
Variations in Intestinal Microbiota Among Three Species in the Cervidae Family Under the Same Feeding Conditions

 ,
,
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and Preservation
2.2. DNA Extraction, PCR Amplification, and Sequencing
2.3. Bioinformatics Analysis
3. Results
3.1. Analysis of 16S rRNA Sequencing Data
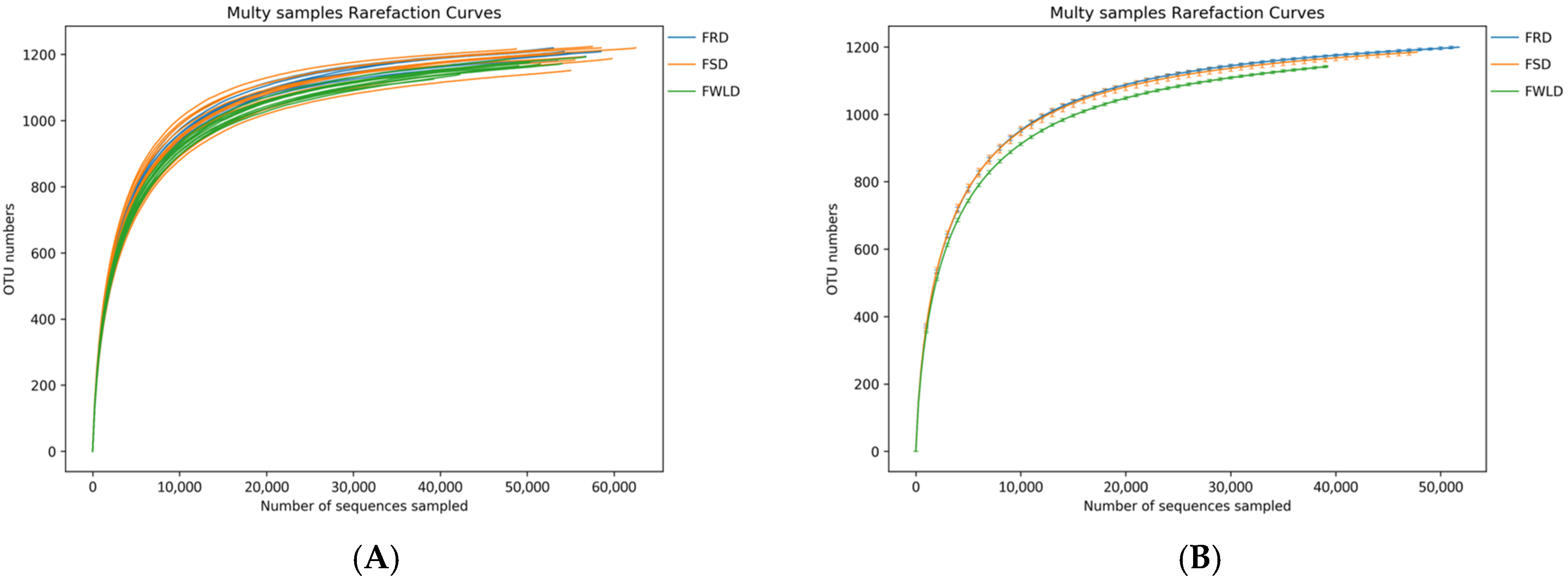
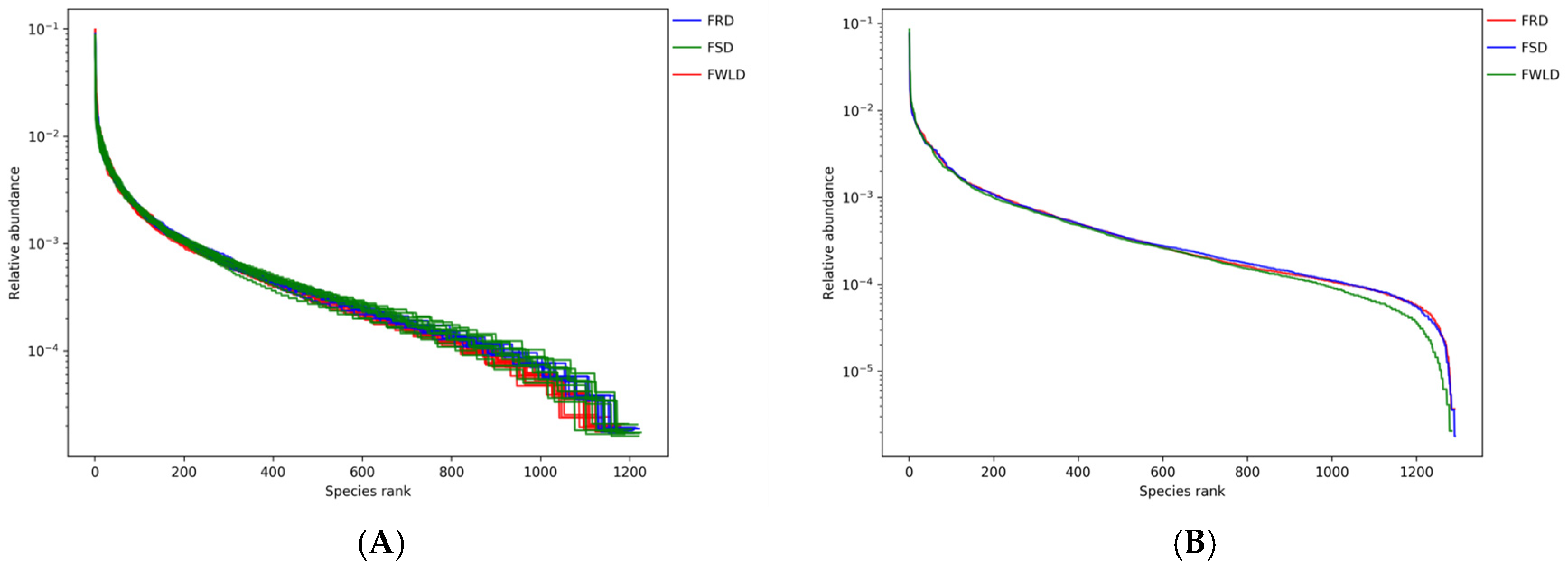
3.2. Analysis of Species Diversity
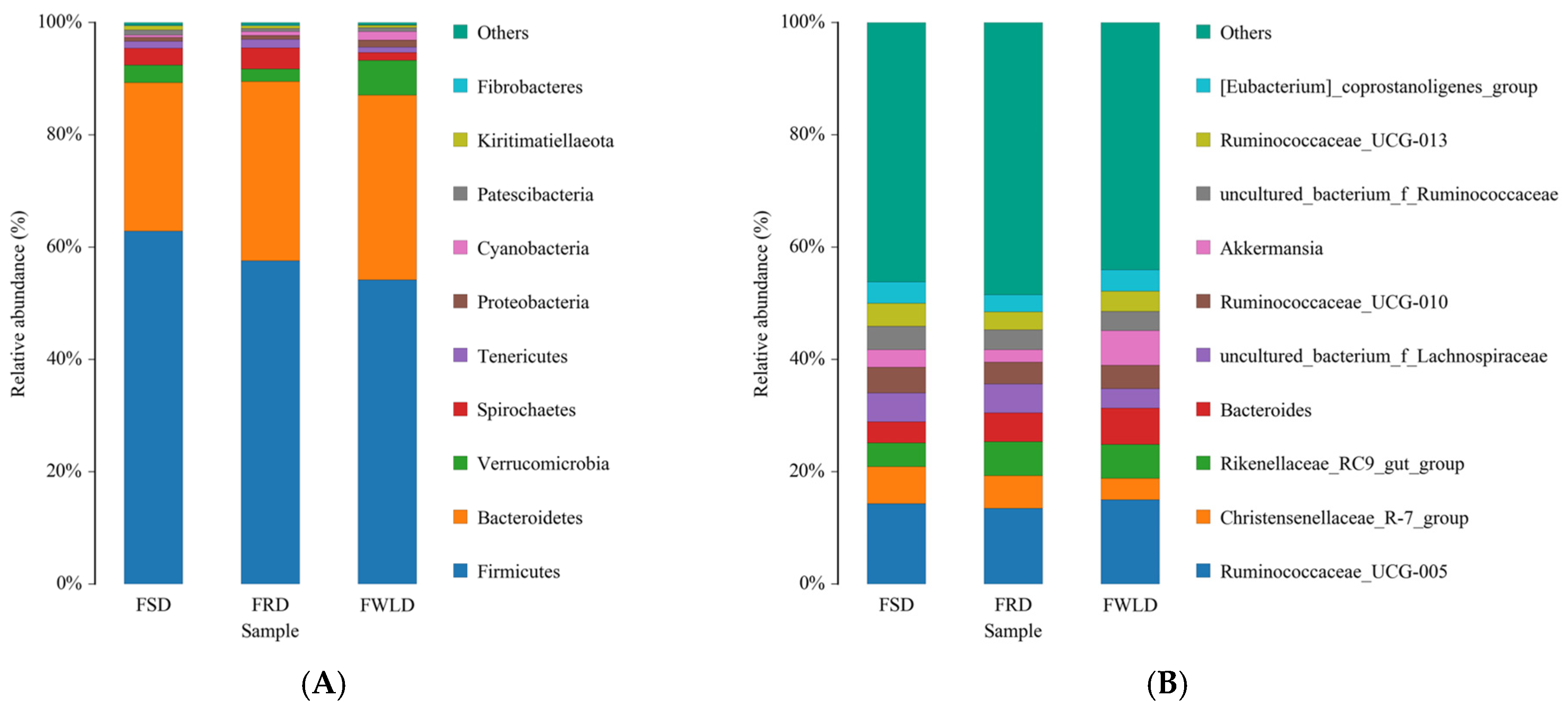
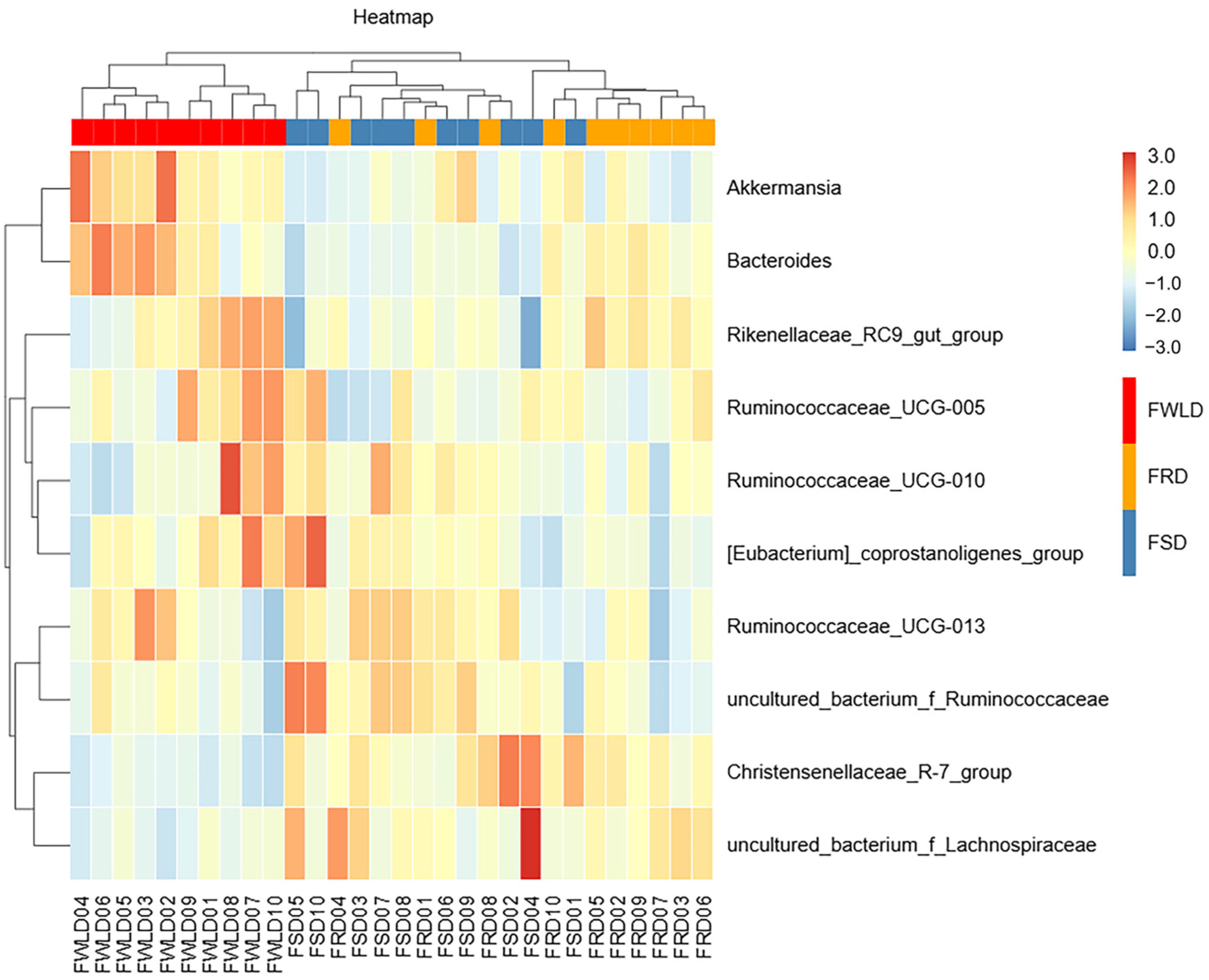
3.3. Comparison of Bacterial Community Composition
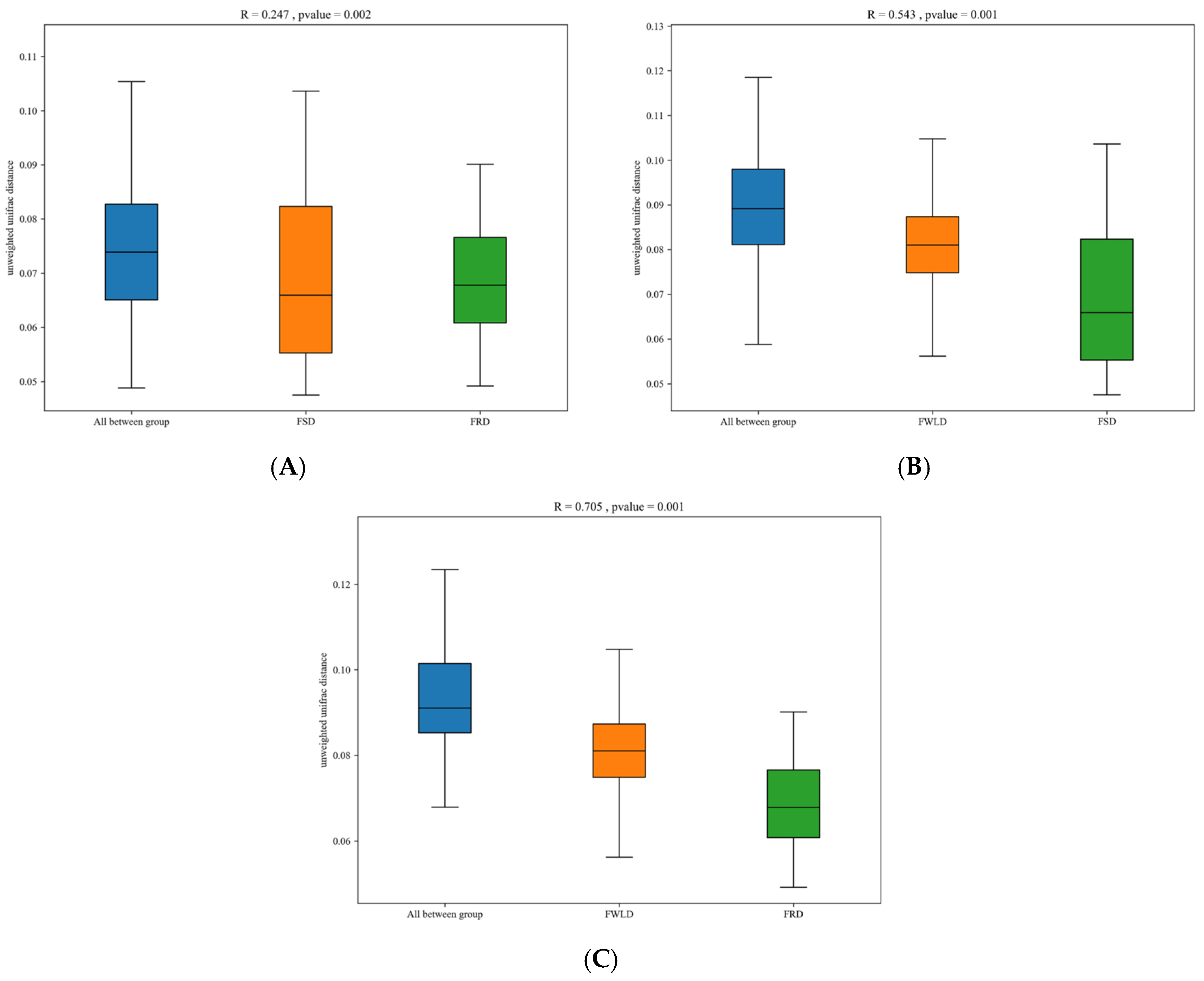
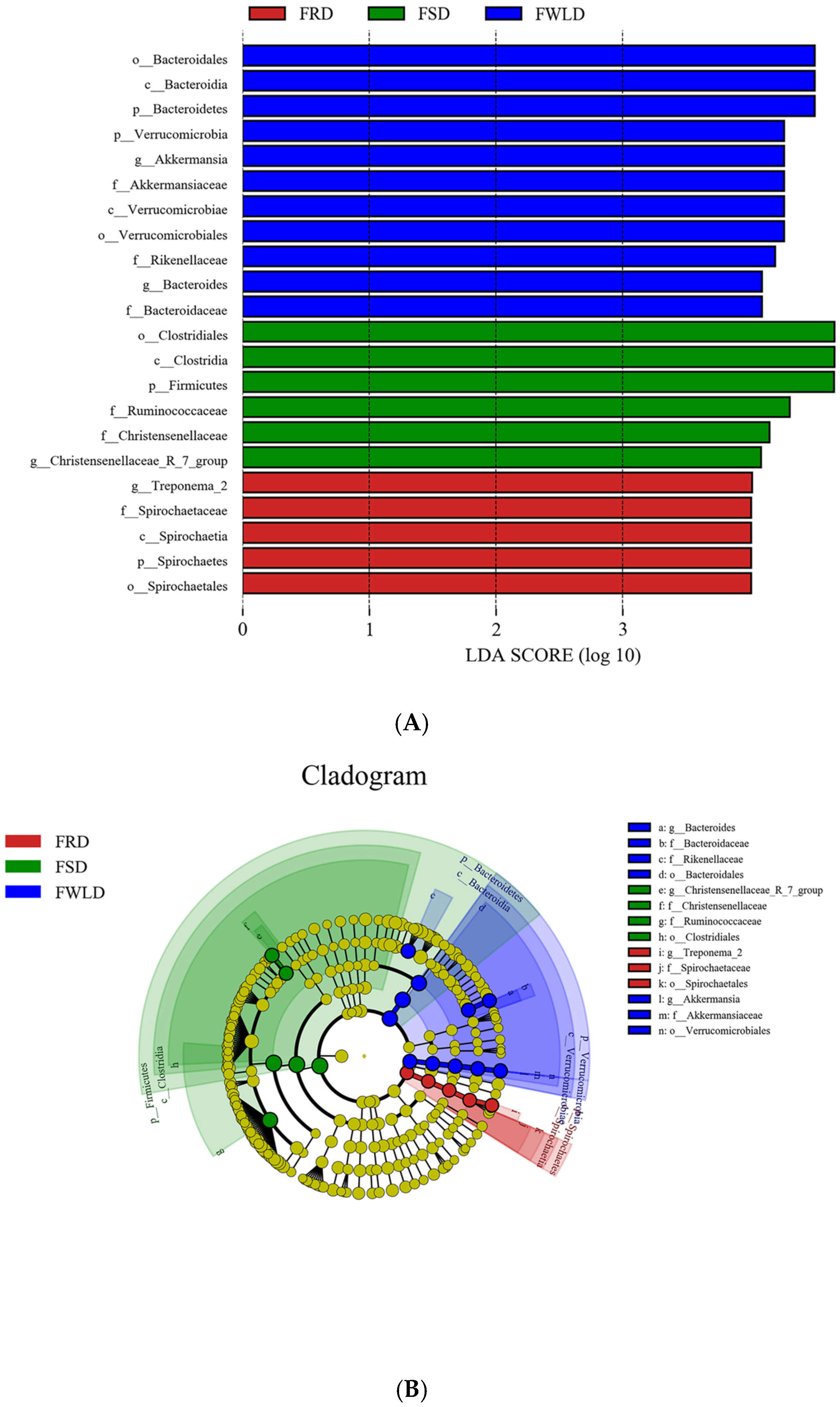
3.4. Comparison of Differences in Intestinal Microbiota in Different Groups
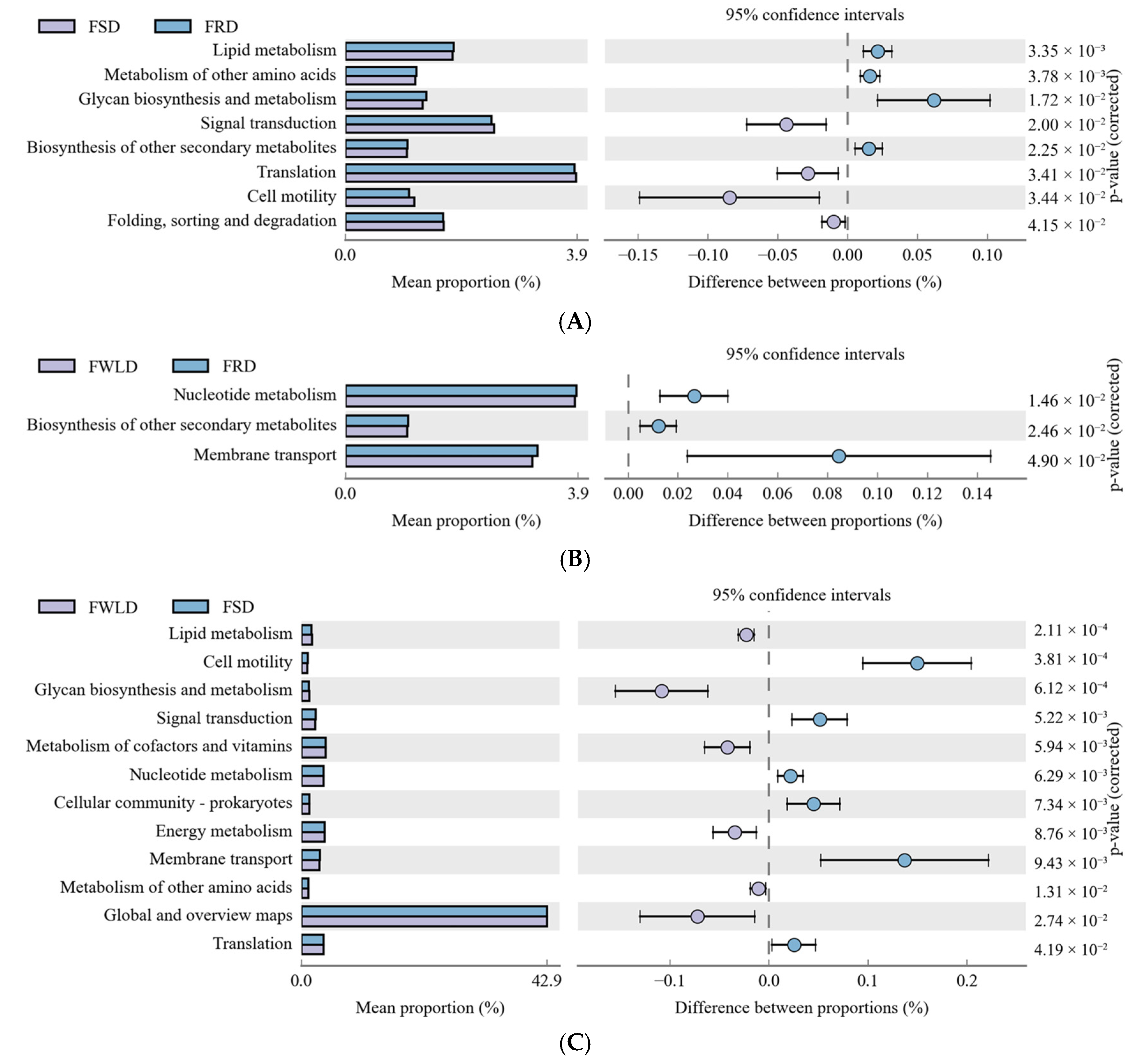
3.5. Analysis of PICRUSt Function Prediction
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Clemente, J.C.; Ursell, L.K.; Parfrey, L.W.; Knight, R. The Impact of the Gut Microbiota on Human Health: An Integrative View. Cell 2012, 148, 1258–1270. [Google Scholar] [CrossRef] [PubMed]
- Song, Q.; Wang, Y.; Huang, L.; Shen, M.; Yu, Y.; Yu, Q.; Chen, Y.; Xie, J. Review of the Relationships among Polysaccharides, Gut Microbiota, and Human Health. Food Res. Int. 2021, 140, 109858. [Google Scholar] [CrossRef] [PubMed]
- Tremaroli, V.; Bäckhed, F. Functional Interactions between the Gut Microbiota and Host Metabolism. Nature 2012, 489, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.Y.; Groer, M.; Dutra, S.V.O.; Sarkar, A.; McSkimming, D.I. Gut Microbiota and Immune System Interactions. Microorganisms 2020, 8, 1587. [Google Scholar] [CrossRef]
- Al Jassim, R.A.M.; Andrews, F.M. The Bacterial Community of the Horse Gastrointestinal Tract and Its Relation to Fermentative Acidosis, Laminitis, Colic, and Stomach Ulcers. Vet. Clin. N. Am. Equine Pract. 2009, 25, 199–215. [Google Scholar] [CrossRef]
- Tardiolo, G.; La Fauci, D.; Riggio, V.; Daghio, M.; Di Salvo, E.; Zumbo, A.; Sutera, A.M. Gut Microbiota of Ruminants and Monogastric Livestock: An Overview. Animals 2025, 15, 758. [Google Scholar] [CrossRef]
- Malmuthuge, N.; Li, M.; Goonewardene, L.A.; Oba, M.; Guan, L.L. Effect of Calf Starter Feeding on Gut Microbial Diversity and Expression of Genes Involved in Host Immune Responses and Tight Junctions in Dairy Calves during Weaning Transition. J. Dairy Sci. 2013, 96, 3189–3200. [Google Scholar] [CrossRef]
- Zierer, J.; Jackson, M.A.; Kastenmüller, G.; Mangino, M.; Long, T.; Telenti, A.; Mohney, R.P.; Small, K.S.; Bell, J.T.; Steves, C.J.; et al. The Fecal Metabolome as a Functional Readout of the Gut Microbiome. Nat. Genet. 2018, 50, 790–795. [Google Scholar] [CrossRef]
- Li, Y.; Li, X.; Wu, Y.; Zhang, W. Effects of Fecal Microbiota Transplantation from Yaks on Weaning Diarrhea, Fecal Microbiota Composition, Microbial Network Structure and Functional Pathways in Chinese Holstein Calves. Front. Microbiol. 2022, 13, 898505. [Google Scholar] [CrossRef]
- Yin, X.; Ji, S.; Duan, C.; Tian, P.; Ju, S.; Yan, H.; Zhang, Y.; Liu, Y. The Succession of Fecal Bacterial Community and Its Correlation with the Changes of Serum Immune Indicators in Lambs from Birth to 4 Months. J. Integr. Agric. 2023, 22, 537–550. [Google Scholar] [CrossRef]
- Sun, G.; Zhang, H.; Wei, Q.; Zhao, C.; Yang, X.; Wu, X.; Xia, T.; Liu, G.; Zhang, L.; Gao, Y.; et al. Comparative Analyses of Fecal Microbiota in European Mouflon (Ovis orientalis musimon) and Blue Sheep (Pseudois nayaur) Living at Low or High Altitudes. Front. Microbiol. 2019, 10, 1735. [Google Scholar] [CrossRef] [PubMed]
- De Filippo, C.; Cavalieri, D.; Di Paola, M.; Ramazzotti, M.; Poullet, J.B.; Massart, S.; Collini, S.; Pieraccini, G.; Lionetti, P. Impact of Diet in Shaping Gut Microbiota Revealed by a Comparative Study in Children from Europe and Rural Africa. Proc. Natl. Acad. Sci. USA 2010, 107, 14691–14696. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Liu, G.; Shafer, A.B.A.; Wei, Y.; Zhou, J.; Lin, S.; Wu, H.; Zhou, M.; Hu, D.; Liu, S. Comparative Analysis of the Gut Microbial Communities in Forest and Alpine Musk Deer Using High-Throughput Sequencing. Front. Microbiol. 2017, 8, 572. [Google Scholar] [CrossRef]
- Xiao, L.; Estellé, J.; Kiilerich, P.; Ramayo-Caldas, Y.; Xia, Z.; Feng, Q.; Liang, S.; Pedersen, A.; Kjeldsen, N.J.; Liu, C.; et al. A Reference Gene Catalogue of the Pig Gut Microbiome. Nat. Microbiol. 2016, 1, 16161. [Google Scholar] [CrossRef]
- Li, Y.; Hu, X.; Yang, S.; Zhou, J.; Qi, L.; Sun, X.; Fan, M.; Xu, S.; Cha, M.; Zhang, M.; et al. Comparison between the Fecal Bacterial Microbiota of Healthy and Diarrheic Captive Musk Deer. Front. Microbiol. 2018, 9, 300. [Google Scholar] [CrossRef]
- Minich, D.; Madden, C.; Evans, M.V.; Ballash, G.A.; Barr, D.J.; Poulsen, K.P.; Dennis, P.M.; Hale, V.L. Alterations in Gut Microbiota Linked to Provenance, Sex, and Chronic Wasting Disease in White-Tailed Deer (Odocoileus virginianus). Sci. Rep. 2021, 11, 13218. [Google Scholar] [CrossRef]
- Zhang, M.; Shi, M.; Fan, M.; Xu, S.; Li, Y.; Zhang, T.; Cha, M.; Liu, Y.; Guo, X.; Chen, Q.; et al. Comparative Analysis of Gut Microbiota Changes in Père David’s Deer Populations in Beijing Milu Park and Shishou, Hubei Province in China. Front. Microbiol. 2018, 9, 1258. [Google Scholar] [CrossRef]
- Sun, G.; Xia, T.; Wei, Q.; Dong, Y.; Zhao, C.; Yang, X.; Zhang, L.; Wang, X.; Sha, W.; Zhang, H. Analysis of Gut Microbiota in Three Species Belonging to Different Genera (Hemitragus, Pseudois, and Ovis) from the Subfamily Caprinae in the Absence of Environmental Variance. Ecol. Evol. 2021, 11, 12129–12140. [Google Scholar] [CrossRef]
- Zeng, Y.; Pu, Y.; Niu, L.L.; Deng, J.B.; Zeng, D.; Amato, K.R.; Li, Y.; Zhou, Y.; Lin, Y.C.; Wang, J.; et al. Comparison of Gastrointestinal Microbiota in Golden Snub-Nosed Monkey (Rhinopithecus roxellanae), Green Monkey (Chlorocebus aethiops sabaeus), and Ring-Tailed Lemur (Lemur catta) by High Throughput Sequencing. Glob. Ecol. Conserv. 2022, 33, e01946. [Google Scholar] [CrossRef]
- Wang, L.; Song, L. Ecological Status of Sika Deer in China. Spec. Econ. Anim. Plants 2014, 17, 12–14. (In Chinese) [Google Scholar]
- Jin, Y.; Zhang, M. Review of Molecular Ecology Research and Prospects of Red Deer (Cervus elaphus) in China Based on Feces Molecular Biology Technology. Chin. J. Wildl. 2012, 33, 152–157+169. (In Chinese) [Google Scholar] [CrossRef]
- Wu, J.; Pei, J. Present Status of Research of White—Lipped Deer and its Conservation Strategy. Chin. J. Wildl. 2007, 28, 36–39. (In Chinese) [Google Scholar] [CrossRef]
- Liu, S.; Zhang, N.; Li, N. Culture Technology and Development Prospect of Sika Deer. Chin. Livest. Poult. Breed. 2022, 18, 129–131. (In Chinese) [Google Scholar]
- Liu, Y.; Zheng, C.; Zhang, X.; Ling, L.; Chang, Q. Research on Current Situation and Development Countermeasures of Antler Deer Breeding in China. Chin. J. Anim. Sci. 2011, 47, 18–21. (In Chinese) [Google Scholar]
- Yamano, H.; Ichimura, Y.; Sawabe, Y.; Koike, S.; Suzuki, Y.; Kobayashi, Y. Seasonal Differences in Rumen Bacterial Flora of Wild Hokkaido Sika Deer and Partial Characterization of an Unknown Bacterial Group Possibly Involved in Fiber Digestion in Winter. Anim. Sci. J. 2019, 90, 790–798. [Google Scholar] [CrossRef]
- Guan, Y.; Yang, H.; Han, S.; Feng, L.; Wang, T.; Ge, J. Comparison of the Gut Microbiota Composition between Wild and Captive Sika Deer (Cervus nippon hortulorum) from Feces by High-Throughput Sequencing. AMB Express 2017, 7, 212. [Google Scholar] [CrossRef]
- Wang, X.; Chen, Y.; Shang, Y.; Wu, X.; Wei, Q.; Chen, J.; Yan, J.; Zhang, H. Comparison of the Gut Microbiome in Red Deer (Cervus elaphus) and Fallow Deer (Dama dama) by High-Throughput Sequencing of the V3–V4 Region of the 16S RRNA Gene. ScienceAsia 2019, 45, 515–524. [Google Scholar] [CrossRef]
- Guo, J.; Jin, Y.; Tian, X.; Bao, H.; Sun, Y.; Gray, T.; Song, Y.; Zhang, M. Diet-Induced Microbial Adaptation Process of Red Deer (Cervus elaphus) under Different Introduced Periods. Front. Microbiol. 2022, 13, 1033050. [Google Scholar] [CrossRef]
- Li, J.G.; Wang, C.D.; Tang, Z.H.; Guo, Y.Q.; Zheng, T.C.; Li, Y.Z.; You, Z.Q. The Gut Bacterial Community Composition of Wild Cervus albirostris (White-Lipped Deer) Detected by the 16S Ribosomal RNA Gene Sequencing. Curr. Microbiol. 2017, 74, 1100–1107. [Google Scholar] [CrossRef]
- Zhao, P.; Xu, H.; Li, D.; Xie, M.; Xu, H.; Wu, J.; Wen, A.; Ni, Q.; Zhang, M.; Wang, Q.; et al. The Complete Mitochondrial Genome Sequence and Phylogenetic Analysis of White-Lipped Deer (Cervus albirostris). Conserv. Genet. Resour. 2018, 10, 741–745. [Google Scholar] [CrossRef]
- Li, B.; Gao, H.; Song, P.; Liang, C.; Jiang, F.; Xu, B.; Liu, D.; Zhang, T. Captivity Shifts Gut Microbiota Communities in White-Lipped Deer (Cervus albirostris). Animals 2022, 12, 431. [Google Scholar] [CrossRef]
- You, Z.; Deng, J.; Liu, J.; Fu, J.; Xiong, H.; Luo, W.; Xiong, J. Seasonal Variations in the Composition and Diversity of Gut Microbiota in White-Lipped Deer (Cervus albirostris). PeerJ 2022, 10, e13753. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt Removes Adapter Sequences from High-throughput Sequencing Reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Edgar, R.C. UPARSE: Highly Accurate OTU Sequences from Microbial Amplicon Reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME Improves Sensitivity and Speed of Chimera Detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Subramanian, S.; Faith, J.J.; Gevers, D.; Gordon, J.I.; Knight, R.; Mills, D.A.; Caporaso, J.G. Quality-Filtering Vastly Improves Diversity Estimates from Illumina Amplicon Sequencing. Nat. Methods 2013, 10, 57–59. [Google Scholar] [CrossRef]
- Li, Y.; Guo, W.; Han, S.; Kong, F.; Wang, C.; Li, D.; Zhang, H.; Yang, M.; Xu, H.; Zeng, B.; et al. The Evolution of the Gut Microbiota in the Giant and the Red Pandas. Sci. Rep. 2015, 5, 10185. [Google Scholar] [CrossRef]
- Suzuki, T.A.; Phifer-Rixey, M.; Mack, K.L.; Sheehan, M.J.; Lin, D.; Bi, K.; Nachman, M.W. Host Genetic Determinants of the Gut Microbiota of Wild Mice. Mol. Ecol. 2019, 28, 3197–3207. [Google Scholar] [CrossRef]
- Knowles, S.C.L.; Eccles, R.M.; Baltrūnaitė, L. Species Identity Dominates over Environment in Shaping the Microbiota of Small Mammals. Ecol. Lett. 2019, 22, 826–837. [Google Scholar] [CrossRef]
- Amato, K.R.; Sanders, J.G.; Song, S.J.; Nute, M.; Metcalf, J.L.; Thompson, L.R.; Morton, J.T.; Amir, A.; McKenzie, V.J.; Humphrey, G.; et al. Evolutionary Trends in Host Physiology Outweigh Dietary Niche in Structuring Primate Gut Microbiomes. ISME J. 2019, 13, 576–587. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.R.; Du, R.F. Evolution of Karyotype of the Genus Cervus. J. Genet. Genom. 1982, 9, 24–31. (In Chinese) [Google Scholar]
- Sheng, H.L. The Deer in China, 1st ed.; East China Normal University Press: Shanghai, China, 1992. (In Chinese) [Google Scholar]
- Wang, Y.L.; Lu, B.Q.; Tao, B.; Xu, D. A Comparative Analysis on the Serum LDH Isozymes in Deers. Acta Ecol. Sin. 1996, 16, 76–77. (In Chinese) [Google Scholar] [CrossRef]
- Li, Y.; Hu, X.; Yang, S.; Zhou, J.; Zhang, T.; Qi, L.; Sun, X.; Fan, M.; Xu, S.; Cha, M.; et al. Comparative Analysis of the Gut Microbiota Composition between Captive and Wild Forest Musk Deer. Front. Microbiol. 2017, 8, 1705. [Google Scholar] [CrossRef]
- Spence, C.; Wells, W.G.; Smith, C.J. Characterization of the Primary Starch Utilization Operon in the Obligate Anaerobe Bacteroides fragilis: Regulation by Carbon Source and Oxygen. J. Bacteriol. 2006, 188, 4663–4672. [Google Scholar] [CrossRef]
- Thoetkiattikul, H.; Mhuantong, W.; Laothanachareon, T.; Tangphatsornruang, S.; Pattarajinda, V.; Eurwilaichitr, L.; Champreda, V. Comparative Analysis of Microbial Profiles in Cow Rumen Fed with Different Dietary Fiber by Tagged 16S RRNA Gene Pyrosequencing. Curr. Microbiol. 2013, 67, 130–137. [Google Scholar] [CrossRef]
- Waters, J.L.; Ley, R.E. The Human Gut Bacteria Christensenellaceae Are Widespread, Heritable, and Associated with Health. BMC Biol. 2019, 17, 83. [Google Scholar] [CrossRef]
- Zhou, L.; Xiao, X.; Zhang, Q.; Zheng, J.; Li, M.; Yu, M.; Wang, X.; Deng, M.; Zhai, X.; Li, R. Improved Glucose and Lipid Metabolism in the Early Life of Female Offspring by Maternal Dietary Genistein Is Associated with Alterations in the Gut Microbiota. Front. Endocrinol. 2018, 9, 516. [Google Scholar] [CrossRef]
- Zafar, H.; Saier, M.H. Gut Bacteroides Species in Health and Disease. Gut Microbes 2021, 13, 1848158. [Google Scholar] [CrossRef]
- Edwards, J.E.; Shetty, S.A.; van den Berg, P.; Burden, F.; van Doorn, D.A.; Pellikaan, W.F.; Dijkstra, J.; Smidt, H. Multi-Kingdom Characterization of the Core Equine Fecal Microbiota Based on Multiple Equine (Sub)Species. Anim. Microbiome 2020, 2, 6. [Google Scholar] [CrossRef]
- Li, Y.; Lan, Y.; Zhang, S.; Wang, X. Comparative Analysis of Gut Microbiota Between Healthy and Diarrheic Horses. Front. Vet. Sci. 2022, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Jia, H.; Li, J.; Yu, M.; Yang, Y.; Tian, D.; Zhang, H.; Zou, Z. Cecal Gut Microbiota and Metabolites Might Contribute to the Severity of Acute Myocardial Ischemia by Impacting the Intestinal Permeability, Oxidative Stress, and Energy Metabolism. Front. Microbiol. 2019, 10, 1745. [Google Scholar] [CrossRef] [PubMed]
- Klitgaard, K.; Strube, M.L.; Isbrand, A.; Jensen, T.K.; Nielsen, M.W. Microbiota Analysis of an Environmental Slurry and Its Potential Role as a Reservoir of Bovine Digital Dermatitis Pathogens. Appl. Environ. Microbiol. 2017, 83, e00244-17. [Google Scholar] [CrossRef] [PubMed]
- Stanton, T.B.; Canale-Parola, E. Treponema bryantii sp. nov., a Rumen Spirochete that Interacts with Cellulolytic Bacteria. Arch. Microbiol. 1980, 127, 145–156. [Google Scholar] [CrossRef]
- Cwyk, W.M.; Canale-Parola, E. Treponema succinifaciens sp. nov., an Anaerobic Spirochete from the Swine Intestine. Arch. Microbiol. 1979, 122, 231–239. [Google Scholar] [CrossRef]
- Derrien, M.; Belzer, C.; de Vos, W.M. Akkermansia muciniphila and Its Role in Regulating Host Functions. Microb. Pathog. 2017, 106, 171–181. [Google Scholar] [CrossRef]
- Oliphant, K.; Allen-Vercoe, E. Macronutrient Metabolism by the Human Gut Microbiome: Major Fermentation by-Products and Their Impact on Host Health. Microbiome 2019, 7, 91. [Google Scholar] [CrossRef]
- Delgado, M.L.; Singh, P.; Funk, J.A.; Moore, J.A.; Cannell, E.M.; Kanesfsky, J.; Manning, S.D.; Scribner, K.T. Intestinal Microbial Community Dynamics of White-Tailed Deer (Odocoileus virginianus) in an Agroecosystem. Microb. Ecol. 2017, 74, 496–506. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Items | Content (%) |
|---|---|
| Composition | |
| Corn meal | 48.9 |
| Soybean meal | 23.0 |
| Heat-treated soybean | 14.0 |
| Wheat bran | 10.0 |
| NaCl | 1.5 |
| Mineral premix | 2.6 |
| Total | 100.0 |
| Nutritional level | |
| GE (MJ/kg) | 16.89 |
| ME (MJ/kg) | 12.25 |
| CP | 20.00 |
| Ca | 0.99 |
| P | 1.02 |
| Taxon | Name | Relative Abundance (%) | ||
|---|---|---|---|---|
| FSD | FRD | FWLD | ||
| Phylum | Firmicutes | 62.87 | 57.57 | 54.19 |
| Bacteroidetes | 26.47 | 31.94 | 32.89 | |
| Verrucomicrobia | 3.13 | 2.27 | 6.17 | |
| Cyanobacteria | 0.42 | 0.66 | 1.54 | |
| Proteobacteria | 0.70 | 0.74 | 1.25 | |
| Spirochaetes | 2.94 | 3.70 | 1.44 | |
| Tenericutes | 1.26 | 1.52 | 0.95 | |
| Patescibacteria | 0.94 | 0.58 | 0.67 | |
| Kiritimatiellaeota | 0.76 | 0.50 | 0.41 | |
| Fibrobacteres | 0.14 | 0.22 | 0.11 | |
| Genus | Ruminococcaceae_UCG-005 | 14.29 | 13.46 | 14.98 |
| Christensenellaceae_R-7_group | 6.61 | 5.87 | 3.85 | |
| Rikenellaceae_RC9_gut_group | 4.27 | 6.01 | 6.03 | |
| Bacteroides | 3.69 | 5.13 | 6.47 | |
| uncultured_bacterium_f_Lachnospiraceae | 5.15 | 5.16 | 3.44 | |
| Ruminococcaceae_UCG-010 | 4.60 | 3.89 | 4.20 | |
| Akkermansia | 3.13 | 2.27 | 6.17 | |
| uncultured_bacterium_f_ Ruminococcaceae | 4.15 | 3.54 | 3.45 | |
| Ruminococcaceae_UCG-013 | 4.15 | 3.13 | 3.60 | |
| [Eubacterium]_coprostanoligenes_group | 3.82 | 3.07 | 3.79 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Shi, M.; Wu, J.; Han, X.; Li, M.; Wu, Y.; Jiang, Y.; Zhang, H.; Liu, S.; Hu, D. Variations in Intestinal Microbiota Among Three Species in the Cervidae Family Under the Same Feeding Conditions. Vet. Sci. 2025, 12, 438. https://doi.org/10.3390/vetsci12050438
Wang Y, Shi M, Wu J, Han X, Li M, Wu Y, Jiang Y, Zhang H, Liu S, Hu D. Variations in Intestinal Microbiota Among Three Species in the Cervidae Family Under the Same Feeding Conditions. Veterinary Sciences. 2025; 12(5):438. https://doi.org/10.3390/vetsci12050438
Chicago/Turabian StyleWang, Yichen, Minghui Shi, Jiahui Wu, Xiangyu Han, Mengqi Li, Yining Wu, Yuanlin Jiang, Haonan Zhang, Shuqiang Liu, and Defu Hu. 2025. "Variations in Intestinal Microbiota Among Three Species in the Cervidae Family Under the Same Feeding Conditions" Veterinary Sciences 12, no. 5: 438. https://doi.org/10.3390/vetsci12050438
APA StyleWang, Y., Shi, M., Wu, J., Han, X., Li, M., Wu, Y., Jiang, Y., Zhang, H., Liu, S., & Hu, D. (2025). Variations in Intestinal Microbiota Among Three Species in the Cervidae Family Under the Same Feeding Conditions. Veterinary Sciences, 12(5), 438. https://doi.org/10.3390/vetsci12050438