
Loss of Myostatin Alters Gut Microbiota and Carbohydrate Metabolism to Influence the Gut–Muscle Axis in Cattle
 , and
, and
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Cecum and Colon Fluid Sampling
2.3. Hematoxylin–Eosin (HE) Staining
2.4. Metagenomic Sequencing and Analysis
2.5. Untargeted Metabolome Analysis
2.6. Serum Glucose and Insulin Measurements
2.7. Transcriptome Analysis
2.8. Data Analysis
3. Results
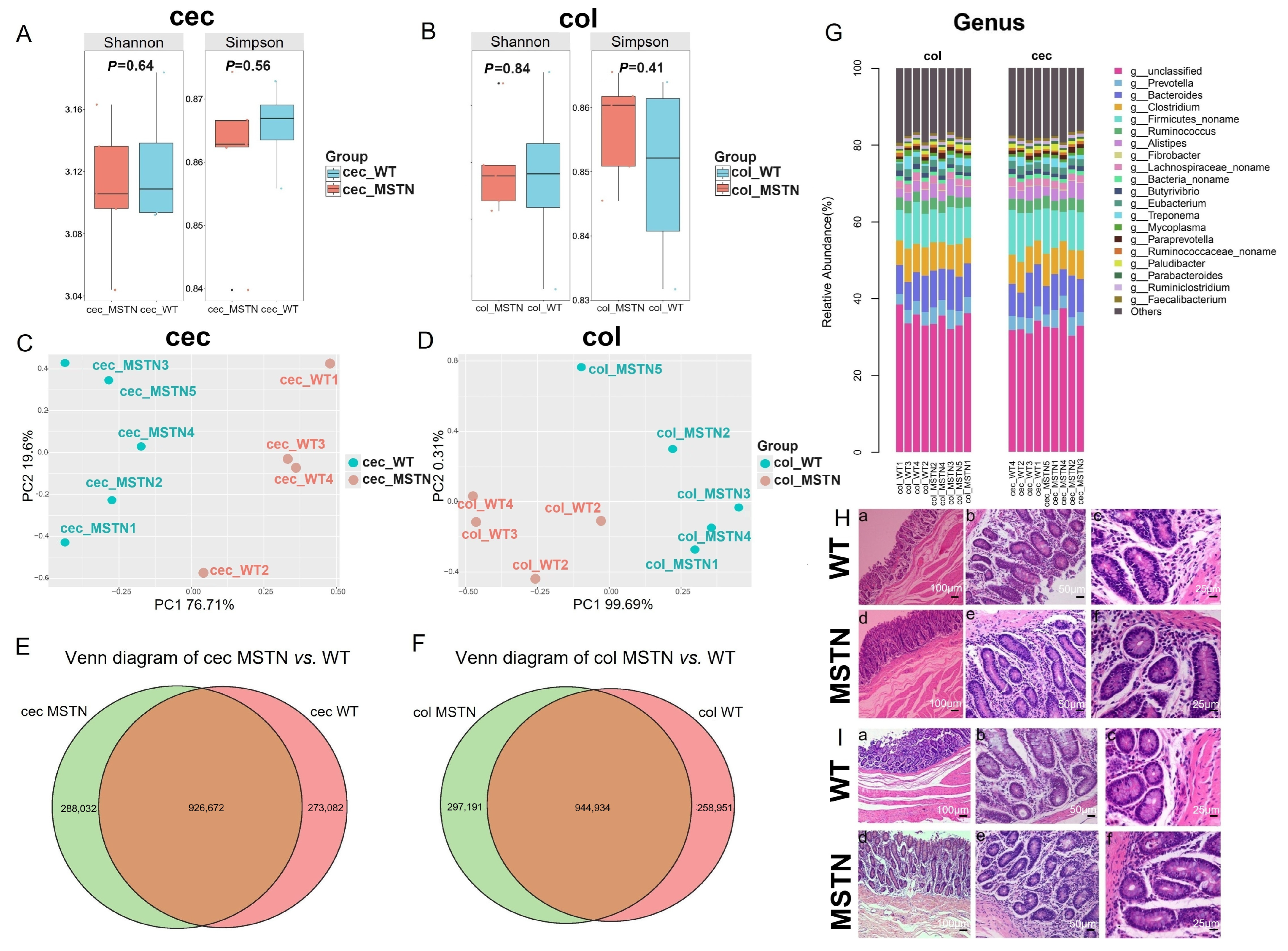
3.1. Characterization of Gut Microbiota and Histological Features in Cecum and Colon
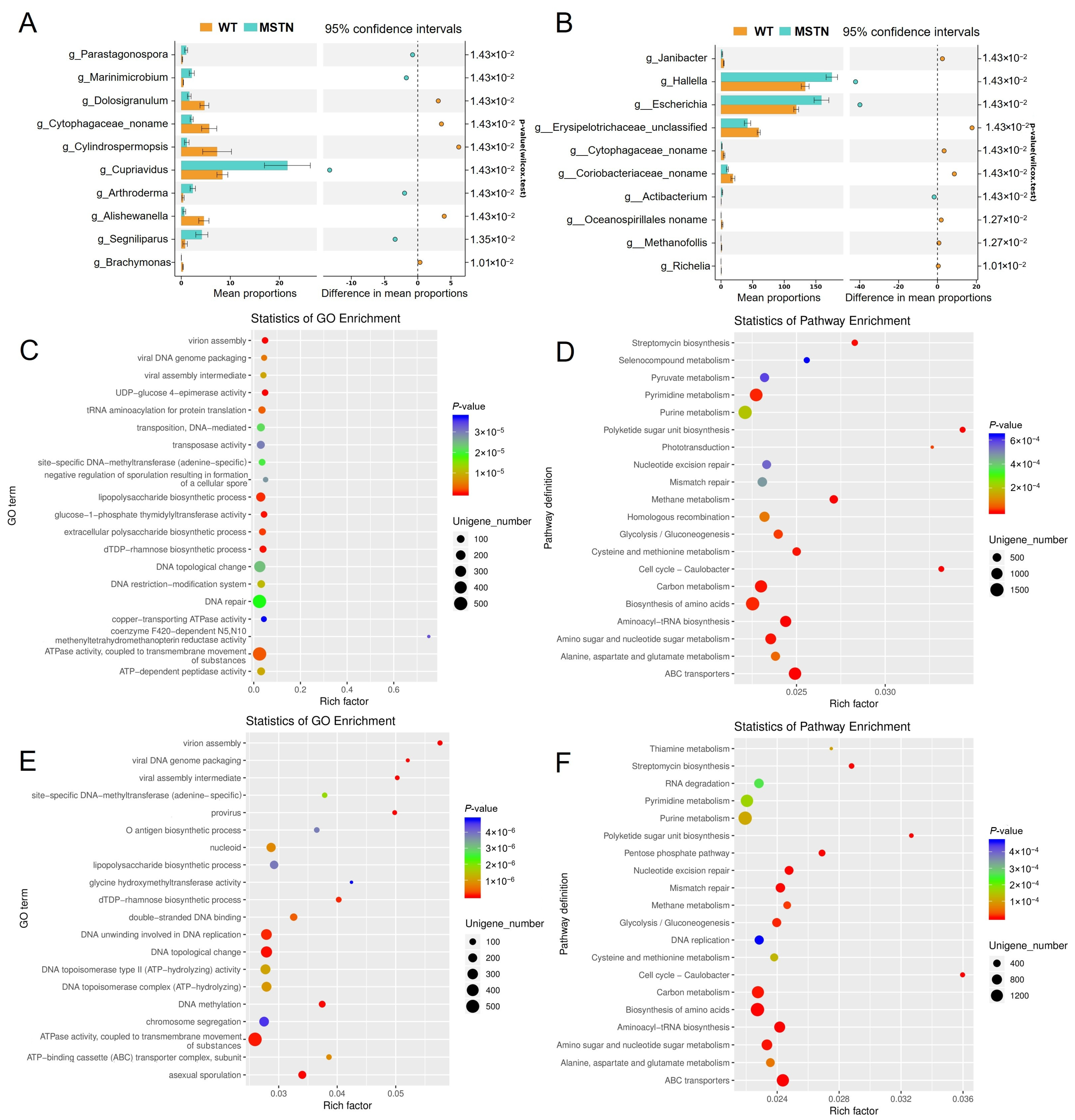
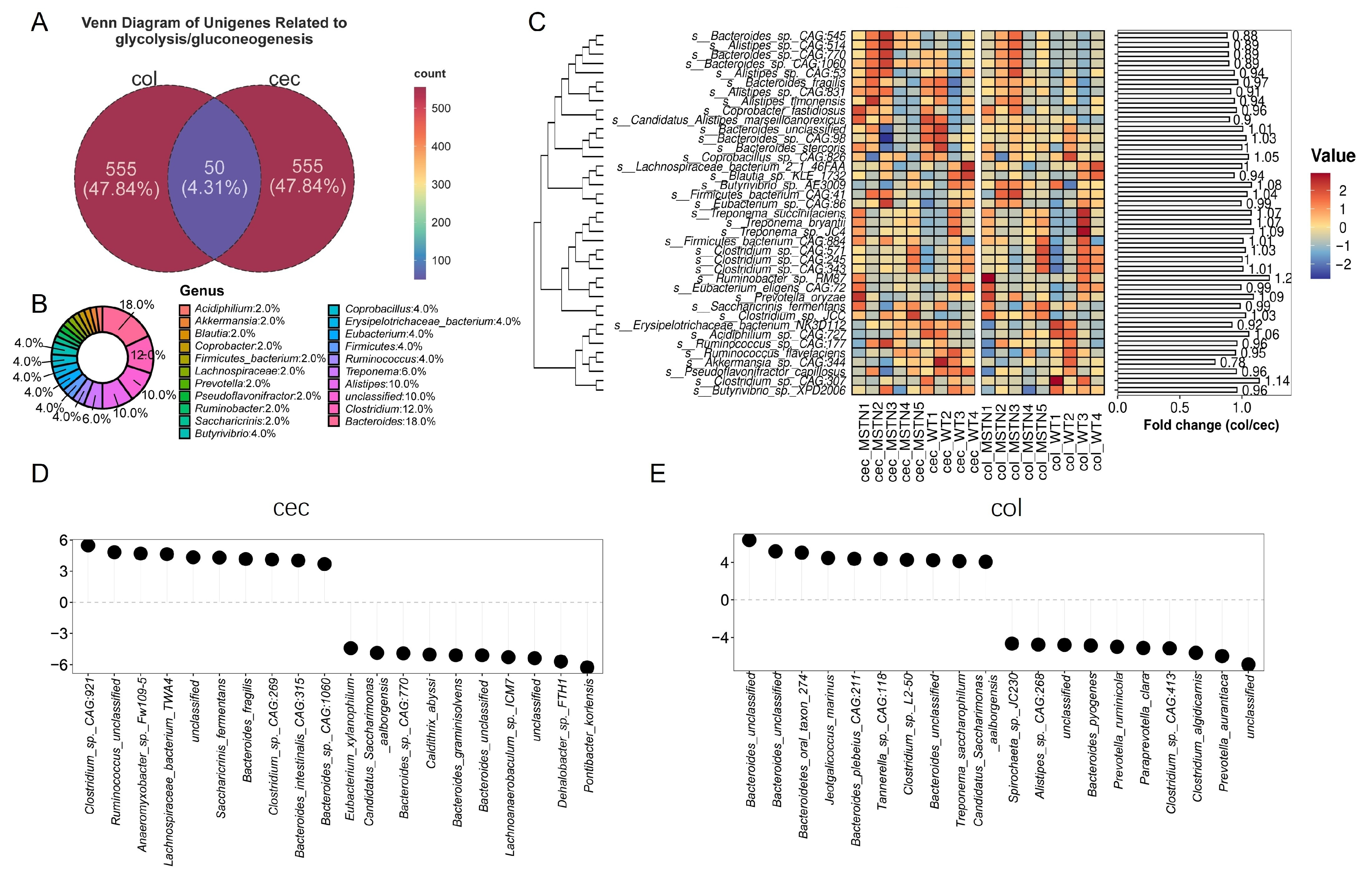
3.2. Differential Microorganisms in the Cecum and Colon Influence the Glycolysis/Gluconeogenesis Pathway
3.3. Bacteroides Is the Key Microbe in the Glycolysis/Gluconeogenesis Pathway
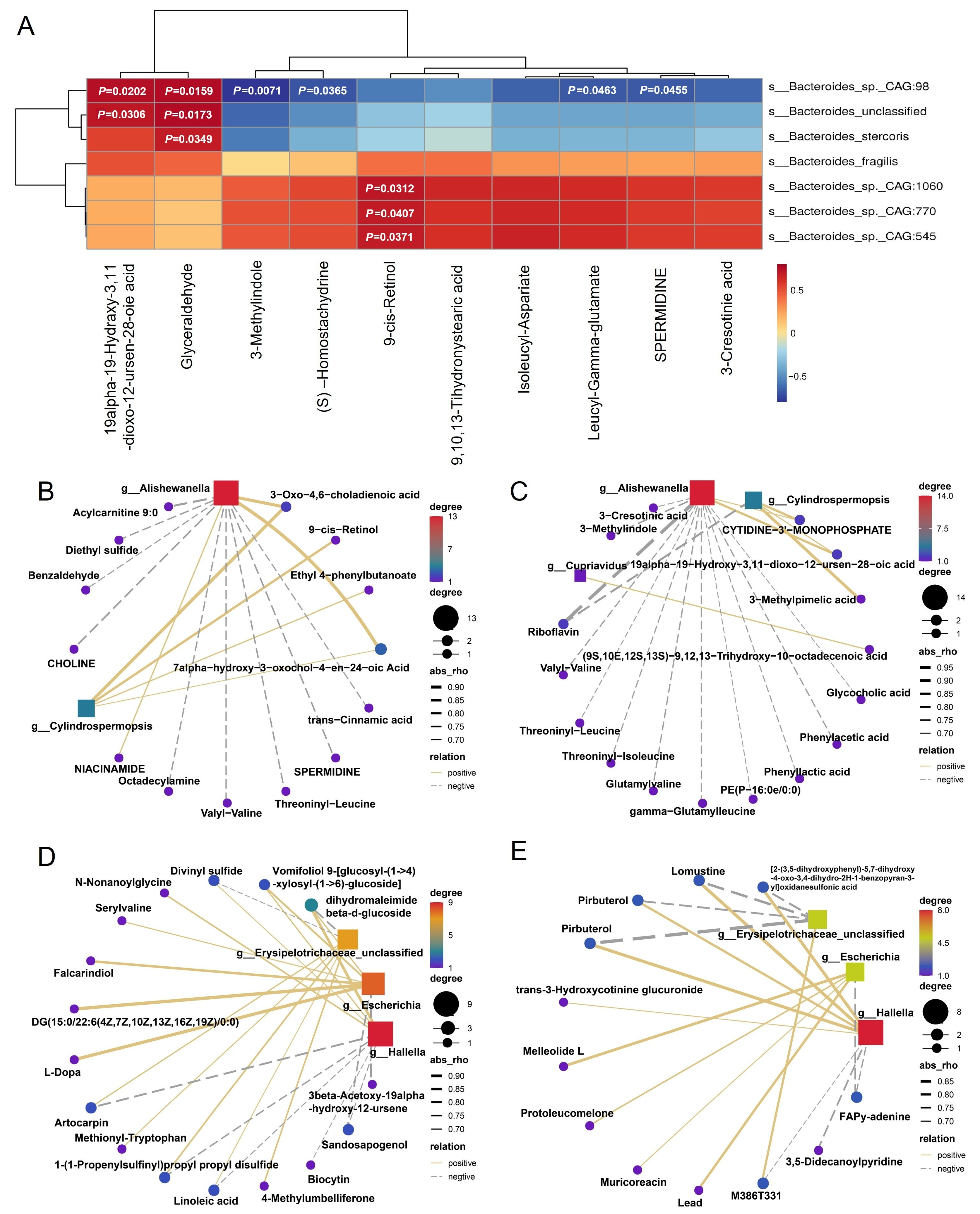
3.4. Metabolomic Profiling and Carbohydrate Metabolism Pathways
3.5. Key Differential Microbes Exhibit Metabolic Links to Carbohydrate Metabolism
3.6. Integration of Transcriptomic Data with Systemic Metabolism
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zafar, H.; Saier, M.H., Jr. Gut Bacteroides species in health and disease. Gut Microbes 2021, 13, 1848158. [Google Scholar] [CrossRef] [PubMed]
- Parker, A.; Fonseca, S.; Carding, S.R. Gut microbes and metabolites as modulators of blood-brain barrier integrity and brain health. Gut Microbes 2020, 11, 135–157. [Google Scholar] [CrossRef]
- Rinninella, E.; Cintoni, M.; Raoul, P.; Lopetuso, L.R.; Scaldaferri, F.; Pulcini, G.; Miggiano, G.A.D.; Gasbarrini, A.; Mele, M.C. Food Components and Dietary Habits: Keys for a Healthy Gut Microbiota Composition. Nutrients 2019, 11, 2393. [Google Scholar] [CrossRef]
- Luo, Z.B.; Han, S.; Yin, X.J.; Liu, H.; Wang, J.; Xuan, M.; Hao, C.; Wang, D.; Liu, Y.; Chang, S.; et al. Fecal transplant from myostatin deletion pigs positively impacts the gut-muscle axis. eLife 2023, 12, e81858. [Google Scholar] [CrossRef]
- Hughes, R.L.; Holscher, H.D. Fueling Gut Microbes: A Review of the Interaction between Diet, Exercise, and the Gut Microbiota in Athletes. Adv. Nutr. 2021, 12, 2190–2215. [Google Scholar] [CrossRef]
- Frampton, J.; Murphy, K.G.; Frost, G.; Chambers, E.S. Short-chain fatty acids as potential regulators of skeletal muscle metabolism and function. Nat. Metab. 2020, 2, 840–848. [Google Scholar] [CrossRef]
- Wang, H.; Feng, L.; Pei, Z.; Zhao, J.; Lu, S.; Lu, W. Gut microbiota metabolism of branched-chain amino acids and their metabolites can improve the physiological function of aging mice. Aging Cell 2025, 24, e14434. [Google Scholar] [CrossRef]
- Boets, E.; Gomand, S.V.; Deroover, L.; Preston, T.; Vermeulen, K.; De Preter, V.; Hamer, H.M.; Van den Mooter, G.; De Vuyst, L.; Courtin, C.M.; et al. Systemic availability and metabolism of colonic-derived short-chain fatty acids in healthy subjects: A stable isotope study. J. Physiol. 2017, 595, 541–555. [Google Scholar] [CrossRef]
- Ma, N.; Tian, Y.; Wu, Y.; Ma, X. Contributions of the Interaction Between Dietary Protein and Gut Microbiota to Intestinal Health. Curr. Protein Pept. Sci. 2017, 18, 795–808. [Google Scholar] [CrossRef]
- Flint, H.J.; Scott, K.P.; Duncan, S.H.; Louis, P.; Forano, E. Microbial degradation of complex carbohydrates in the gut. Gut Microbes 2012, 3, 289–306. [Google Scholar] [CrossRef]
- Yang, M.; Liu, C.; Jiang, N.; Liu, Y.; Luo, S.; Li, C.; Zhao, H.; Han, Y.; Chen, W.; Li, L.; et al. Myostatin: A potential therapeutic target for metabolic syndrome. Front. Endocrinol. 2023, 14, 1181913. [Google Scholar] [CrossRef] [PubMed]
- Yano, S.; Nagai, A.; Isomura, M.; Yamasaki, M.; Kijima, T.; Takeda, M.; Hamano, T.; Nabika, T. Relationship between Blood Myostatin Levels and Kidney Function:Shimane CoHRE Study. PLoS ONE 2015, 10, e0141035. [Google Scholar] [CrossRef]
- Ongaro, L.; Zhou, X.; Wang, Y.; Schultz, H.; Zhou, Z.; Buddle, E.R.S.; Brûlé, E.; Lin, Y.F.; Schang, G.; Hagg, A.; et al. Muscle-derived myostatin is a major endocrine driver of follicle-stimulating hormone synthesis. Science 2025, 387, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Hai, C.; Hao, Z.; Bu, L.; Lei, J.; Liu, X.; Zhao, Y.; Bai, C.; Su, G.; Yang, L.; Li, G. Increased rumen Prevotella enhances BCAA synthesis, leading to synergistically increased skeletal muscle in myostatin-knockout cattle. Commun. Biol. 2024, 7, 1575. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Yang, L.; Su, G.; Wei, Z.; Liu, X.; Song, L.; Hai, C.; Wu, D.; Hao, Z.; Wu, Y.; et al. Growth Traits and Sperm Proteomics Analyses of Myostatin Gene-Edited Chinese Yellow Cattle. Life 2022, 12, 627. [Google Scholar] [CrossRef]
- Feldman, A.T.; Wolfe, D. Tissue processing and hematoxylin and eosin staining. Methods Mol. Biol. 2014, 1180, 31–43. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Pertea, G. fqtrim; v0.9.4 Release; Zenodo: Geneva, Switzerland, 2015. [Google Scholar]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Peng, Y.; Leung, H.C.; Yiu, S.M.; Chin, F.Y. IDBA-UD: A de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 2012, 28, 1420–1428. [Google Scholar] [CrossRef]
- Zhu, W.; Lomsadze, A.; Borodovsky, M. Ab initio gene identification in metagenomic sequences. Nucleic Acids Res. 2010, 38, e132. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.A.; Want, E.J.; O’Maille, G.; Abagyan, R.; Siuzdak, G. XCMS: Processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal. Chem. 2006, 78, 779–787. [Google Scholar] [CrossRef] [PubMed]
- Kuhl, C.; Tautenhahn, R.; Böttcher, C.; Larson, T.R.; Neumann, S. CAMERA: An integrated strategy for compound spectra extraction and annotation of liquid chromatography/mass spectrometry data sets. Anal. Chem. 2012, 84, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Wen, B.; Mei, Z.; Zeng, C.; Liu, S. metaX: A flexible and comprehensive software for processing metabolomics data. BMC Bioinform. 2017, 18, 183. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, D480–D484. [Google Scholar] [CrossRef]
- Drula, E.; Garron, M.L.; Dogan, S.; Lombard, V.; Henrissat, B.; Terrapon, N. The carbohydrate-active enzyme database: Functions and literature. Nucleic Acids Res. 2022, 50, D571–D577. [Google Scholar] [CrossRef]
- Hajishengallis, G.; Reis, E.S.; Mastellos, D.C.; Ricklin, D.; Lambris, J.D. Novel mechanisms and functions of complement. Nat. Immunol. 2017, 18, 1288–1298. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.T.; Xiao, G.J.; Jiang, S.W.; Qian, L.L.; Cai, C.B.; Li, B.; Xie, S.S.; Gao, T.; Li, K. Effect of ZFN-edited myostatin loss-of-function mutation on gut microbiota in Meishan pigs. PLoS ONE 2019, 14, e0210619. [Google Scholar] [CrossRef] [PubMed]
- Du, C.; Zhou, X.; Zhang, K.; Huang, S.; Wang, X.; Zhou, S.; Chen, Y. Inactivation of the MSTN gene expression changes the composition and function of the gut microbiome in sheep. BMC Microbiol. 2022, 22, 273. [Google Scholar] [CrossRef] [PubMed]
- Peterson, L.W.; Artis, D. Intestinal epithelial cells: Regulators of barrier function and immune homeostasis. Nat. Rev. Immunol. 2014, 14, 141–153. [Google Scholar] [CrossRef]
- Cheng, R.; Li, D.; Shi, X.; Gao, Q.; Wei, C.; Li, X.; Li, Y.; Zhou, H. Reduced CX3CL1 Secretion Contributes to the Susceptibility of Oral Leukoplakia-Associated Fibroblasts to Candida albicans. Front. Cell Infect. Microbiol. 2016, 6, 150. [Google Scholar] [CrossRef]
- Fu, X.; Liu, Z.; Zhu, C.; Mou, H.; Kong, Q. Nondigestible carbohydrates, butyrate, and butyrate-producing bacteria. Crit. Rev. Food Sci. Nutr. 2019, 59, S130–S152. [Google Scholar] [CrossRef]
- Mussatto, S.I.; Mancilha, I.M. Non-digestible oligosaccharides: A review. Carbohydr. Polym. 2007, 68, 587–597. [Google Scholar] [CrossRef]
- Morava, E. Galactose supplementation in phosphoglucomutase-1 deficiency; review and outlook for a novel treatable CDG. Mol. Genet. Metab. 2014, 112, 275–279. [Google Scholar] [CrossRef]
- Demirbas, D.; Coelho, A.I.; Rubio-Gozalbo, M.E.; Berry, G.T. Hereditary galactosemia. Metabolism 2018, 83, 188–196. [Google Scholar] [CrossRef]
- Tian, X.; Jiang, H.; Cai, B.; Feng, H.; Wang, X.; Yu, G. Comparative Proteomic Analysis of Fucosylated Glycoproteins Produced by Bacteroides thetaiotaomicron Under Different Polysaccharide Nutrition Conditions. Front. Microbiol. 2022, 13, 826942. [Google Scholar] [CrossRef]
- Shin, J.H.; Tillotson, G.; MacKenzie, T.N.; Warren, C.A.; Wexler, H.M.; Goldstein, E.J.C. Bacteroides and related species: The keystone taxa of the human gut microbiota. Anaerobe 2024, 85, 102819. [Google Scholar] [CrossRef] [PubMed]
- Benítez-Páez, A.; Gómez Del Pulgar, E.M.; Sanz, Y. The Glycolytic Versatility of Bacteroides uniformis CECT 7771 and Its Genome Response to Oligo and Polysaccharides. Front. Cell Infect. Microbiol. 2017, 7, 383. [Google Scholar] [CrossRef] [PubMed]
- Tian, S.; Lei, Y.; Zhao, F.; Che, J.; Wu, Y.; Lei, P.; Kang, Y.E.; Shan, Y. Improving insulin resistance by sulforaphane via activating the Bacteroides and Lactobacillus SCFAs-GPR-GLP1 signal axis. Food Funct. 2024, 15, 8644–8660. [Google Scholar] [CrossRef] [PubMed]
- Nkamga, V.D.; Lotte, R.; Roger, P.M.; Drancourt, M.; Ruimy, R. Methanobrevibacter smithii and Bacteroides thetaiotaomicron cultivated from a chronic paravertebral muscle abscess. Clin. Microbiol. Infect. 2016, 22, 1008–1009. [Google Scholar] [CrossRef]
- Qiu, J.; Zhou, H.; Jing, Y.; Dong, C. Association between blood microbiome and type 2 diabetes mellitus: A nested case-control study. J. Clin. Lab. Anal. 2019, 33, e22842. [Google Scholar] [CrossRef]
- Hai, C.; Bai, C.; Yang, L.; Wei, Z.; Wang, H.; Ma, H.; Ma, H.; Zhao, Y.; Su, G.; Li, G. Effects of Different Generations and Sex on Physiological, Biochemical, and Growth Parameters of Crossbred Beef Cattle by Myostatin Gene-Edited Luxi Bulls and Simmental Cows. Animals 2023, 13, 3216. [Google Scholar] [CrossRef]
- Hitch, T.C.A.; Bisdorf, K.; Afrizal, A.; Riedel, T.; Overmann, J.; Strowig, T.; Clavel, T. A taxonomic note on the genus Prevotella: Description of four novel genera and emended description of the genera Hallella and Xylanibacter. Syst. Appl. Microbiol. 2022, 45, 126354. [Google Scholar] [CrossRef]
- Bai, L.; Paek, J.; Kim, H.; Kim, S.H.; Shin, J.H.; Kook, J.K.; Chang, Y.H. Description and comparative genome analysis of Hallella absiana sp. nov., isolated from pig feces. Anaerobe 2023, 81, 102735. [Google Scholar] [CrossRef]
- Kobaek-Larsen, M.; Nielsen, D.S.; Kot, W.; Krych, Ł.; Christensen, L.P.; Baatrup, G. Effect of the dietary polyacetylenes falcarinol and falcarindiol on the gut microbiota composition in a rat model of colorectal cancer. BMC Res. Notes 2018, 11, 411. [Google Scholar] [CrossRef]
- Nittayananta, W.; Wongwitthayakool, P.; Srichana, T.; Setthanurakkul, C.; Yampuen, P.; Terachinda, P.; Deebunjerd, T.; Tachapiriyakun, J. α-Mangostin and lawsone methyl ether in tooth gel synergistically increase its antimicrobial and antibiofilm formation effects in vitro. BMC Oral. Health 2023, 23, 840. [Google Scholar] [CrossRef]
- Kim, G.S.; Zeng, L.; Alali, F.; Rogers, L.L.; Wu, F.E.; Sastrodihardjo, S.; McLaughlin, J.L. Muricoreacin and murihexocin C, mono-tetrahydrofuran acetogenins, from the leaves of Annona muricata. Phytochemistry 1998, 49, 565–571. [Google Scholar] [CrossRef]
- Min, K.; Ebeler, S.E. Flavonoid effects on DNA oxidation at low concentrations relevant to physiological levels. Food Chem. Toxicol. 2008, 46, 96–104. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Feed Composition | Quality (kg)/Day |
|---|---|
| Silage (kg/head) | 12 |
| Gluten (kg/head) | 2 |
| Hay (bale/head) | 2 |
| Refined feed | 2.5 |
| Nutrients | Composition |
|---|---|
| Crude protein, not less than | 16.0 |
| Crude fat, not more than | 12.0 |
| Crude fiber, not more than | 9.0 |
| Calcium | 0.5–1.8 |
| Total phosphorus, not less than | 0.4 |
| Sodium chloride | 0.8–1.5 |
| Lysine, not less than | 0.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hai, C.; Gong, H.; Xu, Y.; Liu, X.; Bai, C.; Su, G.; Yang, L.; Li, G. Loss of Myostatin Alters Gut Microbiota and Carbohydrate Metabolism to Influence the Gut–Muscle Axis in Cattle. Vet. Sci. 2025, 12, 560. https://doi.org/10.3390/vetsci12060560
Hai C, Gong H, Xu Y, Liu X, Bai C, Su G, Yang L, Li G. Loss of Myostatin Alters Gut Microbiota and Carbohydrate Metabolism to Influence the Gut–Muscle Axis in Cattle. Veterinary Sciences. 2025; 12(6):560. https://doi.org/10.3390/vetsci12060560
Chicago/Turabian StyleHai, Chao, Hongyu Gong, Yanan Xu, Xuefei Liu, Chunling Bai, Guanghua Su, Lei Yang, and Guangpeng Li. 2025. "Loss of Myostatin Alters Gut Microbiota and Carbohydrate Metabolism to Influence the Gut–Muscle Axis in Cattle" Veterinary Sciences 12, no. 6: 560. https://doi.org/10.3390/vetsci12060560
APA StyleHai, C., Gong, H., Xu, Y., Liu, X., Bai, C., Su, G., Yang, L., & Li, G. (2025). Loss of Myostatin Alters Gut Microbiota and Carbohydrate Metabolism to Influence the Gut–Muscle Axis in Cattle. Veterinary Sciences, 12(6), 560. https://doi.org/10.3390/vetsci12060560