
No Genomic Signatures Were Found in Escherichia coli Isolates from Camels With or Without Clinical Endometritis

Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Genome Sequencing
2.2.1. Identification and Typing of Isolates
2.2.2. Mobilome and Virulome
2.2.3. Phylogenomic
2.2.4. Antibiotic Susceptibility Testing
2.2.5. Statistical Analysis
3. Results
3.1. ANI, Phylogroups, Serotype, and MLST Analysis
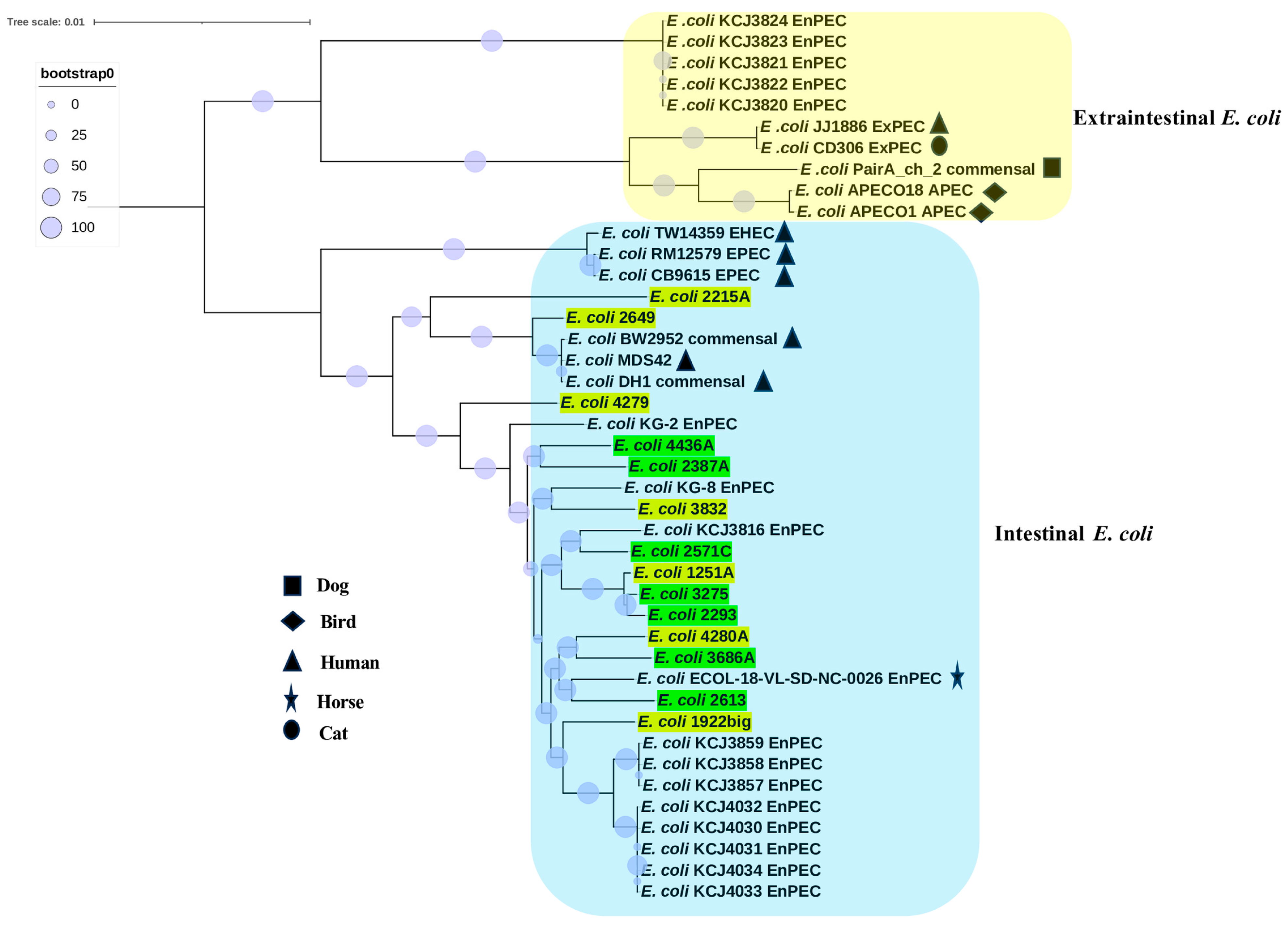
3.2. Phylogenomic Analyses
3.3. Antibacterial Susceptibility Profile
3.4. Virulome
3.5. Comparative Analysis of Functional Characteristics
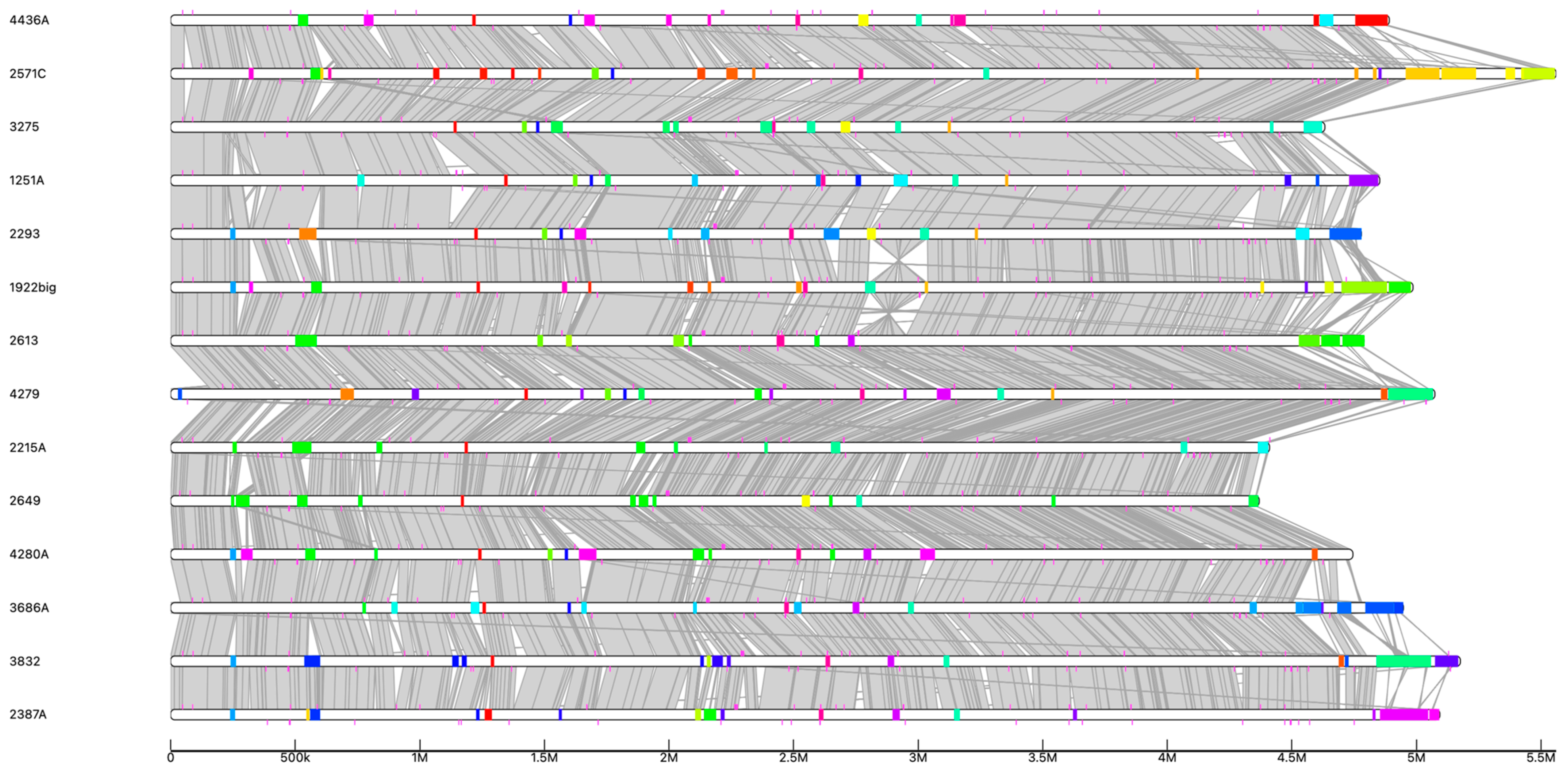
3.6. Mobilome Analysis
4. Discussion
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ali, A.; Derar, D.R. Clinical and subclinical endometritis in dromedary camels: An overview of definition and clinical presentation, etiopathogenesis, diagnostic biomarkers, and treatment protocols. Anim. Reprod. Sci. 2023, 257, 107328. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Derar, D.; Alsharari, A.; Alsharari, A.; Khalil, R.; Almundarij, T.I.; Alboti, Y.; Al-Sobayil, F. Factors affecting reproductive performance in dromedary camel herds in Saudi Arabia. Trop. Anim. Health Prod. 2018, 50, 1155–1160. [Google Scholar] [CrossRef]
- Russo, T.A.; Johnson, J.R. Proposal for a New Inclusive Designation for Extraintestinal Pathogenic Isolates of Escherichia coli: ExPEC. J. Infect. Dis. 2000, 181, 1753–1754. [Google Scholar] [CrossRef]
- Schwidder, M.; Heinisch, L.; Schmidt, H. Genetics, Toxicity, and Distribution of Enterohemorrhagic Escherichia coli Hemolysin. Toxins 2019, 11, 502. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Feuille, C. Enteropathogenic Escherichia coli (EPEC) Causes Chronic Diarrhea and Hyponatremia in an Adult. Clin. Case Rep. 2025, 13, e70288. [Google Scholar] [CrossRef] [PubMed]
- von Mentzer, A.; Svennerholm, A.M. Colonization factors of human and animal-specific enterotoxigenic Escherichia coli (ETEC). Trends Microbiol. 2024, 32, 448–464. [Google Scholar] [CrossRef]
- Terlizzi, M.E.; Gribaudo, G.; Maffei, M.E. UroPathogenic Escherichia coli (UPEC) infections: Virulence factors, bladder responses, antibiotic, and non-antibiotic antimicrobial strategies. Front. Microbiol. 2017, 8, 1566. [Google Scholar] [CrossRef]
- Nawaz, S.; Wang, Z.; Zhang, Y.; Jia, Y.; Jiang, W.; Chen, Z.; Yin, H.; Huang, C.; Han, X. Avian pathogenic Escherichia coli (APEC): Current insights and future challenges. Poult. Sci. 2024, 103, 104359. [Google Scholar] [CrossRef]
- Goldstone, R.J.; Popat, R.; Schuberth, H.J.; Sandra, O.; Sheldon, I.M.; Smith, D.G. Genomic characterisation of an endometrial pathogenic Escherichia coli strain reveals the acquisition of genetic elements associated with extra-intestinal pathogenicity. BMC Genom. 2014, 15, 1075. [Google Scholar] [CrossRef]
- El-Deeb, W.; Abdelghani, M.A.; Alhaider, A.; Fayez, M. Oxidative stress, ceruloplasmin and neopterin biomarkers in dromedary camels with clinical endometritis. Anim. Reprod. 2022, 19, e20220035. [Google Scholar] [CrossRef]
- Elshazly, M.O.; El-Rahman, S.S.A.; Hamza, D.A.; Ali, M.E. Pathological and bacteriological studies on reproductive tract abnormalities of she-camels (Camelus dromedarius), emphasizing on zoonotic importance. J. Adv. Veter-Anim. Res. 2020, 7, 633–646. [Google Scholar] [CrossRef] [PubMed]
- Getahun, A.M.; Hunderra, G.C.; Gebrezihar, T.G.; Boru, B.G.; Desta, N.T.; Ayana, T.D. Comparative study on lesions of reproductive disorders of cows and female dromedary camels slaughtered at Addis Ababa, Adama and Akaki abattoirs with bacterial isolation and characterization. BMC Vet. Res. 2021, 17, 134. [Google Scholar] [CrossRef]
- Belina, D.; Eshetu, A.; Alemu, S.; Shasho, B.; Mohammed, T.; Mohammed, A.; Mummed, B.; Regassa, D. Reproductive Diseases and Disorders of Female Camels: An Assessment and Pathological and Bacteriological Study in Eastern Ethiopia. Veter. Med. Int. 2021, 2021, 6641361. [Google Scholar] [CrossRef]
- Piersanti, R.L.; Zimpel, R.; Molinari, P.C.; Dickson, M.J.; Ma, Z.; Jeong, K.C.; Santos, J.E.; Sheldon, I.M.; Bromfield, J.J. A model of clinical endometritis in Holstein heifers using pathogenic Escherichia coli and Trueperella pyogenes. J. Dairy Sci. 2019, 102, 2686–2697. [Google Scholar] [CrossRef] [PubMed]
- Bicalho, R.C.; Machado, V.S.; Bicalho, M.L.; Gilbert, R.O.; Teixeira, A.G.; Caixeta, L.S.; Pereira, R.V. Molecular and epidemiological characterization of bovine intrauterine Escherichia coli. J. Dairy Sci. 2010, 93, 5818–5830. [Google Scholar] [CrossRef]
- Kassé, F.N.; Fairbrother, J.M.; Dubuc, J. Relationship between Escherichia coli Virulence Factors and Postpartum Metritis in Dairy Cows. J. Dairy Sci. 2016, 99, 4656–4667. [Google Scholar] [CrossRef] [PubMed]
- Garzon, A.; Basbas, C.; Schlesener, C.; Silva-Del-Rio, N.; Karle, B.M.; Lima, F.S.; Weimer, B.C.; Pereira, R.V. WGS of intrauterine E. coli from cows with early postpartum uterine infection reveals a non-uterine specific genotype and virulence factors. mBio 2024, 15, e0102724. [Google Scholar] [CrossRef]
- Sapountzis, P.; Segura, A.; Desvaux, M.; Forano, E. An Overview of the Elusive Passenger in the Gastrointestinal Tract of Cattle: The Shiga Toxin Producing Escherichia coli. Microorganisms 2020, 8, 877. [Google Scholar] [CrossRef]
- Ochman, H.; Lawrence, J.G.; Groisman, E.A. Lateral gene transfer and the nature of bacterial innovation. Nature 2000, 405, 299–304. [Google Scholar] [CrossRef]
- Gyles, C.; Boerlin, P. Horizontally transferred genetic elements and their role in pathogenesis of bacterial disease. Vet. Pathol. 2014, 51, 328–340. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 22 September 2022).
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG Tools for Functional Characterization of Genome and Metagenome Sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.; Ouk Kim, Y.; Park, S.C.; Chun, J. OrthoANI: An improved algorithm and software for calculating average nucleotide identity. Int. J. Syst. Evol. Microbiol. 2016, 66, 1100–1103. [Google Scholar] [CrossRef]
- Chun, J.; Oren, A.; Ventosa, A.; Christensen, H.; Arahal, D.R.; da Costa, M.S.; Rooney, A.P.; Yi, H.; Xu, X.W.; De Meyer, S.; et al. Proposed minimal standards for the use of genome data for the taxonomy of prokaryotes. Int. J. Syst. Evol. Microbiol. 2018, 68, 461–466. [Google Scholar] [CrossRef]
- Jolley, K.A.; Bray, J.E.; Maiden, M.C.J. Open-access bacterial population genomics: BIGSdb software, the PubMLST.org website and their applications. Wellcome Open Res. 2018, 3, 124. [Google Scholar] [CrossRef]
- Waters, N.R.; Abram, F.; Brennan, F.; Holmes, A.; Pritchard, L. Easy phylotyping of Escherichia coli via the EzClermont web app and command-line tool. Access Microbiol. 2020, 2, acmi000143. [Google Scholar] [CrossRef]
- Joensen, K.G.; Tetzschner, A.M.M.; Iguchi, A.; Aarestrup, F.M.; Scheutz, F. Rapid and easy in silico serotyping of Escherichia coli isolates by use of whole-genome sequencing data. J. Clin. Microbiol. 2015, 53, 2410–2426. [Google Scholar] [CrossRef]
- Wishart, D.S.; Han, S.; Saha, S.; Oler, E.; Peters, H.; Grant, J.R.; Stothard, P.; Gautam, V. PHASTEST: Faster than PHASTER, better than PHAST. Nucleic Acids Res. 2023, 51, W443–W450. [Google Scholar] [CrossRef]
- Bertelli, C.; Laird, M.R.; Williams, K.P.; Lau, B.Y.; Hoad, G.; Winsor, G.L.; Brinkman, F.S. IslandViewer 4: Expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 2017, 45, W30–W35. [Google Scholar] [CrossRef]
- Bertelli, C.; Gray, K.L.; Woods, N.; Lim, A.C.; Tilley, K.E.; Winsor, G.L.; Hoad, G.R.; Roudgar, A.; Spencer, A.; Peltier, J.; et al. Enabling genomic island prediction and comparison in multiple genomes to investigate bacterial evolution and outbreaks. Microb. Genom. 2022, 8, 000818. [Google Scholar] [CrossRef] [PubMed]
- Johansson, M.H.K.; Bortolaia, V.; Tansirichaiya, S.; Aarestrup, F.M.; Roberts, A.P.; Petersen, T.N. Detection of Mobile Genetic Elements Associated with Antibiotic Resistance in Salmonella Enterica Using a Newly Developed Web Tool: MobileElementFinder. J. Antimicrob. Chemother. 2020, 76, 101–109. [Google Scholar] [CrossRef]
- Liu, M.; Li, X.; Xie, Y.; Bi, D.; Sun, J.; Li, J.; Tai, C.; Deng, Z.; Ou, H.-Y. ICEberg 2.0: An updated database of bacterial integrative and conjugative elements. Nucleic Acids Res. 2019, 47, D660–D665. [Google Scholar] [CrossRef] [PubMed]
- Contreras-Moreira, B.; Vinuesa, P. GET_HOMOLOGUES, a versatile software package for scalable and robust microbial pangenome analysis. Appl. Environ. Microbiol. 2013, 79, 7696–7701. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; Von Haeseler, A.; Lanfear, R. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree of Life (ITOL) v6: Recent Updates to the Phylogenetic Tree Display and Annotation Tool. Nucleic Acids Res. 2024, 52, W78–W82. [Google Scholar] [CrossRef] [PubMed]
- Matuschek, E.; Brown, D.F.; Kahlmeter, G. Development of the EUCAST disk diffusion antimicrobial susceptibility testing method and its implementation in routine microbiology laboratories. Clin. Microbiol. Infect. 2014, 4, O255–O266. [Google Scholar] [CrossRef]
- Feldgarden, M.; Brover, V.; Gonzalez-Escalona, N.; Frye, J.G.; Haendiges, J.; Haft, D.H.; Hoffmann, M.; Pettengill, J.B.; Prasad, A.B.; Tillman, G.E.; et al. AMRFinderPlus and the Reference Gene Catalog Facilitate Examination of the Genomic Links among Antimicrobial Resistance, Stress Response, and Virulence. Sci. Rep. 2021, 11, 12728. [Google Scholar] [CrossRef]
- Mahony, M.; McMullan, B.; Brown, J.; Kennedy, S.E. Multidrug-resistant organisms in urinary tract infections in children. Pediatr. Nephrol. 2020, 35, 1563–1573. [Google Scholar] [CrossRef]
- Sheldon, I.M.; Rycroft, A.N.; Dogan, B.; Craven, M.; Bromfield, J.J.; Chandler, A.; Roberts, M.H.; Price, S.B.; Gilbert, R.O.; Simpson, K.W. Specific strains of Escherichia coli are pathogenic for the endometrium of cattle and cause pelvic inflammatory disease in cattle and mice. PLoS ONE 2010, 5, e9192. [Google Scholar] [CrossRef] [PubMed]
- Henriques, S.; Silva, E.; Lemsaddek, A.; Lopes-da-Costa, L.; Mateus, L. Genotypic and phenotypic comparison of Escherichia coli from uterine infections with different outcomes: Clinical metritis in the cow and pyometra in the bitch. Vet. Microbiol. 2014, 170, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Nocera, F.P.; Maurizi, L.; Masullo, A.; Nicoletti, M.; Conte, A.L.; Brunetti, F.; De Martino, L.; Zagaglia, C.; Longhi, C. Genotypic and Phenotypic Characterization of Escherichia coli Isolates Recovered from the Uterus of Mares with Fertility Problems. Animals 2023, 13, 1639. [Google Scholar] [CrossRef]
- Lopes, C.E.; De Carli, S.; Weber, M.N.; Fonseca, A.C.V.; Tagliari, N.J.; Foresti, L.; Cibulski, S.P.; Mayer, F.Q.; Canal, C.W.; Siqueira, F.M. Insights on the Genetic Features of Endometrial Pathogenic Escherichia coli Strains from Pyometra in Companion Animals: Improving the Knowledge about Pathogenesis. Infect. Genet. Evol. 2020, 85, 104453. [Google Scholar] [CrossRef]
- Picard, B.; Garcia, J.S.; Gouriou, S.; Duriez, P.; Brahimi, N.; Bingen, E.; Elion, J.; Denamur, E. The link between phylogeny and virulence in Escherichia coli extraintestinal infection. Infect. Immun. 1999, 67, 546–553. [Google Scholar] [CrossRef]
- Beghain, J.; Bridier-Nahmias, A.; Le Nagard, H.; Denamur, E.; Clermont, O. ClermonTyping: An easy-to-use and accurate in silico method for Escherichia genus strain phylotyping. Microb. Genom. 2018, 4, e000192. [Google Scholar] [CrossRef]
- Lawrence, J.G.; Ochman, H. Molecular archaeology of the Escherichia coli genome. Proc. Natl. Acad. Sci. USA 1998, 95, 9413–9417. [Google Scholar] [CrossRef]
- Hershberg, R.; Tang, H.; Petrov, D.A. Reduced selection leads to accelerated gene loss in Shigella. Genome Biol. 2007, 8, R164. [Google Scholar] [CrossRef] [PubMed]
- McKinnon, J.; Chowdhury, P.R.; Djordjevic, S.P. Genomic analysis of multidrug-resistant Escherichia coli ST58 causing urosepsis. Int. J. Antimicrob. Agents 2018, 52, 430–435. [Google Scholar] [CrossRef]
- Flores-Oropeza, M.A.; Ochoa, S.A.; Cruz-Córdova, A.; Chavez-Tepecano, R.; Martínez-Peñafiel, E.; Rembao-Bojórquez, D.; Zavala-Vega, S.; Hernández-Castro, R.; Flores-Encarnacion, M.; Arellano-Galindo, J.; et al. Comparative genomic analysis of uropathogenic Escherichia coli strains from women with recurrent urinary tract infection. Front. Microbiol. 2024, 14, 1340427. [Google Scholar] [CrossRef]
- Chiluisa-Guacho, C.; Escobar-Perez, J.; Dutra-Asensi, M. First detection of the CTXM-15 producing Escherichia coli O25-ST131 pandemic clone in Ecuador. Pathogens 2018, 7, 42. [Google Scholar] [CrossRef] [PubMed]
- Poh, C.Y.; Rodwell, E.V.; Godbole, G.; Jenkins, C. Genotypic analysis of Shiga toxin-producing Escherichia coli clonal complex 17 in England and Wales. J. Med. Microbiol. 2024, 73, 001928. [Google Scholar] [CrossRef] [PubMed]
- Bando, S.Y.; Iamashita, P.; Guth, B.E.; Dos Santos, L.F.; Fujita, A.; Abe, C.M.; Ferreira, L.R.; Moreira-Filho, C.A. A hemolytic-uremic syndrome-associated strain O113:H21 Shiga toxin-producing Escherichia coli specifically expresses a transcriptional module containing dicA and is related to gene network dysregulation in Caco-2 cells. PLoS ONE 2017, 12, e0189613. [Google Scholar] [CrossRef]
- Uhlich, G.A.; Gunther, N.W.; Bayles, D.O.; Mosier, D.A. The CsgA and Lpp Proteins of an Escherichia coli O157:H7 Strain Affect HEp-2 Cell Invasion, Motility, and Biofilm Formation. Infect. Immun. 2009, 77, 1543–1552. [Google Scholar] [CrossRef]
- Barnich, N.; Bringer, M.A.; Claret, L.; Darfeuille-Michaud, A. Involvement of lipoprotein NlpI in the virulence of adherent invasive Escherichia coli strain LF82 isolated from a patient with Crohn’s disease. Infect. Immun. 2004, 72, 2484–2493. [Google Scholar] [CrossRef] [PubMed]
- Lai, X.H.; Arencibia, I.; Johansson, A.; Wai, S.N.; Oscarsson, J.; Kalfas, S.; Sundqvist, K.G.; Mizunoe, Y.; Sjostedt, A.; Uhlin, B.E. Cytocidal and apoptotic effects of the ClyA protein from Escherichia coli on primary and cultured monocytes and macrophages. Infect. Immun. 2000, 68, 4363–4367. [Google Scholar] [CrossRef]
- May, K.; Sames, L.; Scheper, C.; König, S. Genomic loci and genetic parameters for uterine diseases in first-parity Holstein cows and associations with milk production and fertility. J. Dairy Sci. 2022, 105, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Moore, S.; Ericsson, A.; Poock, S.; Melendez, P.; Lucy, M. Hot topic: 16S rRNA gene sequencing reveals the microbiome of the virgin and pregnant bovine uterus. J. Dairy Sci. 2017, 100, 4953–4960. [Google Scholar] [CrossRef]
- Galvão, K.N.; Bicalho, R.C.; Jeon, S.J. Symposium review: The uterine microbiome associated with the development of uterine disease in dairy cows. J. Dairy Sci. 2019, 102, 11786–11797. [Google Scholar] [CrossRef]
- Wang, M.L.; Liu, M.C.; Xu, J.; An, L.G.; Wang, J.F.; Zhu, Y.H. Uterine microbiota of dairy cows with clinical and subclinical endometritis. Front. Microbiol. 2018, 9, 2691. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Strains | OrthoANI | Genome Size (bp) | Genome CDS | Plasmidome Size (bp) | Plasmidome CDS | Serogroup | ST | Phylogroup | Accession Number |
|---|---|---|---|---|---|---|---|---|---|
| CE isolates | |||||||||
| 2649 | 99.6 | 4,369,294 | 4040 | None | None | H33 | 13358 | A | JBKIBG000000000 |
| 2215A | 98.5 | 4,410,531 | 4069 | 16,186 | 20 | O51:H15 | 5044 | A | JBKIBF000000000 |
| 4280A | 98.5 | 4,745,178 | 4418 | None | None | O188:H7 | 180 | B1 | JBKIBJ000000000 |
| 1251A | 98.5 | 4,853,119 | 4545 | 39,940 | 38 | O8:H21 | 58 | B1 | JBKIBE000000000 |
| 1922big | 98.5 | 4,986,463 | 4648 | 182,430 | 202 | O167:H7 | 707 | B1 | JBKIBD000000000 |
| 4279 | 98.7 | 5,074,171 | 4764 | 129,640 | 142 | O8:H4 | 88 | C | JBKIBI000000000 |
| 3832 | 98.6 | 5,176,518 | 4898 | 144,257 | 155 | O181:H49 | 173 | B1 | JBKIBH000000000 |
| Non-CE isolates | |||||||||
| 3275 | 98.5 | 4,632,655 | 4302 | 21,459 | 32 | O50:H8 | 58 | B1 | JBKIBO000000000 |
| 2293 | 98.6 | 4,779,125 | 4481 | 74,255 | 83 | O9:H12 | 58 | B1 | JBKIBK000000000 |
| 2613 | 98.6 | 4,790,025 | 4440 | 136,941 | 167 | O188:H40 | 2520 | B1 | JBKIBN000000000 |
| 4436A | 98.5 | 4,891,287 | 4577 | 6966 | 7 | H11 | 295 | B1 | JBKIBQ000000000 |
| 3686A | 98.5 | 4,947,877 | 4674 | 178,641 | 219 | O74:H28 | 2164 | B1 | JBKIBP000000000 |
| 2387A | 98.5 | 5,166,692 | 4925 | 64,099 | 71 | O103:H2 | 17 | B1 | JBKIBL000000000 |
| 2571C | 98.5 | 5,560,896 | 5213 | 69,119 | 86 | O113:H21 | 3695 | B1 | JBKIBM000000000 |
| Isolates | Typing | Disk Diffusion Method | Resistance Genes | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Serogroup | ST | Phylogroup | Antibiotics | Ceftazidime | Streptomycin | Tetracycline | strA | strB | dfrA1 | dfrA5 | sul2 | tetA | |
| CE isolates | |||||||||||||
| 2649 | H33 | 13358 | A | S | S | S | S | ||||||
| 2215A | O51:H15 | 5044 | A | S | S | S | S | ||||||
| 4280A | O188:H7 | 180 | B1 | S | R | R | R | + | + | + | + | + | |
| 1251A | O8:H21 | 58 | B1 | S | S | S | S | ||||||
| 1922big | O167:H7 | 707 | B1 | S | S | S | R | + | |||||
| 4279 | O8:H4 | 88 | C | S | S | R | R | + | + | + | + | ||
| 3832 | O181:H49 | 173 | B1 | S | S | S | R | + | + | ||||
| Non-CE isolates | |||||||||||||
| 3275 | O50:H8 | 58 | B1 | S | S | S | S | ||||||
| 2293 | O9:H12 | 58 | B1 | S | R | S | S | ||||||
| 2613 | O188:H40 | 2520 | B1 | S | R | R | R | + | + | + | + | + | |
| 4436A | H11 | 295 | B1 | S | S | S | S | ||||||
| 3686A | O74:H28 | 2164 | B1 | S | S | S | S | ||||||
| 2387A | O103:H2 | 17 | B1 | S | S | S | S | ||||||
| 2571C | O113:H21 | 3695 | B1 | S | S | S | S | ||||||
| Strain | 4436A | 2571C | 2613 | 3686 | 2293 | 3275 | 2387A | 3832 | 4280A | 1922big | 1251A | 2215a | 4279 | 2649 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Group | Non-CE isolates | CE isolates | |||||||||||||
| MST typing | 295 | 3695 | 2520 | 2164 | 58 | 58 | 17 | 173 | 180 | 707 | 58 | 5044 | 88 | 13358 | |
| Phylogroup | B1 | B1 | B1 | B1 | B1 | B1 | B1 | B1 | B1 | B1 | B1 | A | C | A | |
| Adhesin | csgA | ||||||||||||||
| fdeC | |||||||||||||||
| fimH | |||||||||||||||
| hra | |||||||||||||||
| iha | |||||||||||||||
| lpfA | |||||||||||||||
| nlpI | |||||||||||||||
| tir | |||||||||||||||
| yehA | |||||||||||||||
| yehB | |||||||||||||||
| yehC | |||||||||||||||
| yehD | |||||||||||||||
| Invasion | AslA | ||||||||||||||
| tia | |||||||||||||||
| Iron uptake | fyuA | ||||||||||||||
| ireA | |||||||||||||||
| iroN | |||||||||||||||
| irp2 | |||||||||||||||
| iucC | |||||||||||||||
| iutA | |||||||||||||||
| sitA | |||||||||||||||
| Toxins | astA | ||||||||||||||
| eae-e01-epsilon | |||||||||||||||
| hlyE | |||||||||||||||
| hlyF | |||||||||||||||
| senB | |||||||||||||||
| stx2b-O174-031 | |||||||||||||||
| Protectins | capU | ||||||||||||||
| cba | |||||||||||||||
| cea | |||||||||||||||
| cia | |||||||||||||||
| cma | |||||||||||||||
| colE7 | |||||||||||||||
| cvaC | |||||||||||||||
| espA | |||||||||||||||
| espB | |||||||||||||||
| espF | |||||||||||||||
| espJ | |||||||||||||||
| etpD | |||||||||||||||
| etsC | |||||||||||||||
| hha | |||||||||||||||
| iss | |||||||||||||||
| mchB | |||||||||||||||
| mchC | |||||||||||||||
| mchF | |||||||||||||||
| mcmA | |||||||||||||||
| ompT | |||||||||||||||
| shiA | |||||||||||||||
| shiB | |||||||||||||||
| traJ | |||||||||||||||
| traT | |||||||||||||||
| Others | aamR:FN554766 | ||||||||||||||
| anr | |||||||||||||||
| gad | |||||||||||||||
| nleA | |||||||||||||||
| nleB | |||||||||||||||
| tccP | |||||||||||||||
| terC | |||||||||||||||
| % genes in GIs | 40 | 42.9 | 53.8 | 37.5 | 50 | 30.8 | 65.4 | 78.8 | 46.2 | 63.2 | 58.8 | 57.1 | 64.3 | 33.3 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elbir, H. No Genomic Signatures Were Found in Escherichia coli Isolates from Camels With or Without Clinical Endometritis. Vet. Sci. 2025, 12, 457. https://doi.org/10.3390/vetsci12050457
Elbir H. No Genomic Signatures Were Found in Escherichia coli Isolates from Camels With or Without Clinical Endometritis. Veterinary Sciences. 2025; 12(5):457. https://doi.org/10.3390/vetsci12050457
Chicago/Turabian StyleElbir, Haitham. 2025. "No Genomic Signatures Were Found in Escherichia coli Isolates from Camels With or Without Clinical Endometritis" Veterinary Sciences 12, no. 5: 457. https://doi.org/10.3390/vetsci12050457
APA StyleElbir, H. (2025). No Genomic Signatures Were Found in Escherichia coli Isolates from Camels With or Without Clinical Endometritis. Veterinary Sciences, 12(5), 457. https://doi.org/10.3390/vetsci12050457