
Metagenomic Insights into the Diverse Antibiotic Resistome of Non-Migratory Corvidae Species on the Qinghai–Tibetan Plateau

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Samples Collection
2.3. DNA Extraction and Metagenomic Sequencing
2.4. Raw Data Processing and Taxonomy Profiling
2.5. ARG and MGE Annotation and Quantification
2.6. Host Identification Analysis
2.7. Identification of High-Risk ARGs
2.8. Statistical Analysis
2.9. Data Availability Statement
3. Results
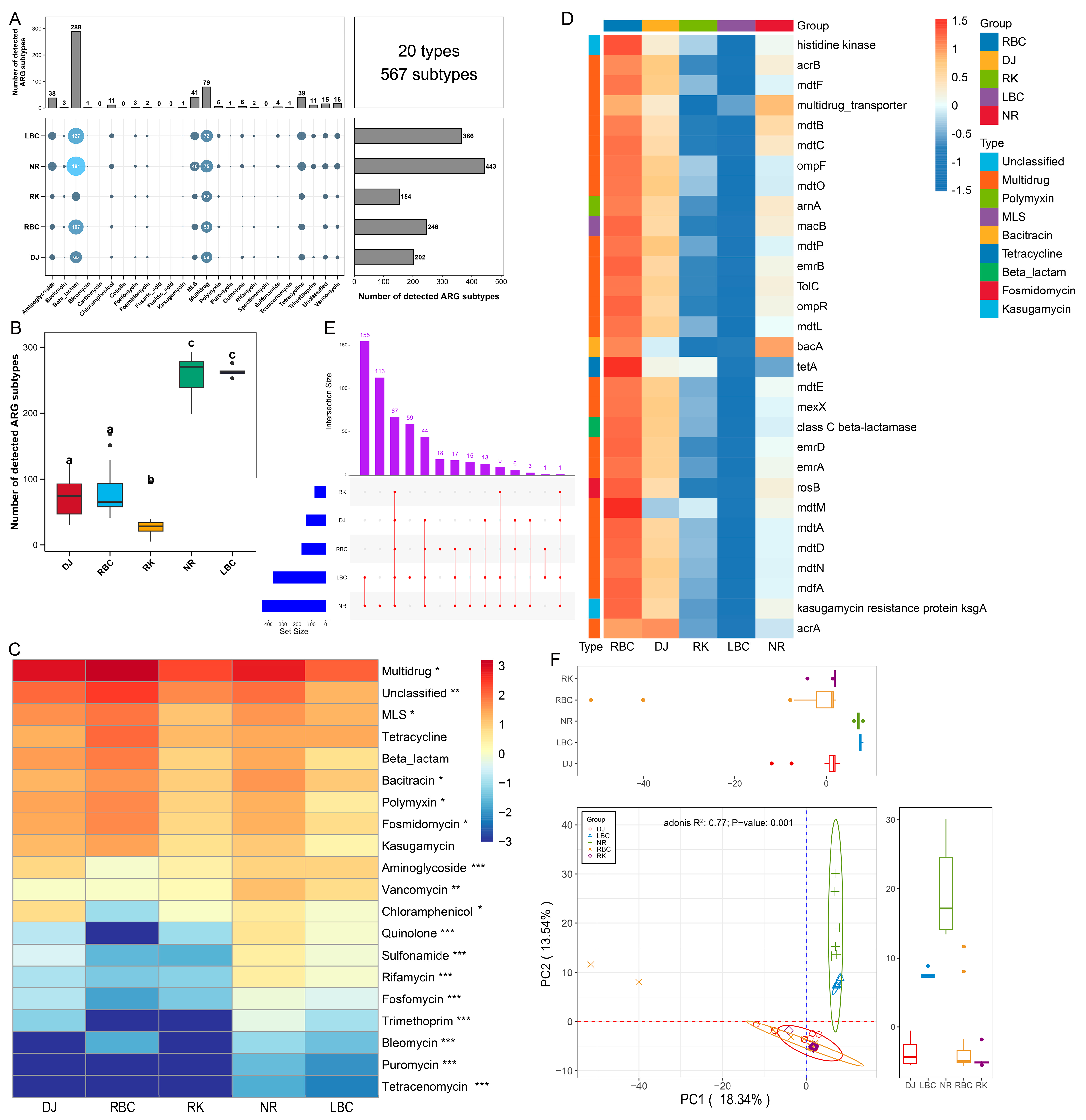
3.1. The Profiles of ARGs Involving Five Groups of Crows
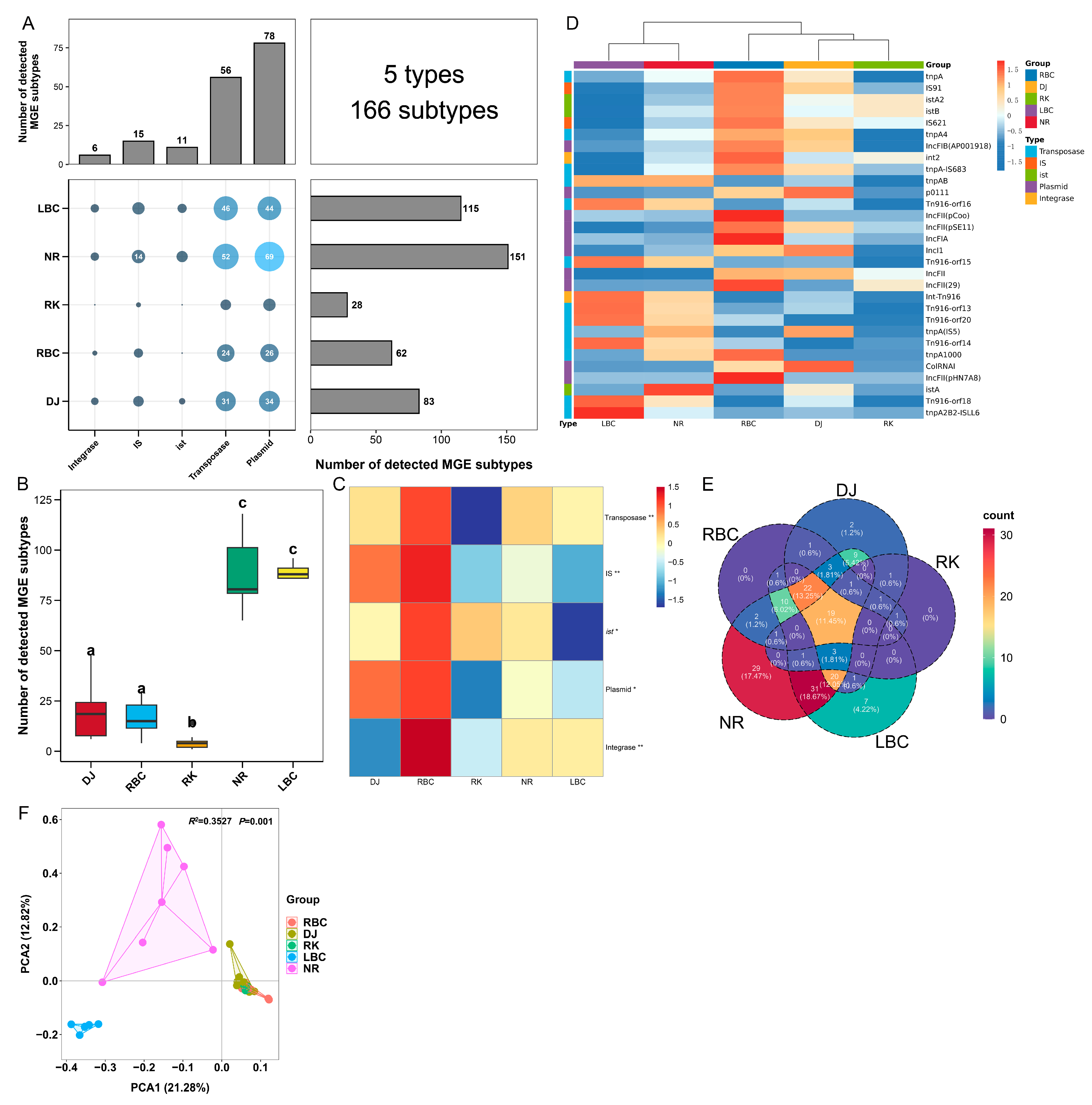
3.2. The Profiles of MGEs Involving Five Groups of Crows
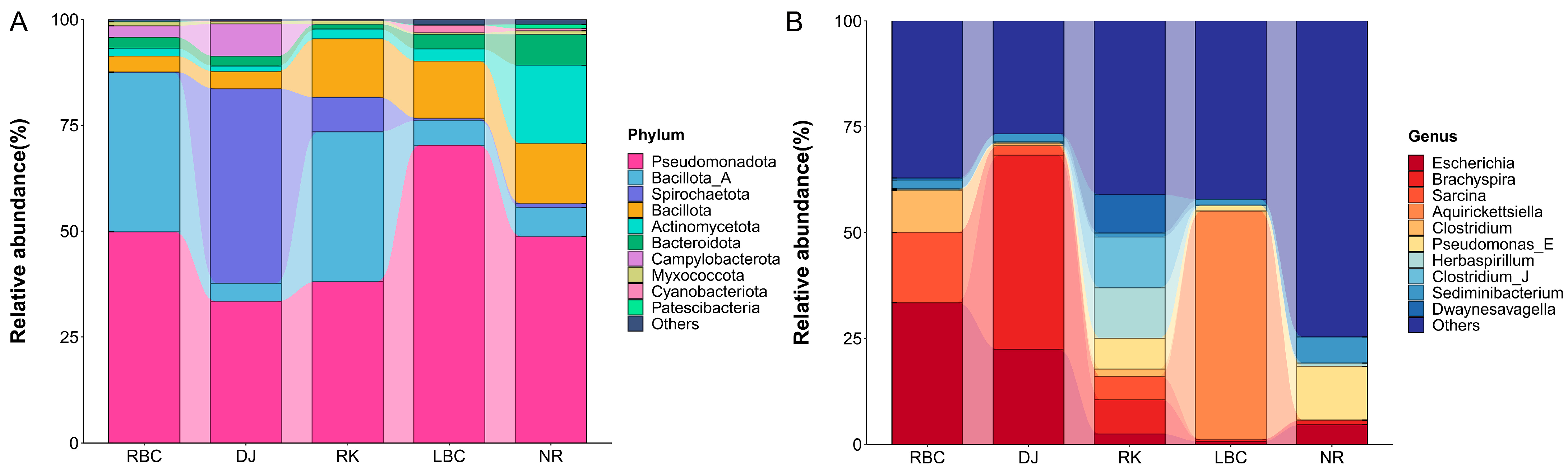
3.3. The Profiles of Microbial Communities Involving Five Groups of Crows
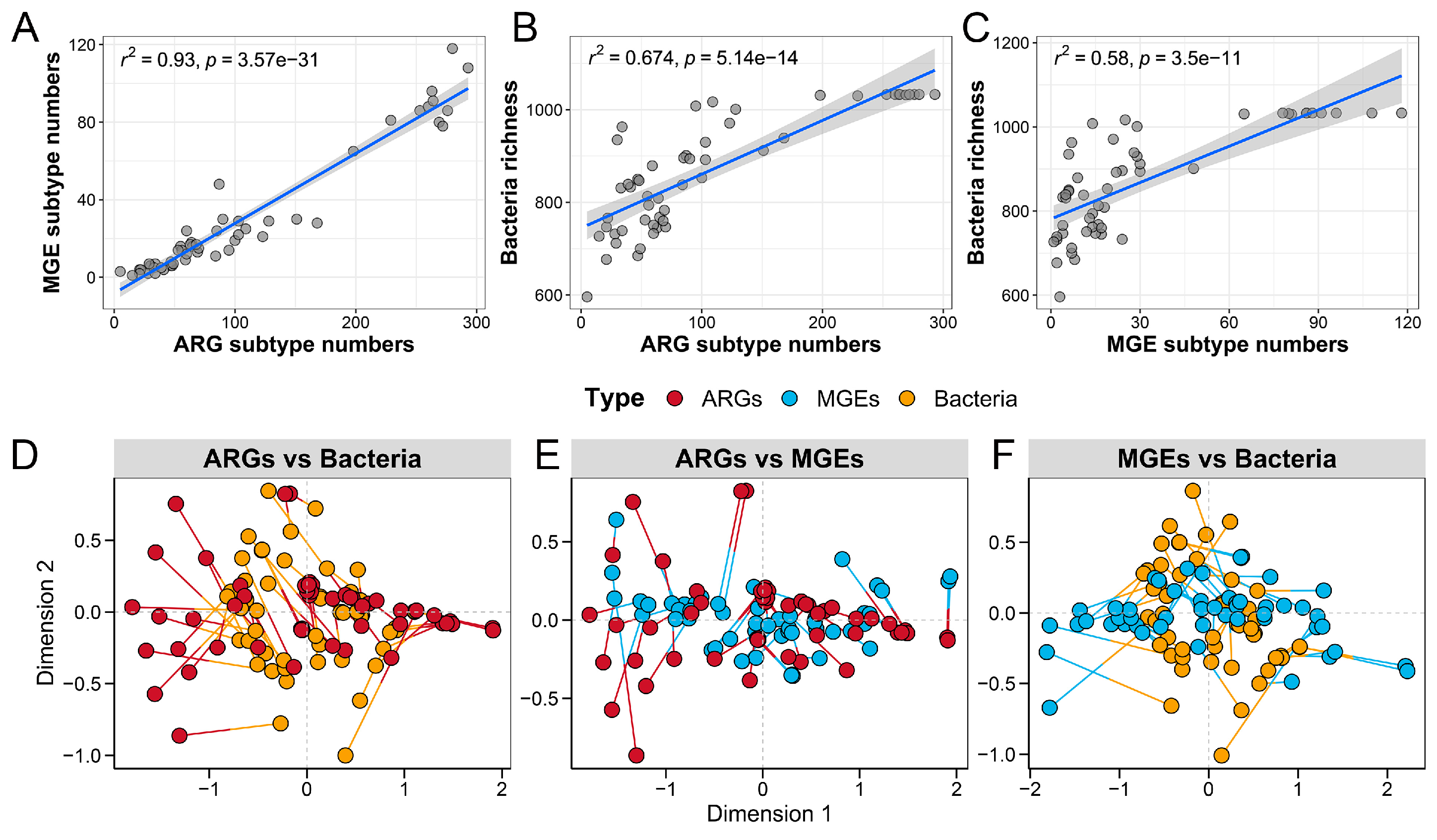
3.4. Correlations and Co-Occurrence Patterns Between Microbial Communities, ARGs, and MGEs
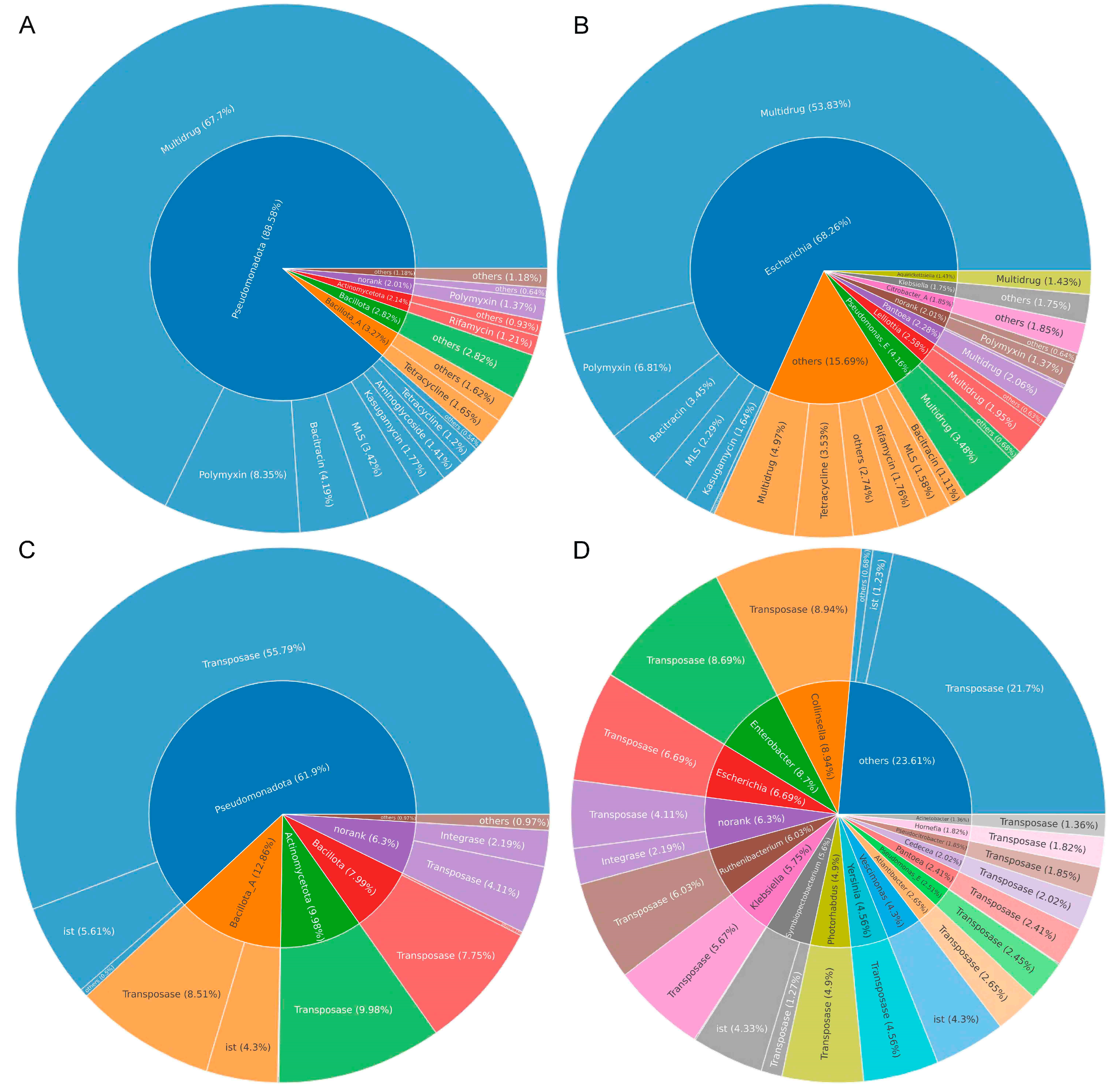
3.5. Identification of Bacterial Hosts of ARGs and MGEs
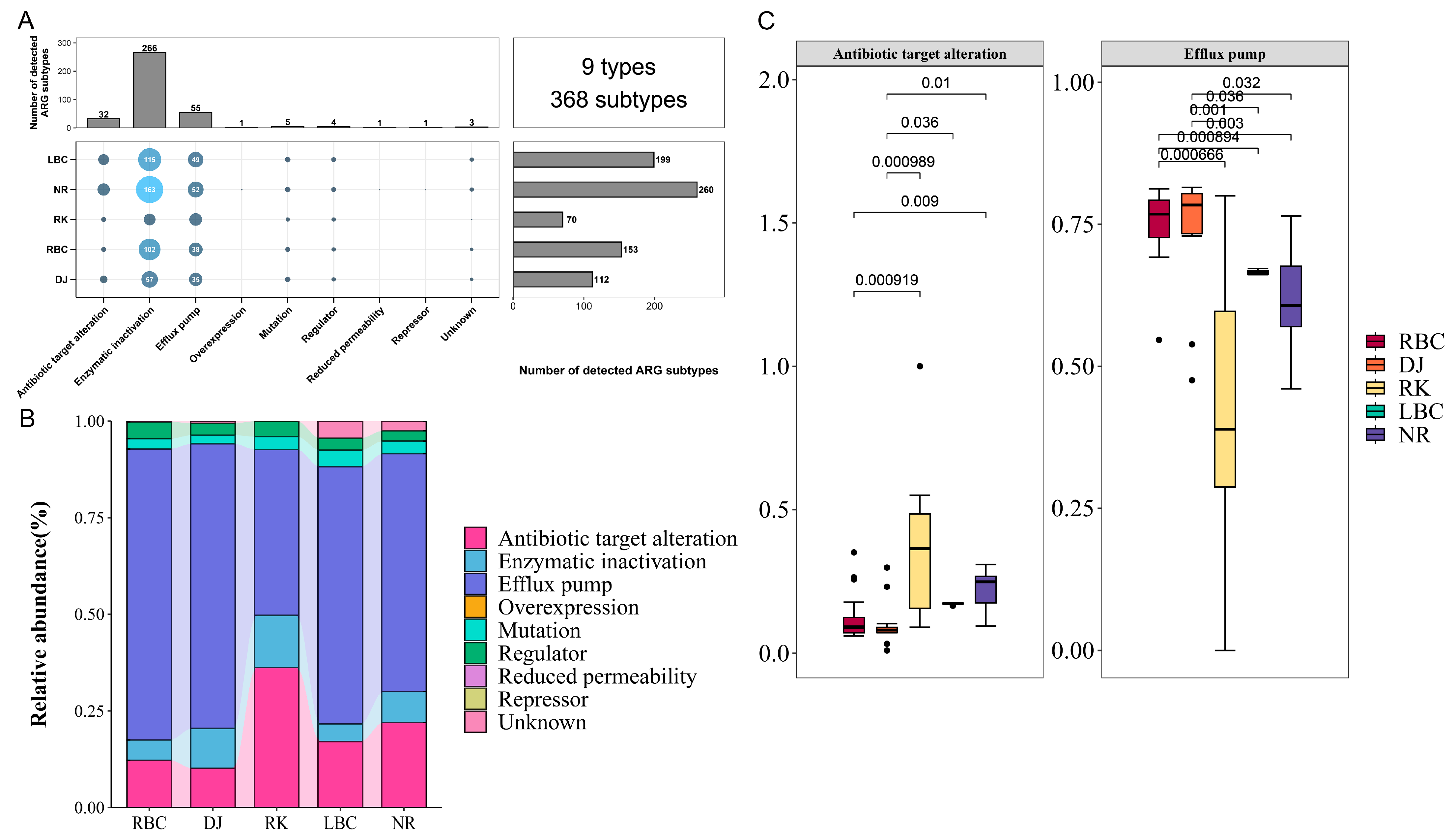
3.6. Resistance Mechanisms of ARGs in Crows
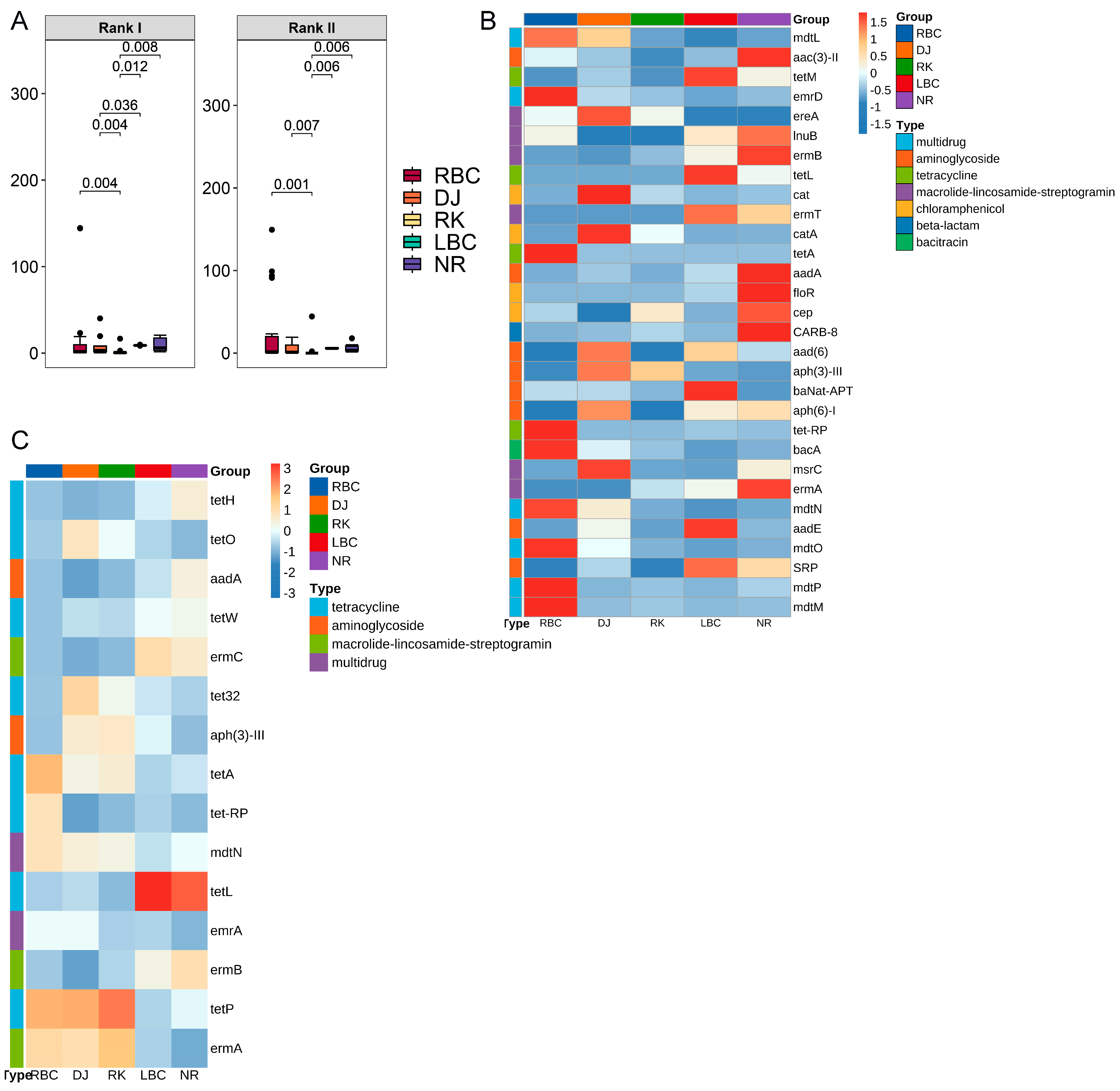
3.7. Detection of High-Risk ARGs in Crows
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Roope, L.S.J.; Smith, R.D.; Pouwels, K.B.; Buchanan, J.; Abel, L.; Eibich, P.; Butler, C.C.; Tan, P.S.; Walker, A.S.; Robotham, J.V.; et al. The challenge of antimicrobial resistance: What economics can contribute. Science 2019, 364, eaau4679. [Google Scholar] [CrossRef]
- Zhou, B.; Wang, C.; Zhao, Q.; Wang, Y.; Huo, M.; Wang, J.; Wang, S. Prevalence and dissemination of antibiotic resistance genes and coselection of heavy metals in Chinese dairy farms. J. Hazard. Mater. 2016, 320, 10–17. [Google Scholar] [CrossRef]
- Islam, M.A.; Bose, P.; Rahman, M.Z.; Muktaruzzaman, M.; Sultana, P.; Ahamed, T.; Khatun, M.M. A review of antimicrobial usage practice in livestock and poultry production and its consequences on human and animal health. J. Adv. Vet. Anim. Res. 2024, 11, 675–685. [Google Scholar] [CrossRef]
- Husna, A.; Rahman, M.M.; Badruzzaman, A.T.M.; Sikder, M.H.; Islam, M.R.; Rahman, M.T.; Alam, J.; Ashour, H.M. Extended-Spectrum β-Lactamases (ESBL): Challenges and Opportunities. Biomedicines 2023, 11, 2937. [Google Scholar] [CrossRef] [PubMed]
- Lyu, J.; Yang, L.; Zhang, L.; Ye, B.; Wang, L. Antibiotics in soil and water in China-a systematic review and source analysis. Environ. Pollut. 2020, 266, 115147. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, Q.; Ji, Z.; Andom, O.; Wang, X.; Guo, X.; Li, Z. Current Status and Spatiotemporal Evolution of Antibiotic Residues in Livestock and Poultry Manure in China. Agriculture 2023, 13, 1877. [Google Scholar] [CrossRef]
- Zhu, D.; Xiang, Q.; Yang, X.R.; Ke, X.; O’Connor, P.; Zhu, Y.G. Trophic Transfer of Antibiotic Resistance Genes in a Soil Detritus Food Chain. Environ. Sci. Technol. 2019, 53, 7770–7781. [Google Scholar] [CrossRef]
- Marcelino, V.R.; Wille, M.; Hurt, A.C.; González-Acuña, D.; Klaassen, M.; Schlub, T.E.; Eden, J.S.; Shi, M.; Iredell, J.R.; Sorrell, T.C.; et al. Meta-transcriptomics reveals a diverse antibiotic resistance gene pool in avian microbiomes. BMC Biol. 2019, 17, 31. [Google Scholar] [CrossRef]
- Rolland, R.M.; Hausfater, G.; Marshall, B.; Levy, S.B. Antibiotic-resistant bacteria in wild primates: Increased prevalence in baboons feeding on human refuse. Appl. Environ. Microbiol. 1985, 49, 791–794. [Google Scholar] [CrossRef]
- Rwego, I.B.; Isabirye-Basuta, G.; Gillespie, T.R.; Goldberg, T.L. Gastrointestinal bacterial transmission among humans, mountain gorillas, and livestock in Bwindi Impenetrable National Park, Uganda. Conserv. Biol. 2008, 22, 1600–1607. [Google Scholar] [CrossRef]
- Li, T.; Long, M.; Ji, C.; Shen, Z.; Gatesoupe, F.J.; Zhang, X.; Zhang, Q.; Zhang, L.; Zhao, Y.; Liu, X.; et al. Alterations of the gut microbiome of largemouth bronze gudgeon (Coreius guichenoti) suffering from furunculosis. Sci. Rep. 2016, 6, 30606. [Google Scholar] [CrossRef] [PubMed]
- Cevidanes, A.; Esperón, F.; Di Cataldo, S.; Neves, E.; Sallaberry-Pincheira, N.; Millán, J. Antimicrobial resistance genes in Andean foxes inhabiting anthropized landscapes in central Chile. Sci. Total. Environ. 2020, 724, 138247. [Google Scholar] [CrossRef]
- Hernando-Amado, S.; Coque, T.M.; Baquero, F.; Martínez, J.L. Defining and combating antibiotic resistance from One Health and Global Health perspectives. Nat. Microbiol. 2019, 4, 1432–1442. [Google Scholar] [CrossRef]
- Liu, G.; Xu, N.; Feng, J. Metagenomic analysis of gut microbiota and antibiotic-resistant genes in Anser erythropus wintering at Shengjin and Caizi Lakes in China. Front. Microbiol. 2023, 13, 1081468. [Google Scholar] [CrossRef] [PubMed]
- Ruzauskas, M.; Vaskeviciute, L. Detection of the mcr-1 gene in Escherichia coli prevalent in the migratory bird species Larus argentatus. J. Antimicrob. Chemother. 2016, 71, 2333–2334. [Google Scholar] [CrossRef] [PubMed]
- Dolejská, M.; Senk, D.; Cízek, A.; Rybaríková, J.; Sychra, O.; Literák, I. Antimicrobial resistant Escherichia coli isolates in cattle and house sparrows on two Czech dairy farms. Res. Vet. Sci. 2008, 85, 491–494. [Google Scholar] [CrossRef]
- Oravcová, V.; Peixe, L.; Coque, T.M.; Novais, C.; Francia, M.V.; Literák, I.; Freitas, A.R. Wild corvid birds colonized with vancomycin-resistant Enterococcus faecium of human origin harbor epidemic vanA plasmids. Environ. Int. 2018, 118, 125–133. [Google Scholar] [CrossRef]
- Miller, E.A.; Ponder, J.B.; Willette, M.; Johnson, T.J.; VanderWaal, K.L. Merging Metagenomics and Spatial Epidemiology to Understand the Distribution of Antimicrobial Resistance Genes from Enterobacteriaceae in Wild Owls. Appl. Environ. Microbiol. 2020, 86, e00571-20. [Google Scholar] [CrossRef]
- Song, G.; Qu, Y.H. Environmental changes and uplift of the Qinghai-Tibet Plateau drive genetic diversification and speciation of the birds. Yi Chuan 2025, 47, 133–145. [Google Scholar]
- Wu, D.; Luo, R.; Gong, G.; Zhang, L.; Huang, J.; Cai, C.; Li, Y.; Irshad, I.; Song, R.; Suolang, S. Antimicrobial susceptibility and multilocus sequence typing of Clostridium perfringens isolated from yaks in Qinghai-Tibet plateau, China. Front. Vet. Sci. 2022, 9, 1022215. [Google Scholar] [CrossRef]
- Benmazouz, I.; Jokimäki, J.; Lengyel, S.; Juhász, L.; Kaisanlahti-Jokimäki, M.L.; Kardos, G.; Paládi, P.; Kövér, L. Corvids in Urban Environments: A Systematic Global Literature Review. Animals 2021, 11, 3226. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.H. Corvid cognition. Wiley Interdiscip. Rev. Cogn. Sci. 2014, 5, 361–372. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Li, D.; Liu, C.M.; Luo, R.; Sadakane, K.; Lam, T.W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef]
- Hyatt, D.; Chen, G.L.; Locascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 2010, 11, 119. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Jiang, X.T.; Chai, B.; Li, L.; Yang, Y.; Cole, J.R.; Tiedje, J.M.; Zhang, T. ARGs-OAP v2.0 with an expanded SARG database and Hidden Markov Models for enhancement characterization and quantification of antibiotic resistance genes in environmental metagenomes. Bioinformatics 2018, 34, 2263–2270. [Google Scholar] [CrossRef] [PubMed]
- Pärnänen, K.; Karkman, A.; Hultman, J.; Lyra, C.; Bengtsson-Palme, J.; Larsson, D.G.J.; Rautava, S.; Isolauri, E.; Salminen, S.; Kumar, H.; et al. Maternal gut and breast milk microbiota affect infant gut antibiotic resistome and mobile genetic elements. Nat. Commun. 2018, 9, 3891. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.N.; Gaston, J.M.; Dai, C.L.; Zhao, S.; Poyet, M.; Groussin, M.; Yin, X.; Li, L.G.; van Loosdrecht, M.C.M.; Topp, E.; et al. An omics-based framework for assessing the health risk of antimicrobial resistance genes. Nat. Commun. 2021, 12, 4765. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Ma, Z.B.; Zeng, Z.L.; Yang, X.W.; Huang, Y.; Liu, J.H. The role of wildlife (wild birds) in the global transmission of antimicrobial resistance genes. Zool. Res. 2017, 38, 55–80. [Google Scholar] [CrossRef]
- Brito, I.L. Examining horizontal gene transfer in microbial communities. Nat. Rev. Microbiol. 2021, 19, 442–453. [Google Scholar] [CrossRef]
- Li, L.G.; Huang, Q.; Yin, X.; Zhang, T. Source tracking of antibiotic resistance genes in the environment—Challenges, progress, and prospects. Water. Res. 2020, 185, 116127. [Google Scholar] [CrossRef]
- Zhai, J.; Wang, Y.; Tang, B.; Zheng, S.; He, S.; Zhao, W.; Lin, J.; Li, F.; Bao, Y.; Lancuo, Z.; et al. A comparison of antibiotic resistance genes and mobile genetic elements in wild and captive Himalayan vultures. PeerJ 2024, 12, e17710. [Google Scholar] [CrossRef]
- Fu, Y.; Dou, Q.; Smalla, K.; Wang, Y.; Johnson, T.A.; Brandt, K.K.; Mei, Z.; Liao, M.; Hashsham, S.A.; Schäffer, A.; et al. Gut microbiota research nexus: One Health relationship between human, animal, and environmental resistomes. mLife 2023, 2, 350–364. [Google Scholar] [CrossRef] [PubMed]
- Shi, B.; Zhao, R.; Su, G.; Liu, B.; Liu, W.; Xu, J.; Li, Q.; Meng, J. Metagenomic surveillance of antibiotic resistome in influent and effluent of wastewater treatment plants located on the Qinghai-Tibetan Plateau. Sci. Total. Environ. 2023, 870, 162031. [Google Scholar] [CrossRef]
- Song, M.; Wang, K.; Xie, Y.; Wen, X.; Tu, Y.; Teng, T.; Luo, C.; Zhang, D. Impacts of anthropogenic disturbances on antibiotic resistomes in biological soil crusts on the Qinghai-Tibetan Plateau. Environ. Pollut. 2025, 367, 125582. [Google Scholar] [CrossRef]
- Zhao, J.; Feng, T.; An, X.; Chen, X.; Han, N.; Wang, J.; Chang, G.; Hou, X. Livestock grazing is associated with the gut microbiota and antibiotic resistance genes in sympatric plateau pika (Ochotona curzoniae). Integr. Zool. 2024, 19, 646–661. [Google Scholar] [CrossRef]
- Rohrer, S.D.; Jiménez-Uzcátegui, G.; Parker, P.G.; Chubiz, L.M. Composition and function of the Galapagos penguin gut microbiome vary with age, location, and a putative bacterial pathogen. Sci. Rep. 2023, 13, 5358. [Google Scholar] [CrossRef]
- Li, W.; Wang, Y.; Gao, J.; Wang, A. Antimicrobial resistance and its risks evaluation in wetlands on the Qinghai-Tibetan Plateau. Ecotoxicol. Environ. Saf. 2024, 282, 116699. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Park, J.; Kang, M.; Yang, J.; Park, W. Gain and loss of antibiotic-resistant genes in multidrug resistant bacteria: One Health perspective. J. Microbiol. 2021, 59, 535–545. [Google Scholar] [CrossRef] [PubMed]
- El-Demerdash, A.S.; Kamel, S.A.; Elariny, E.Y.T.; Henidi, H.; Mahran, Y.; Alahdal, H.; Saleh, A.M.; Ibrahim, R.A. Natural Inhibitors of Salmonella MDR Efflux Pumps AcrAB and AcrD: An Integrated in Silico, Molecular, and In Vitro Investigation. Int. J. Mol. Sci. 2024, 25, 12949. [Google Scholar] [CrossRef] [PubMed]
- Zgurskaya, H.I.; Nikaido, H. Cross-linked complex between oligomeric periplasmic lipoprotein AcrA and the inner-membrane-associated multidrug efflux pump AcrB from Escherichia coli. J. Bacteriol. 2000, 182, 4264–4267. [Google Scholar] [CrossRef] [PubMed]
- Bagdad, Y.; Sisquellas, M.; Arthur, M.; Miteva, M.A. Machine Learning and Deep Learning Models for Predicting Noncovalent Inhibitors of AmpC β-Lactamase. ACS Omega 2024, 9, 41334–41342. [Google Scholar] [CrossRef]
- Blanco, P.; Hernando-Amado, S.; Reales-Calderon, J.A.; Corona, F.; Lira, F.; Alcalde-Rico, M.; Bernardini, A.; Sanchez, M.B.; Martinez, J.L. Bacterial Multidrug Efflux Pumps: Much More Than Antibiotic Resistance Determinants. Microorganisms 2016, 4, 14. [Google Scholar] [CrossRef]
- Uddin, T.M.; Chakraborty, A.J.; Khusro, A.; Zidan, B.R.M.; Mitra, S.; Emran, T.B.; Dhama, K.; Ripon, M.K.H.; Gajdács, M.; Sahibzada, M.U.K.; et al. Antibiotic resistance in microbes: History, mechanisms, therapeutic strategies and future prospects. J. Infect. Public Health 2021, 14, 1750–1766. [Google Scholar] [CrossRef]
- Rajput, P.; Nahar, K.S.; Rahman, K.M. Evaluation of Antibiotic Resistance Mechanisms in Gram-Positive Bacteria. Antibiotics 2024, 13, 1197. [Google Scholar] [CrossRef]
- Fernandez, M.; Shkumatov, A.V.; Liu, Y.; Stulemeijer, C.; Derclaye, S.; Efremov, R.G.; Hallet, B.; Alsteens, D. AFM-based force spectroscopy unravels stepwise formation of the DNA transposition complex in the widespread Tn3 family mobile genetic elements. Nucleic. Acids. Res. 2023, 51, 4929–4941. [Google Scholar] [CrossRef]
- Garcillán-Barcia, M.P.; de la Cruz, F. Distribution of IS91 family insertion sequences in bacterial genomes: Evolutionary implications. FEMS. Microbiol. Ecol. 2002, 42, 303–313. [Google Scholar] [CrossRef]
- Lucatelli, A.; Monte, D.F.M.; Alvares, P.P.; Guth, B.E.C.; Destro, M.T.; Franco, B.D.G.M.; Landgraf, M. Virulent shiga toxin-producing Escherichia coli (STEC) O157:H7 ST11 isolated from ground beef in Brazil. Braz. J. Microbiol. 2024, 55, 3513–3520. [Google Scholar] [CrossRef] [PubMed]
- Adator, E.H.; Walker, M.; Narvaez-Bravo, C.; Zaheer, R.; Goji, N.; Cook, S.R.; Tymensen, L.; Hannon, S.J.; Church, D.; Booker, C.W.; et al. Whole Genome Sequencing Differentiates Presumptive Extended Spectrum Beta-Lactamase Producing Escherichia coli along Segments of the One Health Continuum. Microorganisms 2020, 8, 448. [Google Scholar] [CrossRef] [PubMed]
- Vincent, J.L. Nosocomial infections in adult intensive-care units. Lancet 2003, 361, 2068–2077. [Google Scholar] [CrossRef] [PubMed]
- Frank, C.; Werber, D.; Cramer, J.P.; Askar, M.; Faber, M.; an der Heiden, M.; Bernard, H.; Fruth, A.; Prager, R.; Spode, A.; et al. Epidemic profile of Shiga-toxin-producing Escherichia coli O104:H4 outbreak in Germany. N. Engl. J. Med. 2011, 365, 1771–1780. [Google Scholar] [CrossRef]
- Rezazadegan, M.; Forootani, B.; Hoveyda, Y.; Rezazadegan, N.; Amani, R. Major heavy metals and human gut microbiota composition: A systematic review with nutritional approach. J. Health Popul. Nutr. 2025, 44, 21. [Google Scholar] [CrossRef]
- Cani, P.D.; Possemiers, S.; Van de Wiele, T.; Guiot, Y.; Everard, A.; Rottier, O.; Geurts, L.; Naslain, D.; Neyrinck, A.; Lambert, D.M.; et al. Changes in gut microbiota control inflammation in obese mice through a mechanism involving GLP-2-driven improvement of gut permeability. Gut 2009, 58, 1091–1103. [Google Scholar] [CrossRef]
- Munk, P.; Knudsen, B.E.; Lukjancenko, O.; Duarte, A.S.R.; Van Gompel, L.; Luiken, R.E.C.; Smit, L.A.M.; Schmitt, H.; Garcia, A.D.; Hansen, R.B.; et al. Abundance and diversity of the faecal resistome in slaughter pigs and broilers in nine European countries. Nat. Microbiol. 2018, 3, 898–908. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Cui, Q.; Hou, Y.; He, S.; Zhao, W.; Lancuo, Z.; Sharshov, K.; Wang, W. Metagenomic Insights into the Diverse Antibiotic Resistome of Non-Migratory Corvidae Species on the Qinghai–Tibetan Plateau. Vet. Sci. 2025, 12, 297. https://doi.org/10.3390/vetsci12040297
Wang Y, Cui Q, Hou Y, He S, Zhao W, Lancuo Z, Sharshov K, Wang W. Metagenomic Insights into the Diverse Antibiotic Resistome of Non-Migratory Corvidae Species on the Qinghai–Tibetan Plateau. Veterinary Sciences. 2025; 12(4):297. https://doi.org/10.3390/vetsci12040297
Chicago/Turabian StyleWang, You, Quanchao Cui, Yuliang Hou, Shunfu He, Wenxin Zhao, Zhuoma Lancuo, Kirill Sharshov, and Wen Wang. 2025. "Metagenomic Insights into the Diverse Antibiotic Resistome of Non-Migratory Corvidae Species on the Qinghai–Tibetan Plateau" Veterinary Sciences 12, no. 4: 297. https://doi.org/10.3390/vetsci12040297
APA StyleWang, Y., Cui, Q., Hou, Y., He, S., Zhao, W., Lancuo, Z., Sharshov, K., & Wang, W. (2025). Metagenomic Insights into the Diverse Antibiotic Resistome of Non-Migratory Corvidae Species on the Qinghai–Tibetan Plateau. Veterinary Sciences, 12(4), 297. https://doi.org/10.3390/vetsci12040297