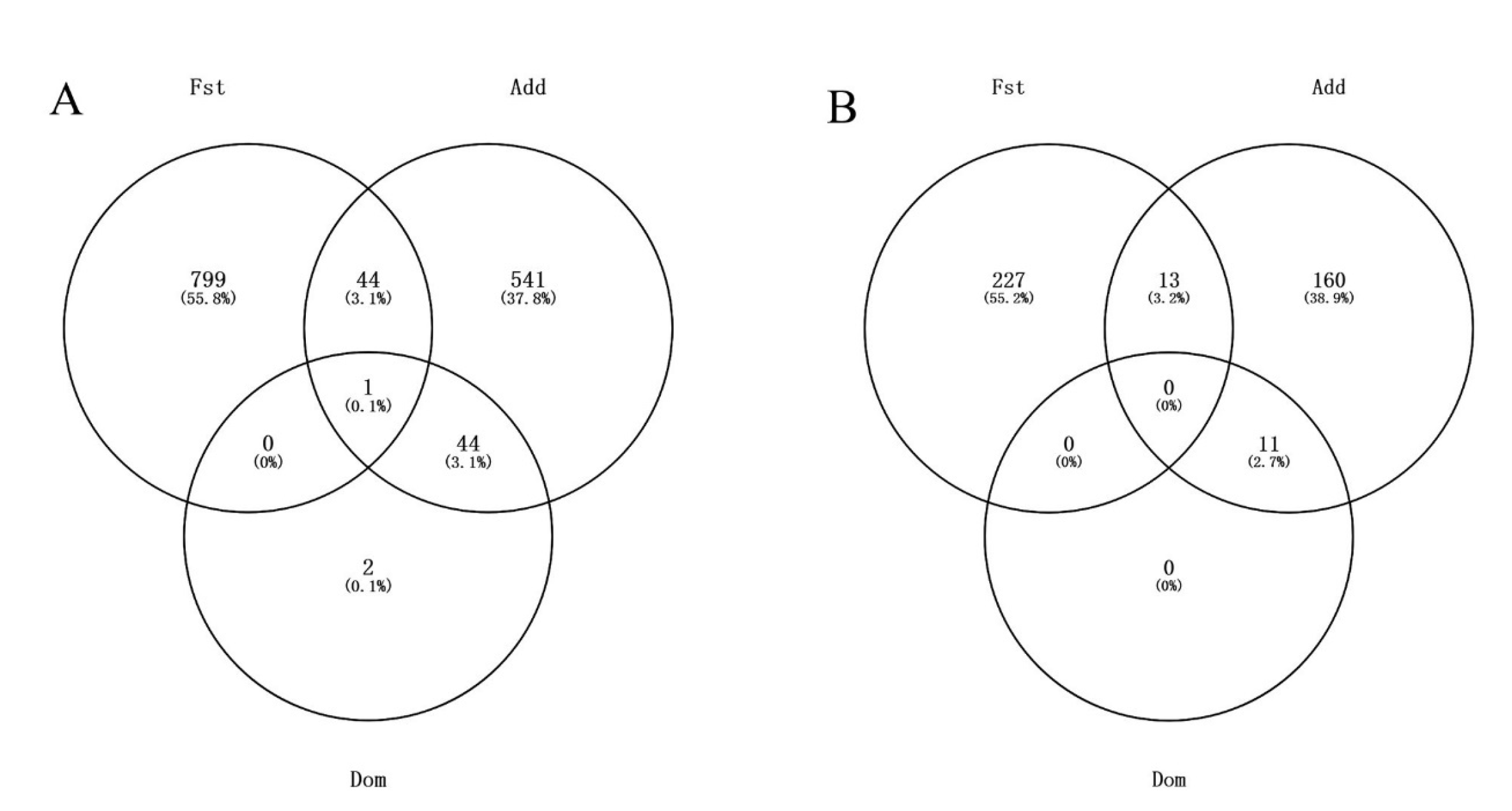
3.3. Comparison of LMM + ADDO Framework with FST Analysis
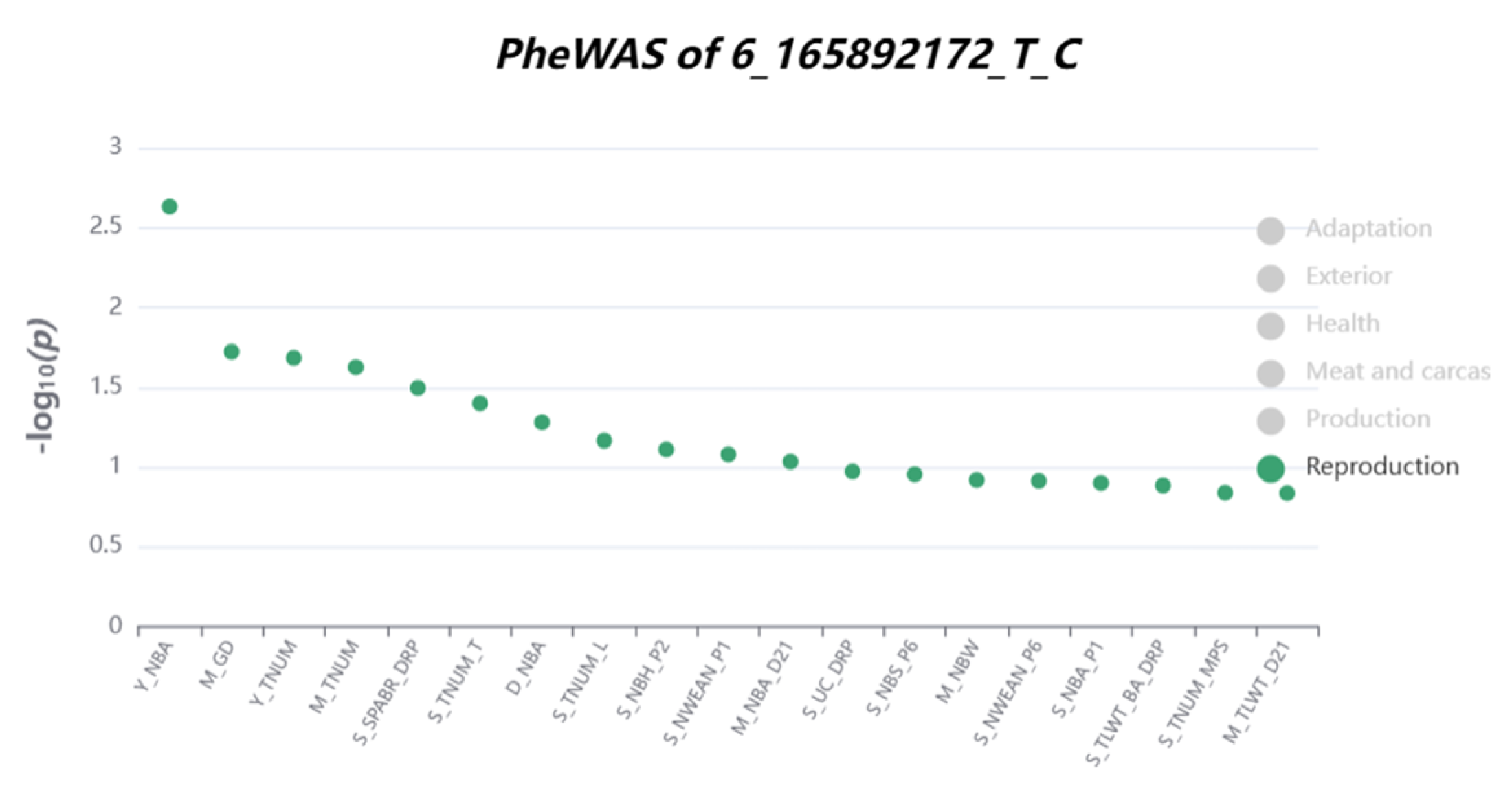
3.4. Functional Annotation Results of Significant Loci
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, K.; Wu, P.; Yang, Q.; Chen, D.; Zhou, J.; Jiang, A.; Ma, J.; Tang, Q.; Xiao, W.; Jiang, Y.; et al. Detection of Selection Signatures in Chinese Landrace and Yorkshire Pigs Based on Genotyping-by-Sequencing Data. Front. Genet. 2018, 9, 119. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Wang, Z.; Zhang, Z.; Xiao, Q.; Mawed, S.; Xu, Z.; Zhang, X.; Yang, H.; Zhu, M.; Xue, M.; et al. Genomic signatures reveal selection of characteristics within and between Meishan pig populations. Anim. Genet. 2018, 49, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Alves, K.; Brito, L.F.; Baes, C.F.; Sargolzaei, M.; Robinson, J.A.B.; Schenkel, F.S. Estimation of additive and non-additive genetic effects for fertility and reproduction traits in North American Holstein cattle using genomic information. J. Anim. Breed. Genet. 2020, 137, 316–330. [Google Scholar] [CrossRef] [PubMed]
- Bolormaa, S.; Pryce, J.E.; Zhang, Y.; Reverter, A.; Barendse, W.; Hayes, B.J.; Goddard, M.E. Non-additive genetic variation in growth, carcass and fertility traits of beef cattle. Genet. Sel. Evol. 2015, 47, 26. [Google Scholar] [CrossRef]
- Cortés, O.; Martinez, A.M.; Cañon, J.; Sevane, N.; Gama, L.T.; Ginja, C.; Landi, V.; Zaragoza, P.; Carolino, N.; Vicente, A.; et al. Conservation priorities of Iberoamerican pig breeds and their ancestors based on microsatellite information. Heredity 2016, 117, 14–24. [Google Scholar] [CrossRef]
- Cui, L.; Yang, B.; Pontikos, N.; Mott, R.; Huang, L. ADDO: A comprehensive toolkit to detect, classify and visualize additive and non-additive quantitative trait loci. Bioinformatics 2020, 36, 1517–1521. [Google Scholar] [CrossRef]
- Li, Q.; Yuan, X.; Chen, Z.; Zhang, A.; Zhang, Z.; Zhang, H.; Li, J. Heritability estimates and effect on lifetime reproductive performance of age at puberty in sows. Anim. Reprod. Sci. 2018, 195, 207–215. [Google Scholar] [CrossRef]
- Ruan, D.; Yang, J.; Zhuang, Z.; Ding, R.; Huang, J.; Quan, J.; Gu, T.; Hong, L.; Zheng, E.; Li, Z.; et al. Assessment of Heterozygosity and Genome-Wide Analysis of Heterozygosity Regions in Two Duroc Pig Populations. Front. Genet. 2021, 12, 812456. [Google Scholar] [CrossRef]
- Deng, M.; Qiu, Z.; Liu, C.; Zhong, L.; Fan, X.; Han, Y.; Wang, R.; Li, P.; Huang, R.; Zhao, Q. Genome-wide association analysis revealed new QTL and candidate genes affecting the teat number in Dutch Large White pigs. Anim. Genet. 2024, 55, 206–216. [Google Scholar] [CrossRef]
- Browning, B.L.; Tian, X.; Zhou, Y.; Browning, S.R. Fast two-stage phasing of large-scale sequence data. Am. J. Hum. Genet. 2021, 108, 1880–1890. [Google Scholar] [CrossRef]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Zhang, H.; Tang, Z.; Yin, D.; Fu, Y.; Yuan, X.; Li, X.; Liu, X.; Zhao, S. HIBLUP: An integration of statistical models on the BLUP framework for efficient genetic evaluation using big genomic data. Nucleic Acids Res. 2023, 51, 3501–3512. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 2015, 4, 7. [Google Scholar] [CrossRef]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef]
- Dyer, S.C.; Austine-Orimoloye, O.; Azov, A.G.; Barba, M.; Barnes, I.; Barrera-Enriquez, V.P.; Becker, A.; Bennett, R.; Beracochea, M.; Berry, A.; et al. Ensembl 2025. Nucleic Acids Res. 2025, 53, D948–D957. [Google Scholar] [CrossRef]
- Fu, Y.; Xu, J.; Tang, Z.; Wang, L.; Yin, D.; Fan, Y.; Zhang, D.; Deng, F.; Zhang, Y.; Zhang, H.; et al. A gene prioritization method based on a swine multi-omics knowledgebase and a deep learning model. Commun. Biol. 2020, 3, 502. [Google Scholar] [CrossRef]
- Zeng, H.; Zhang, W.; Lin, Q.; Gao, Y.; Teng, J.; Xu, Z.; Cai, X.; Zhong, Z.; Wu, J.; Liu, Y.; et al. PigBiobank: A valuable resource for understanding genetic and biological mechanisms of diverse complex traits in pigs. Nucleic Acids Res. 2024, 52, D980–D989. [Google Scholar] [CrossRef]
- Hu, Z.L.; Park, C.A.; Reecy, J.M. Bringing the Animal QTLdb and CorrDB into the future: Meeting new challenges and providing updated services. Nucleic Acids Res. 2022, 50, D956–D961. [Google Scholar] [CrossRef]
- Sun, H.; Cheng, Y.; He, Y.; Cheng, C.; Zhao, H.; Yang, S.; Wei, M.; Yang, J.; Liang, S.; Bai, C.; et al. Genome-wide association studies for the number of piglets born alive and dead in Dongliao black pigs. Anim. Genet. 2024, 55, 282–285. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2024; Available online: https://www.R-project.org/ (accessed on 14 June 2024).
- Wang, X.; Wang, L.; Shi, L.; Zhang, P.; Li, Y.; Li, M.; Tian, J.; Wang, L.; Zhao, F. GWAS of Reproductive Traits in Large White Pigs on Chip and Imputed Whole-Genome Sequencing Data. Int. J. Mol. Sci. 2022, 23, 13338. [Google Scholar] [CrossRef] [PubMed]
- Duijvesteijn, N.; Veltmaat, J.M.; Knol, E.F.; Harlizius, B. High-resolution association mapping of number of teats in pigs reveals regions controlling vertebral development. BMC Genom. 2014, 15, 542. [Google Scholar] [CrossRef] [PubMed]
- Bovo, S.; Ballan, M.; Schiavo, G.; Ribani, A.; Tinarelli, S.; Utzeri, V.J.; Dall’Olio, S.; Gallo, M.; Fontanesi, L. Single-marker and haplotype-based genome-wide association studies for the number of teats in two heavy pig breeds. Anim. Genet. 2021, 52, 440–450. [Google Scholar] [CrossRef] [PubMed]
- Chalkias, H.; Jonas, E.; Andersson, L.S.; Jacobson, M.; de Koning, D.J.; Lundeheim, N.; Lindgren, G. Identification of novel candidate genes for the inverted teat defect in sows using a genome-wide marker panel. J. Appl. Genet. 2017, 58, 249–259. [Google Scholar] [CrossRef]
- He, L.C.; Li, P.H.; Ma, X.; Sui, S.P.; Gao, S.; Kim, S.W.; Gu, Y.Q.; Huang, Y.; Ding, N.S.; Huang, R.H. Identification of new single nucleotide polymorphisms affecting total number born and candidate genes related to ovulation rate in Chinese Erhualian pigs. Anim. Genet. 2017, 48, 48–54. [Google Scholar] [CrossRef]
- Schneider, J.F.; Miles, J.R.; Brown-Brandl, T.M.; Nienaber, J.A.; Rohrer, G.A.; Vallet, J.L. Genomewide association analysis for average birth interval and stillbirth in swine. J. Anim. Sci. 2015, 93, 529–540. [Google Scholar] [CrossRef]
- Chen, J.; Dou, T.; Wu, Z.; Bai, L.; Xu, M.; Zhang, Y.; Yang, S.; Xu, S.; Han, X.; Qiao, R.; et al. Genomic prediction accounting for dominance and epistatic genetic effects on litter size traits in Large White pigs. J. Anim. Sci. 2025, 103, skaf004. [Google Scholar] [CrossRef]
- Esfandyari, H.; Bijma, P.; Henryon, M.; Christensen, O.F.; Sørensen, A.C. Genomic prediction of crossbred performance based on purebred Landrace and Yorkshire data using a dominance model. Genet. Sel. Evol. 2016, 48, 40. [Google Scholar] [CrossRef]
- Zeng, J.; Toosi, A.; Fernando, R.L.; Dekkers, J.C.; Garrick, D.J. Genomic selection of purebred animals for crossbred performance in the presence of dominant gene action. Genet. Sel. Evol. 2013, 45, 11. [Google Scholar] [CrossRef]
- Sun, C.; VanRaden, P.M.; Cole, J.B.; O’Connell, J.R. Improvement of prediction ability for genomic selection of dairy cattle by including dominance effects. PLoS ONE 2014, 9, e103934. [Google Scholar] [CrossRef]
- Vitezica, Z.G.; Varona, L.; Elsen, J.M.; Misztal, I.; Herring, W.; Legarra, A. Genomic BLUP including additive and dominant variation in purebreds and F1 crossbreds, with an application in pigs. Genet. Sel. Evol. 2016, 48, 6. [Google Scholar] [CrossRef] [PubMed]
- Mei, Q.; Vitezica, Z.G.; Li, J.; Zhao, S.; Legarra, A.; Xiang, T. Impacts of additive, dominance, and inbreeding depression effects on genomic evaluation by combining two SNP chips in Canadian Yorkshire pigs bred in China. Genet. Sel. Evol. 2022, 54, 69. [Google Scholar] [CrossRef]
- Zhang, J.; Xiong, Y.; Zuo, B.; Lei, M.; Jiang, S.; Li, F.; Zheng, R.; Li, J.; Xu, D. Detection of quantitative trait loci associated with several internal organ traits and teat number trait in a pig population. J. Genet. Genom. 2007, 34, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Tortereau, F.; Gilbert, H.; Heuven, H.C.; Bidanel, J.P.; Groenen, M.A.; Riquet, J. Combining two Meishan F2 crosses improves the detection of QTL on pig chromosomes 2, 4 and 6. Genet. Sel. Evol. 2010, 42, 42. [Google Scholar] [CrossRef] [PubMed]
- Ogunbawo, A.R.; Hidalgo, J.; Mulim, H.A.; Carrara, E.R.; Ventura, H.T.; Souza, N.O.; Lourenco, D.; Oliveira, H.R. Applying the algorithm for Proven and young in GWAS Reveals high polygenicity for key traits in Nellore cattle. Front. Genet. 2025, 16, 1549284. [Google Scholar] [CrossRef]
- Zachut, M.; Sood, P.; Levin, Y.; Moallem, U. Proteomic analysis of preovulatory follicular fluid reveals differentially abundant proteins in less fertile dairy cows. J. Proteom. 2016, 139, 122–129. [Google Scholar] [CrossRef]
- Ma, Y.; Jin, J.; Tong, X.; Yang, W.; Ren, P.; Dai, Y.; Pan, Y.; Zhang, Y.; Zhang, S. ADAMTS1 and HSPG2 mRNA levels in cumulus cells are related to human oocyte quality and controlled ovarian hyperstimulation outcomes. J. Assist. Reprod. Genet. 2020, 37, 657–667. [Google Scholar] [CrossRef]
- Ghoreishifar, M.; Vahedi, S.M.; Salek Ardestani, S.; Khansefid, M.; Pryce, J.E. Genome-wide assessment and mapping of inbreeding depression identifies candidate genes associated with semen traits in Holstein bulls. BMC Genom. 2023, 24, 230. [Google Scholar] [CrossRef]
- Jia, Y.; Liu, Y.; Li, Y.; Guan, X.; Xu, J.; Yan, Z.; Liu, K.; Zhang, Y.; Bai, D.; Xiang, J.; et al. Persistent Wnt signaling affects IVF embryo implantation and offspring metabolism. Sci. Bull. 2025. [Google Scholar] [CrossRef]
- Zhang, T.; Peng, Z.; Meng, F.; Li, Z.; Chen, J.; Zhou, Q.; Leng, L.; Bo, H.; Lu, G.; Deng, Y.; et al. Maternal transcription factor OTX2 directly induces SETD1A and promotes embryonic genome activation in human pre-implantation embryos. Sci. China Life Sci. 2025. [Google Scholar] [CrossRef]
- Wu, S.J.; Cheng, Y.S.; Liu, H.L.; Wang, H.H.; Huang, H.L. Global transcriptional expression in ovarian follicles from Tsaiya ducks (Anas platyrhynchos) with a high-fertilization rate. Theriogenology 2016, 85, 1439–1445.e1. [Google Scholar] [CrossRef] [PubMed]
- Lang, M.R.; Patterson, L.B.; Gordon, T.N.; Johnson, S.L.; Parichy, D.M. Basonuclin-2 requirements for zebrafish adult pigment pattern development and female fertility. PLoS Genet. 2009, 5, e1000744. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Gong, Y.; Wang, X.; He, X.; He, X.; Chu, M.; Di, R. Screening of Litter-Size-Associated SNPs in NOX4, PDE11A and GHR Genes of Sheep. Animals 2024, 14, 767. [Google Scholar] [CrossRef] [PubMed]
- Petersen, T.S.; Stahlhut, M.; Andersen, C.Y. Phosphodiesterases in the rat ovary: Effect of cAMP in primordial follicles. Reproduction 2015, 150, 11–20. [Google Scholar] [CrossRef]
- Wang, X.Q.; Wang, L.G.; Shi, L.Y.; Tian, J.J.; Li, M.Y.; Wang, L.X.; Zhao, F.P. Imputation strategies for low-coverage whole-genome sequencing data and their effects on genomic prediction and genome-wide association studies in pigs. Animal 2024, 18, 101258. [Google Scholar] [CrossRef]
- Wada, Y.; Akita, T.; Awata, T.; Furukawa, T.; Sugai, N.; Inage, Y.; Ishii, K.; Ito, Y.; Kobayashi, E.; Kusumoto, H.; et al. Quantitative trait loci (QTL) analysis in a Meishan x Göttingen cross population. Anim. Genet. 2000, 31, 376–384. [Google Scholar] [CrossRef]
- Faggion, S.; Bonfatti, V.; Carnier, P. Genome-Wide Association Study for Weight Loss at the End of Dry-Curing of Hams Produced from Purebred Heavy Pigs. Animals 2024, 14, 1983. [Google Scholar] [CrossRef]
- Ahmad, S.F.; Gangwar, M.; Kumar, A.; Kumar, A.; Dige, M.S.; Jha, G.K.; Gaur, G.K.; Dutt, T. Dissecting genomes of multiple yak populations: Unveiling ancestry and high-altitude adaptation through whole-genome resequencing analysis. BMC Genom. 2025, 26, 214. [Google Scholar] [CrossRef]
- Du, H.; Zhou, L.; Liu, Z.; Zhuo, Y.; Zhang, M.; Huang, Q.; Lu, S.; Xing, K.; Jiang, L.; Liu, J.F. The 1000 Chinese Indigenous Pig Genomes Project provides insights into the genomic architecture of pigs. Nat. Commun. 2024, 15, 10137. [Google Scholar] [CrossRef]
- Ma, Y.F.; Han, X.M.; Huang, C.P.; Zhong, L.; Adeola, A.C.; Irwin, D.M.; Xie, H.B.; Zhang, Y.P. Population Genomics Analysis Revealed Origin and High-altitude Adaptation of Tibetan Pigs. Sci. Rep. 2019, 9, 11463. [Google Scholar] [CrossRef]
- Edea, Z.; Dadi, H.; Dessie, T.; Uzzaman, M.R.; Rothschild, M.F.; Kim, E.S.; Sonstegard, T.S.; Kim, K.S. Genome-wide scan reveals divergent selection among taurine and zebu cattle populations from different regions. Anim. Genet. 2018, 49, 550–563. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Zhang, L.; Lin, Q.; Ren, W.; Xu, G. Data on the association of CMPK1 with clinicopathological features and biological effect in human epithelial ovarian cancer. Data Brief. 2017, 13, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Chang, C.; Baiyin, B.; Liu, Z.; Guo, L.; Zhou, L.; Liu, B.; Shi, C.; Zhang, W. Blood Transcriptome Analysis of Beef Cow with Different Parity Revealed Candidate Genes and Gene Networks Regulating the Postpartum Diseases. Genes. 2022, 13, 1671. [Google Scholar] [CrossRef] [PubMed]
- Gòdia, M.; Castelló, A.; Rocco, M.; Cabrera, B.; Rodríguez-Gil, J.E.; Balasch, S.; Lewis, C.; Sánchez, A.; Clop, A. Identification of circular RNAs in porcine sperm and evaluation of their relation to sperm motility. Sci. Rep. 2020, 10, 7985. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Loci | Additive (p) | Dominant (p) | Reproduction QTL (QTL ID) | References | |
|---|---|---|---|---|---|
| 16_156647853 | 5.24 × 10−7 | 0.720151 | 0.096352 | Gestation length (261,966) | Wang et al., 2017 [22] |
| 7_81948941 | 1.64 × 10−7 | 0.843502 | 0.19554 | Teat number (37,446) | Duijvesteijn et al., 2014 [23] |
| 8_4451319 | 2.58 × 10−15 | 0.00373 | 0.004865 | Teat number (223,271) | Bovo et al., 2021 [24] |
| 10_32822679 | 3.64 × 10−7 | 0.424028 | 0.258834 | Nonfunctional nipples (124,209) | Chalkias et al., 2017 [25] |
| 14_107377294 | 1.52 × 10−11 | 0.005087 | 0.002753 | Litter size (130,290) | He et al., 2017 [26] |
| 15_75063953 | 3.91 × 10−9 | 0.049719 | 0.516495 | Number of stillborn (57,518) | Schneider et al., 2015 [27] |
| 16_68542670 | 1.92 × 10−8 | 0.803131 | 0.378008 | Teat number (223,295) | Bovo et al., 2021 [24] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, C.; He, Y.; Ke, J.; Zhang, X.; Fei, J.; Sun, B.; Sun, H.; Bai, C. Genomic Analysis of Reproductive Trait Divergence in Duroc and Yorkshire Pigs: A Comparison of Mixed Models and Selective Sweep Detection. Vet. Sci. 2025, 12, 657. https://doi.org/10.3390/vetsci12070657
Chen C, He Y, Ke J, Zhang X, Fei J, Sun B, Sun H, Bai C. Genomic Analysis of Reproductive Trait Divergence in Duroc and Yorkshire Pigs: A Comparison of Mixed Models and Selective Sweep Detection. Veterinary Sciences. 2025; 12(7):657. https://doi.org/10.3390/vetsci12070657
Chicago/Turabian StyleChen, Changyi, Yu He, Juan Ke, Xiaoran Zhang, Junwen Fei, Boxing Sun, Hao Sun, and Chunyan Bai. 2025. "Genomic Analysis of Reproductive Trait Divergence in Duroc and Yorkshire Pigs: A Comparison of Mixed Models and Selective Sweep Detection" Veterinary Sciences 12, no. 7: 657. https://doi.org/10.3390/vetsci12070657
APA StyleChen, C., He, Y., Ke, J., Zhang, X., Fei, J., Sun, B., Sun, H., & Bai, C. (2025). Genomic Analysis of Reproductive Trait Divergence in Duroc and Yorkshire Pigs: A Comparison of Mixed Models and Selective Sweep Detection. Veterinary Sciences, 12(7), 657. https://doi.org/10.3390/vetsci12070657