
Individualized Pooled CRISPR/Cas9 Screenings Identify CDK2 as a Druggable Vulnerability in a Canine Mammary Carcinoma Patient

 , , , , and
, , , , and
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and Tissue Processing
2.2. Histology
2.3. Maintenance of CMT Organoid Cultures
2.4. Lentivirus Production, Lentiviral Transduction
2.5. Pooled CRISPR/Cas9 Screening
2.6. Proliferation Assay
2.7. Drug Testing and Cell Viability Assays
2.8. RNA Isolation and Sequencing of the Primary Tumors and Non-Neoplastic Mammary Tissues
2.9. RNA Sequencing of the Organoids (Paired Carcinoma and Non-Neoplastic Mammary Tissue)
2.10. Statistical Analysis and Data Representation
3. Results
3.1. Design and Lentiviral Transduction of Two Custom Canine CRISPR/Cas9 Libraries to Identify Therapeutic Vulnerabilities in CMTs
3.2. Pooled CRISPR/Cas9 Screening Targeting the Canine Epigenome in PDOs to Better Understand CMT Biology and Tumorigenesis
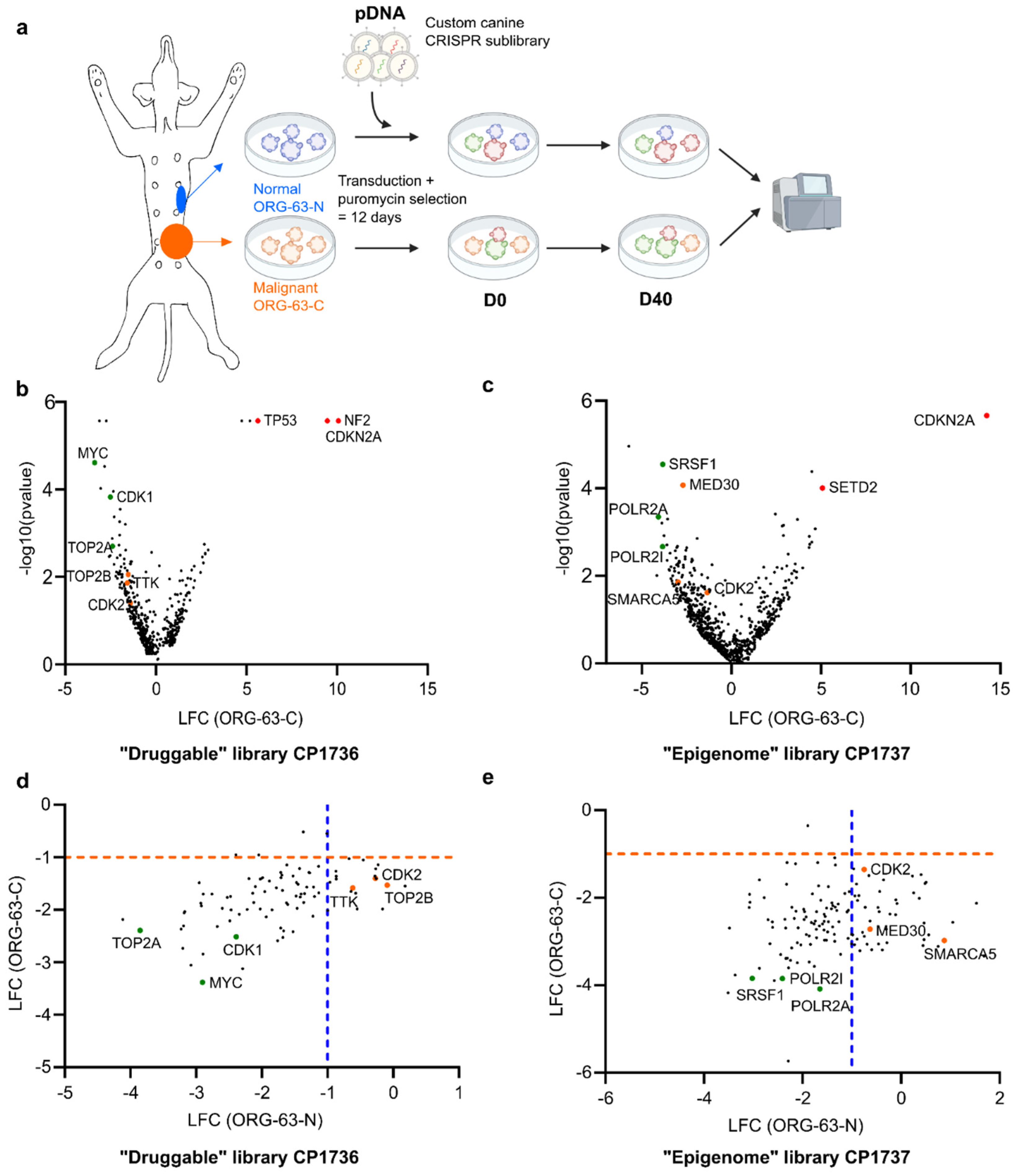
- Outline of the screens performed with a custom canine CRISPR/Cas9 library
- Volcano plot representing depleted (LFC < 0) and enriched (LFC > 0) genes for ORG-63-C forty days (D40) after D0 for the druggable screen. LFC and p-values were calculated from two technical replicates with MAGeCK analysis. Each dot represents one gene for which at least four sgRNAs (out of six) were enrolled in the analysis. Selected hits are color-coded.
- Volcano plot representing depleted (LFC < 0) and enriched (LFC > 0) genes for ORG-63-C forty days (D40) after D0 for the epigenome screen. LFC and p-values were calculated from two technical replicates with MAGeCK analysis. Each dot represents one gene for which at least four sgRNAs (out of six) were enrolled in the analysis. Selected hits are color-coded.
- Scatter plot of the log fold change (LFC) of ORG-63-C and ORG-63-N for the druggable screen. Each dot represents one gene for which at least four sgRNAs (out of six) were enrolled in the analysis, and the p-value < 0.05 for at least one of the two ORG lines. Selected hits are color-coded.
- Scatter plot of the log fold change (LFC) of ORG-63-C and ORG-63-N for the epigenome screen. Each dot represents one gene for which at least four sgRNAs (out of six) were enrolled in the analysis, and the p-value < 0.05 for at least one of the two ORG lines. Selected hits are color-coded.

{kind=link}
{kind=link}
{kind=link}
| Gene | Gene Description | p-Value_N | lfc_N | p-Value_C | lfc_C |
|---|---|---|---|---|---|
| SPEN | Msx2-interacting protein | 0.9141 | 1.531 | 0.02454 | −2.126 |
| VPS72 | Vacuolar protein sorting-associated protein 72 homolog | 0.8979 | 0.4675 | 0.01602 | −2.577 |
| PLOD2 | Procollagen-lysine,2-oxoglutarate 5-dioxygenase 2 | 0.8599 | 1.6872 | 0.01308 | −3.333 |
| NCOR1 | Nuclear receptor co-repressor 1 | 0.7682 | 0.25754 | 0.021244 | −1.889 |
| SRSF2 | Serine/arginine-rich splicing factor 2 | 0.6809 | 0.46804 | 0.015419 | −2.955 |
| HDAC1 | Histone deacetylase 1 | 0.6639 | 0.50999 | 0.045327 | −1.647 |
| ACTL6A | Actin-like protein 6A | 0.60347 | 0.48882 | 0.009406 | −1.670 |
| TBL1X | F-box-like/WD repeat-containing protein TBL1X | 0.60168 | −0.4438 | 0.046566 | −2.318 |
| BAZ1A | Bromodomain adjacent to zinc finger domain protein 1A | 0.59139 | −0.2259 | 0.014092 | −2.371 |
| NOC2L | Nucleolar complex protein 2 homolog | 0.58843 | −0.4693 | 0.018451 | −2.221 |
| TAF8 | Transcription initiation factor TFIID subunit 8 | 0.58046 | −0.3521 | 0.018177 | −1.594 |
| TFDP1 | Transcription factor Dp-1 | 0.46931 | 0.3727 | 0.033807 | −2.012 |
| CBX5 | Chromobox protein homolog 5 | 0.45174 | 0.40584 | 0.039584 | −1.473 |
| SMARCA5 | SWI/SNF-related matrix-associated actin-dependent | 0.44409 | 0.87841 | 0.013878 | −2.98 |
| SRRM1 | Serine/arginine repetitive matrix protein 1 | 0.43516 | −0.4318 | 0.019006 | −1.859 |
| COPS5 | Information COP9 signalosome complex subunit 5 | 0.43285 | 0.75487 | 0.015724 | −3.111 |
| NAP1L1 | Nucleosome assembly protein 1-like 1 | 0.40623 | −0.3009 | 0.024866 | −2.719 |
| HASPIN | Serine/threonine-protein kinase haspin | 0.40398 | −0.5946 | 0.007778 | −3.185 |
| RCC1 | Regulator of chromosome condensation | 0.39469 | 0.23547 | 0.01764 | −1.503 |
| OIP5 | Protein Mis18-beta | 0.38418 | −0.7262 | 0.018399 | −2.218 |
| SRSF5 | Splicing factor, arginine/serine-rich 4/5/6 | 0.34631 | −0.8543 | 0.04839 | −1.951 |
| MIS18BP1 | Mis18-binding protein 1 | 0.34095 | −1.0051 | 0.019045 | −2.84 |
| RBPMS | RNA binding protein, mRNA processing factor | 0.33435 | −0.9347 | 0.047674 | −1.959 |
| SETD1A | Histone-lysine N-methyltransferase SETD1A | 0.32603 | −0.6491 | 0.020014 | −1.490 |
| CDK2 | Cyclin-dependent kinase 2 | 0.31995 | −0.7495 | 0.024024 | −1.354 |
| USP7 | Ubiquitin carboxyl-terminal hydrolase 7 | 0.31791 | −1.0529 | 0.027406 | −1.860 |
| SUZ12 | SUZ12 Polycomb repressive complex 2 subunit | 0.31684 | −0.4537 | 0.019363 | −3.057 |
| DHX38 | Pre-mRNA-splicing factor ATP-dependent RNA helicase PRP16 | 0.31248 | −0.6269 | 0.025534 | −2.534 |
| KDM8 | JmjC-domain-containing protein 5 | 0.30885 | −0.8832 | 0.007473 | −2.061 |
| OTUB1 | Ubiquitin thioesterase | 0.30217 | −0.9042 | 0.017099 | −1.689 |
| GTF3C4 | General transcription factor 3C polypeptide 4 | 0.29667 | −0.1029 | 0.048652 | −2.046 |
| CENPA | Histone H3-like centromeric protein A | 0.2798 | −0.8915 | 0.008441 | −2.949 |
| ZZZ3 | ZZ-type zinc finger-containing protein 3 | 0.26212 | −0.8331 | 0.017243 | −3.213 |
| CHEK1 | Serine/threonine-protein kinase Chk1 | 0.25733 | 1.0502 | 0.02631 | −2.563 |
| DDX17 | Probable ATP-dependent RNA helicase DDX17 | 0.24814 | 0.56402 | 0.031922 | −2.85 |
| SNAPC4 | snRNA-activating protein complex subunit 4 | 0.24499 | −0.9796 | 0.021048 | −2.847 |
| MED30 | Mediator Complex Subunit 30 | 0.17747 | −0.6267 | 8.51 × 10−5 | −2.719 |
| PCNA | Proliferating cell nuclear antigen | 0.14632 | −0.6846 | 0.012683 | −3.062 |
| ECD | Protein ecdysoneless homolog | 0.12707 | −0.2861 | 0.026677 | −3.026 |
| RSF1 | Remodeling and spacing factor 1 | 0.11897 | −0.7464 | 0.032301 | −2.881 |
| UBE2A | Ubiquitin-conjugating enzyme E2 A | 0.11589 | −0.9619 | 0.010972 | −1.337 |
| GTF2B | Transcription initiation factor IIB | 0.1149 | −0.7796 | 0.023854 | −2.791 |
| SMC2 | Structural maintenance of chromosomes protein 2 | 0.10679 | −0.3809 | 0.006639 | −3.191 |
| RUVBL1 | RuvB-like 1 | 0.096328 | 0.44603 | 0.0039294 | −3.093 |
| AURKB | Aurora kinase B | 0.068751 | −0.9429 | 0.0055614 | −3.071 |
3.3. Pooled CRISPR/Cas9 Screening in PDOs to Identify Potential Drug Targets for CMTs
3.4. Candidates from the CRISPR/Cas9 Screens Are Expressed in Primary Tumor Tissues and PDOs but Very Rarely Differentially Expressed Compared to the Non-Neoplastic Mammary Tissues
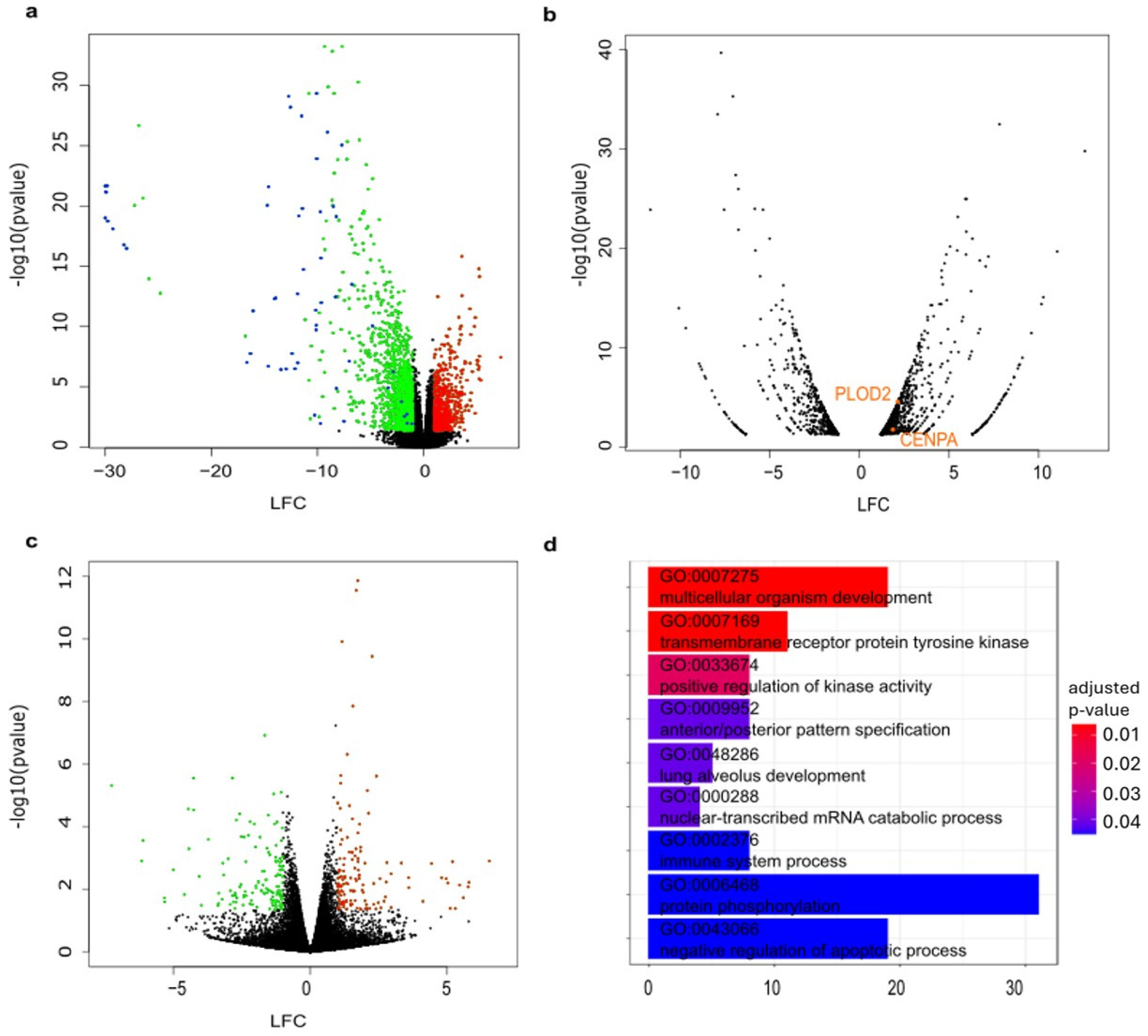
- Volcano plot representing downregulated (log fold change = LFC < 0) and upregulated (LFC 0) genes between all CMT samples compared with all normal mammary tissue samples. Significantly (p-value < 0.05 and |LFC|> = 1.0) downregulated (green) and upregulated (red) are color coded. Keratins and keratin-associated proteins are labeled in blue.
- Volcano plot representing significantly downregulated and upregulated (p-value < 0.05 and |LFC| > 1.0) genes between D63_3_CMT and D63_nor. Candidates from the CRISPR/Cas9 screens that are differentially expressed between D63_3_CMT and D63_nor are labeled in orange.
- Volcano plot representing downregulated (LFC < 0) and upregulated (LFC > 0) genes between three organoid lines derived from carcinomas compared with their matched organoids derived from normal mammary tissues. Significantly (p-value < 0.05 and |LFC| > 1.0) downregulated (green) and upregulated (red) are color coded.
- Bar plot showing Gene Ontology (biological processes) analysis results of all differentially expressed genes between three organoid lines derived from carcinomas compared with their matched organoids derived from normal mammary tissues. The color of each bar represents the p-value of each term involved in the analysis, and the bar size represents the gene counts for this term.
3.5. CDK2 May Be a Druggable Vulnerability for CMTs
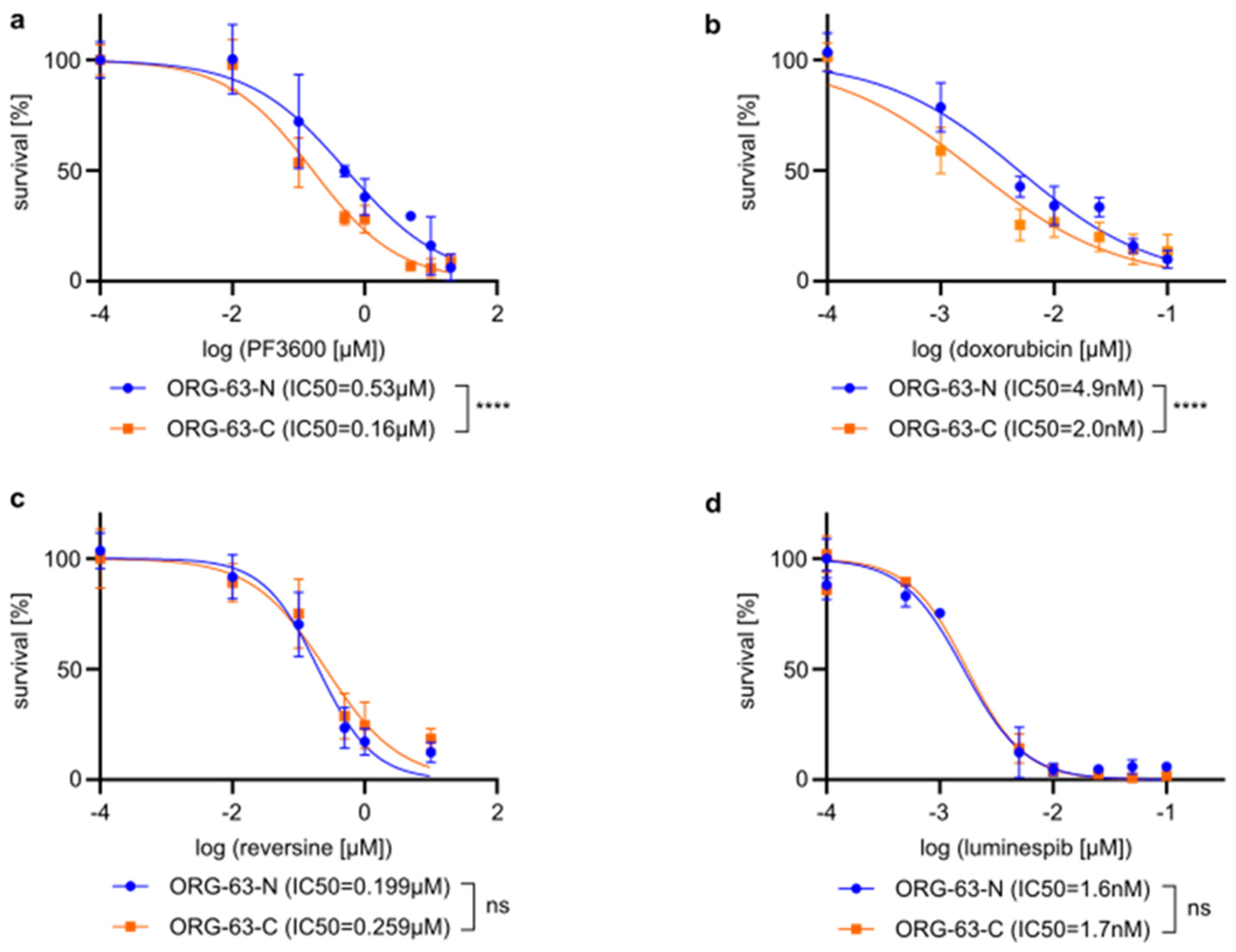
- Dose-response curves illustrating the effect of PF3600 on cell viability in organoid lines ORG-63-C and ORG-63-N. Error bars represent the standard deviation (SD) of three independent experiments. Statistical analysis of the log-transformed IC50 values was performed using an unpaired t-test, **** p < 0.0001. IC50s are indicated in the figure legend.
- Dose-response curves illustrating the effect of doxorubicin on cell viability in organoid lines ORG-63-C and ORG-63-N. Error bars represent the standard deviation (SD) of three independent experiments. Statistical analysis of the log-transformed IC50 values was performed using an unpaired t-test, **** p < 0.0001. IC50s are indicated in the figure legend.
- Dose-response curves depicting the effect of reversine on cell viability in organoid lines ORG-63-C and ORG-63-N. Error bars represent the standard deviation (SD) of three independent experiments. Statistical analysis of the log-transformed IC50 values was performed using an unpaired t-test, ns = non-significant. IC50s are indicated in the figure legend.
- Dose-response curves depicting the effect of luminespib on cell viability in organoid lines ORG-63-C and ORG-63-N. Error bars represent the standard deviation (SD) of three independent experiments. Statistical analysis of the log-transformed IC50 values was performed using an unpaired t-test, ns = non-statistical. IC50s are indicated in the figure legend.
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CMT | Canine Mammary Tumor |
| CRISPR | Clustered Regularly Interspaced Short Palindromic Repeats |
| D0 | Day 0 |
| D40 | Day 40 |
| HBC | Human Breast Cancer |
| IC50 | Half Maximal Inhibitory Concentration |
| LFC | Log Fold Change |
| mRNA | Messenger RNA |
| pDNA | Plasmid DNA |
| PDO | Patient-Derived Organoid |
| sgRNA | Single Guide RNA |
| TCGA | The Cancer Genome Atlas |
References
- Rowell, J.L.; McCarthy, D.O.; Alvarez, C.E. Dog Models of Naturally Occurring Cancer. Trends Mol. Med. 2011, 17, 380–388. [Google Scholar] [CrossRef]
- Grüntzig, K.; Graf, R.; Hässig, M.; Welle, M.; Meier, D.; Lott, G.; Erni, D.; Schenker, N.S.; Guscetti, F.; Boo, G.; et al. The Swiss Canine Cancer Registry: A Retrospective Study on the Occurrence of Tumours in Dogs in Switzerland from 1955 to 2008. J. Comp. Pathol. 2015, 152, 161–171. [Google Scholar] [CrossRef]
- Pinello, K.; Pires, I.; Castro, A.F.; Carvalho, P.T.; Santos, A.; de Matos, A.; Queiroga, F.; Canadas-Sousa, A.; Dias-Pereira, P.; Catarino, J.; et al. Cross Species Analysis and Comparison of Tumors in Dogs and Cats, by Age, Sex, Topography and Main Morphologies. Data from Vet-OncoNet. Vet. Sci. 2022, 9, 167. [Google Scholar] [CrossRef]
- Goldschmidt, M.H.; Peña, L.; Rasotto, R.; Zappulli, V. Classification and Grading of Canine Mammary Tumors. Vet. Pathol. 2011, 48, 117–131. [Google Scholar] [CrossRef]
- Sorenmo, K.U.; Worley, D.R.; Zappulli, V. Tumors of the Mammary Gland. In Withrow and MacEwen’s Small Animal Clinical Oncology; Saunders: Birmingham, AL, USA, 2019; pp. 604–625. [Google Scholar] [CrossRef]
- Sorenmo, K.U.; Kristiansen, V.M.; Cofone, M.A.; Shofer, F.S.; Breen, A.-M.; Langeland, M.; Mongil, C.M.; Grondahl, A.M.; Teige, J.; Goldschmidt, M.H. Canine Mammary Gland Tumours; a Histological Continuum from Benign to Malignant; Clinical and Histopathological Evidence. Vet. Comp. Oncol. 2009, 7, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Sorenmo, K.U.; Shofer, F.S.; Goldschmidt, M.H. Effect of Spaying and Timing of Spaying on Survival of Dogs with Mammary Carcinoma. J. Vet. Intern. Med. 2000, 14, 266–270. [Google Scholar] [CrossRef]
- Sleeckx, N.; de Rooster, H.; Veldhuis Kroeze, E.; Van Ginneken, C.; Van Brantegem, L. Canine Mammary Tumours, an Overview. Reprod. Domest. Anim. 2011, 46, 1112–1131. [Google Scholar] [CrossRef]
- Borst, P.; Wessels, L. Do Predictive Signatures Really Predict Response to Cancer Chemotherapy? Cell Cycle 2010, 9, 4836–4840. [Google Scholar] [CrossRef]
- Alsaihati, B.A.; Ho, K.L.; Watson, J.; Feng, Y.; Wang, T.; Dobbin, K.K.; Zhao, S. Canine Tumor Mutational Burden Is Correlated with TP53 Mutation across Tumor Types and Breeds. Nat. Commun. 2021, 12, 1–13. [Google Scholar] [CrossRef]
- Inglebert, M.; Dettwiler, M.; Hahn, K.; Letko, A.; Drogemuller, C.; Doench, J.; Brown, A.; Memari, Y.; Davies, H.R.; Degasperi, A.; et al. A Living Biobank of Canine Mammary Tumor Organoids as a Comparative Model for Human Breast Cancer. Sci. Rep. 2022, 12, 1–14. [Google Scholar] [CrossRef]
- Kim, T.-M.; Yang, I.S.; Seung, B.-J.; Lee, S.; Kim, D.; Ha, Y.-J.; Seo, M.; Kim, K.-K.; Kim, H.S.; Cheong, J.-H.; et al. Cross-Species Oncogenic Signatures of Breast Cancer in Canine Mammary Tumors. Nat. Commun. 2020, 11, 3616. [Google Scholar] [CrossRef]
- Pereira, B.; Chin, S.-F.; Rueda, O.M.; Vollan, H.-K.M.; Provenzano, E.; Bardwell, H.A.; Pugh, M.; Jones, L.; Russell, R.; Sammut, S.-J.; et al. The Somatic Mutation Profiles of 2,433 Breast Cancers Refine Their Genomic and Transcriptomic Landscapes. Nat. Commun. 2016, 7, 11479. [Google Scholar] [CrossRef]
- Nik-Zainal, S.; Davies, H.; Staaf, J.; Ramakrishna, M.; Glodzik, D.; Zou, X.; Martincorena, I.; Alexandrov, L.B.; Martin, S.; Wedge, D.C.; et al. Landscape of Somatic Mutations in 560 Breast Cancer Whole-Genome Sequences. Nature 2016, 534, 47–54. [Google Scholar] [CrossRef]
- Zappulli, V.; Kiupel, M. Surgical pathology of tumors of domestic animals. In Surgical Pathology of Tumors of Domestic Animals: Mammary Tumors; Davis-Thompson DVM Foundation: Baton Rouge, LA, USA, 2019; ISBN 9781733749114. [Google Scholar]
- Varallo, G.; Gelaleti, G.; Maschio-Signorini, L.; Moschetta, M.; Lopes, J.; De Nardi, A.; Tinucci-Costa, M.; Rocha, R.; de Campos Zuccari, D. Prognostic Phenotypic Classification for Canine Mammary Tumors. Oncol. Lett. 2019, 18, 6545–6553. [Google Scholar] [CrossRef]
- Peña, L.; Gama, A.; Goldschmidt, M.H.; Abadie, J.; Benazzi, C.; Castagnaro, M.; Díez, L.; Gärtner, F.; Hellmén, E.; Kiupel, M.; et al. Canine Mammary Tumors. Vet. Pathol. 2014, 51, 127–145. [Google Scholar] [CrossRef]
- Hartwell, L.H.; Szankasi, P.; Roberts, C.J.; Murray, A.W.; Friend, S.H. Integrating Genetic Approaches into the Discovery of Anticancer Drugs. Science 1997, 278, 1064–1068. [Google Scholar] [CrossRef]
- Hart, T.; Tong, A.H.Y.; Chan, K.; Van Leeuwen, J.; Seetharaman, A.; Aregger, M.; Chandrashekhar, M.; Hustedt, N.; Seth, S.; Noonan, A.; et al. Evaluation and Design of Genome-Wide CRISPR/SpCas9 Knockout Screens. G3 Genes Genomes Genet. 2017, 7, 2719–2727. [Google Scholar] [CrossRef]
- Schuster, A.; Erasimus, H.; Fritah, S.; Nazarov, P.V.; van Dyck, E.; Niclou, S.P.; Golebiewska, A. RNAi/CRISPR Screens: From a Pool to a Valid Hit. Trends Biotechnol. 2019, 37, 38–55. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhu, S.; Cai, C.; Yuan, P.; Li, C.; Huang, Y.; Wei, W. High-Throughput Screening of a CRISPR/Cas9 Library for Functional Genomics in Human Cells. Nature 2014, 509, 487–491. [Google Scholar] [CrossRef]
- Behan, F.M.; Iorio, F.; Picco, G.; Gonçalves, E.; Beaver, C.M.; Migliardi, G.; Santos, R.; Rao, Y.; Sassi, F.; Pinnelli, M.; et al. Prioritization of Cancer Therapeutic Targets Using CRISPR–Cas9 Screens. Nature 2019, 568, 511–516. [Google Scholar] [CrossRef]
- Ungricht, R.; Guibbal, L.; Lasbennes, M.-C.; Orsini, V.; Beibel, M.; Waldt, A.; Cuttat, R.; Carbone, W.; Basler, A.; Roma, G.; et al. Genome-Wide Screening in Human Kidney Organoids Identifies Developmental and Disease-Related Aspects of Nephrogenesis. Cell Stem Cell 2022, 29, 160–175.e7. [Google Scholar] [CrossRef]
- Gerhards, N.M.; Rottenberg, S. New Tools for Old Drugs: Functional Genetic Screens to Optimize Current Chemotherapy. Drug Resist. Updates 2018, 36, 30–46. [Google Scholar] [CrossRef]
- Michels, B.E.; Mosa, M.H.; Streibl, B.I.; Zhan, T.; Menche, C.; Abou-El-Ardat, K.; Darvishi, T.; Członka, E.; Wagner, S.; Winter, J.; et al. Pooled In Vitro and In Vivo CRISPR-Cas9 Screening Identifies Tumor Suppressors in Human Colon Organoids. Cell Stem Cell 2020, 26, 782–792.e7. [Google Scholar] [CrossRef]
- Duarte, A.A.; Gogola, E.; Sachs, N.; Barazas, M.; Annunziato, S.; R De Ruiter, J.; Velds, A.; Blatter, S.; Houthuijzen, J.M.; Van De Ven, M.; et al. BRCA-Deficient Mouse Mammary Tumor Organoids to Study Cancer-Drug Resistance. Nat. Methods 2018, 15, 134–140. [Google Scholar] [CrossRef]
- Francica, P.; Mutlu, M.; Blomen, V.A.; Oliveira, C.; Nowicka, Z.; Trenner, A.; Gerhards, N.M.; Bouwman, P.; Stickel, E.; Hekkelman, M.L.; et al. Functional Radiogenetic Profiling Implicates ERCC6L2 in Non-Homologous End Joining. Cell Rep. 2020, 32, 108068. [Google Scholar] [CrossRef]
- Koo, B.-K.; Stange, D.E.; Sato, T.; Karthaus, W.; Farin, H.F.; Huch, M.; van Es, J.H.; Clevers, H. Controlled Gene Expression in Primary Lgr5 Organoid Cultures. Nat. Methods 2012, 9, 81–83. [Google Scholar] [CrossRef]
- Li, W.; Xu, H.; Xiao, T.; Cong, L.; Love, M.I.; Zhang, F.; Irizarry, R.A.; Liu, J.S.; Brown, M.; Liu, X.S. MAGeCK Enables Robust Identification of Essential Genes from Genome-Scale CRISPR/Cas9 Knockout Screens. Genome Biol. 2014, 15, 554. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. In Proceedings of the Bioinformatics, Oxford, UK, 1 September 2018; Oxford University Press: Oxford, UK, 2018; Volume 34, pp. i884–i890. [Google Scholar]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie Enables Improved Reconstruction of a Transcriptome from RNA-Seq Reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-Optimal Probabilistic RNA-Seq Quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Doench, J.G.; Fusi, N.; Sullender, M.; Hegde, M.; Vaimberg, E.W.; Donovan, K.F.; Smith, I.; Tothova, Z.; Wilen, C.; Orchard, R.; et al. Optimized SgRNA Design to Maximize Activity and Minimize Off-Target Effects of CRISPR-Cas9. Nat. Biotechnol. 2016, 34, 184–191. [Google Scholar] [CrossRef]
- Sanson, K.R.; Hanna, R.E.; Hegde, M.; Donovan, K.F.; Strand, C.; Sullender, M.E.; Vaimberg, E.W.; Goodale, A.; Root, D.E.; Piccioni, F.; et al. Optimized Libraries for CRISPR-Cas9 Genetic Screens with Multiple Modalities. Nat. Commun. 2018, 9, 5416. [Google Scholar] [CrossRef]
- Henser-Brownhill, T.; Monserrat, J.; Scaffidi, P. Generation of an Arrayed CRISPR-Cas9 Library Targeting Epigenetic Regulators: From High-Content Screens to in Vivo Assays. Epigenetics 2017, 12, 1065–1075. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.T.; Guarecuco, R.; Gates, L.A.; Barrows, D.; Passarelli, M.C.; Carey, B.; Baudrier, L.; Jeewajee, S.; La, K.; Prizer, B.; et al. ZBTB1 Regulates Asparagine Synthesis and Leukemia Cell Response to L-Asparaginase. Cell Metab. 2020, 31, 852–861.e6. [Google Scholar] [CrossRef]
- Gokirmak, T.; Sharp, J.; Revankar, C.; Piper, D.R. Abstract LB-067: Identifying Novel and Druggable Targets in a Triple Negative Breast Cancer Cell Line Using the Invitrogen LentiArray Human Kinase CRISPR Library. Cancer Res. 2018, 78, LB-067. [Google Scholar] [CrossRef]
- Hu, M.; Lei, X.Y.; Larson, J.D.; McAlonis, M.; Ford, K.; McDonald, D.; Mach, K.; Rusert, J.M.; Wechsler-Reya, R.J.; Mali, P. Integrated Genome and Tissue Engineering Enables Screening of Cancer Vulnerabilities in Physiologically Relevant Perfusable Ex Vivo Cultures. Biomaterials 2022, 280, 121276. [Google Scholar] [CrossRef]
- Luo, H.; Lin, Y.; Gao, F.; Zhang, C.-T.; Zhang, R. DEG 10, an Update of the Database of Essential Genes That Includes Both Protein-Coding Genes and Noncoding Genomic Elements: Table 1. Nucleic Acids Res. 2014, 42, D574–D580. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING V11: Protein–Protein Association Networks with Increased Coverage, Supporting Functional Discovery in Genome-Wide Experimental Datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a Reference Resource for Gene and Protein Annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef] [PubMed]
- Coussens, L.M.; Werb, Z. Inflammation and Cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Tadesse, S.; Anshabo, A.T.; Portman, N.; Lim, E.; Tilley, W.; Caldon, C.E.; Wang, S. Targeting CDK2 in Cancer: Challenges and Opportunities for Therapy. Drug Discov. Today 2020, 25, 406–413. [Google Scholar] [CrossRef]
- Satyanarayana, A.; Kaldis, P. Mammalian Cell-Cycle Regulation: Several Cdks, Numerous Cyclins and Diverse Compensatory Mechanisms. Oncogene 2009, 28, 2925–2939. [Google Scholar] [CrossRef] [PubMed]
- Freeman-Cook, K.D.; Hoffman, R.L.; Behenna, D.C.; Boras, B.; Carelli, J.; Diehl, W.; Ferre, R.A.; He, Y.A.; Hui, A.; Huang, B.; et al. Discovery of PF-06873600, a CDK2/4/6 Inhibitor for the Treatment of Cancer. J. Med. Chem. 2021, 64, 9056–9077. [Google Scholar] [CrossRef]
- Freeman-Cook, K.; Hoffman, R.L.; Miller, N.; Almaden, J.; Chionis, J.; Zhang, Q.; Eisele, K.; Liu, C.; Zhang, C.; Huser, N.; et al. Expanding Control of the Tumor Cell Cycle with a CDK2/4/6 Inhibitor. Cancer Cell 2021, 39, 1404–1421.e11. [Google Scholar] [CrossRef]
- Pommier, Y.; Leo, E.; Zhang, H.; Marchand, C. DNA Topoisomerases and Their Poisoning by Anticancer and Antibacterial Drugs. Chem. Biol. 2010, 17, 421–433. [Google Scholar] [CrossRef]
- Libouban, M.A.A.; de Roos, J.A.D.M.; Uitdehaag, J.C.M.; Willemsen-Seegers, N.; Mainardi, S.; Dylus, J.; de Man, J.; Tops, B.; Meijerink, J.P.P.; Storchová, Z.; et al. Stable Aneuploid Tumors Cells Are More Sensitive to TTK Inhibition than Chromosomally Unstable Cell Lines. Oncotarget 2017, 8, 38309–38325. [Google Scholar] [CrossRef] [PubMed]
- D’Alise, A.M.; Amabile, G.; Iovino, M.; Di Giorgio, F.P.; Bartiromo, M.; Sessa, F.; Villa, F.; Musacchio, A.; Cortese, R. Reversine, a Novel Aurora Kinases Inhibitor, Inhibits Colony Formation of Human Acute Myeloid Leukemia Cells. Mol. Cancer Ther. 2008, 7, 1140–1149. [Google Scholar] [CrossRef]
- Augello, G.; Emma, M.R.; Cusimano, A.; Azzolina, A.; Mongiovì, S.; Puleio, R.; Cassata, G.; Gulino, A.; Belmonte, B.; Gramignoli, R.; et al. Targeting HSP90 with the Small Molecule Inhibitor AUY922 (Luminespib) as a Treatment Strategy against Hepatocellular Carcinoma. Int. J. Cancer 2019, 144, 2613–2624. [Google Scholar] [CrossRef]
- Antolin, A.A.; Clarke, P.A.; Collins, I.; Workman, P.; Al-Lazikani, B. Evolution of Kinase Polypharmacology across HSP90 Drug Discovery. Cell Chem. Biol. 2021, 28, 1433–1445.e3. [Google Scholar] [CrossRef] [PubMed]
- Matano, M.; Date, S.; Shimokawa, M.; Takano, A.; Fujii, M.; Ohta, Y.; Watanabe, T.; Kanai, T.; Sato, T. Modeling Colorectal Cancer Using CRISPR-Cas9-Mediated Engineering of Human Intestinal Organoids. Nat. Med. 2015, 21, 256–262. [Google Scholar] [CrossRef]
- Drost, J.; van Boxtel, R.; Blokzijl, F.; Mizutani, T.; Sasaki, N.; Sasselli, V.; de Ligt, J.; Behjati, S.; Grolleman, J.E.; van Wezel, T.; et al. Use of CRISPR-Modified Human Stem Cell Organoids to Study the Origin of Mutational Signatures in Cancer. Science 2017, 358, 234–238. [Google Scholar] [CrossRef]
- Malumbres, M. Cyclin-Dependent Kinases. Genome Biol. 2014, 15, 122. [Google Scholar] [CrossRef]
- Schwartz, G.K.; Shah, M.A. Targeting the Cell Cycle: A New Approach to Cancer Therapy. J. Clin. Oncol. 2005, 23, 9408–9421. [Google Scholar] [CrossRef]
- Finn, R.S.; Martin, M.; Rugo, H.S.; Jones, S.; Im, S.-A.; Gelmon, K.; Harbeck, N.; Lipatov, O.N.; Walshe, J.M.; Moulder, S.; et al. Palbociclib and Letrozole in Advanced Breast Cancer. N. Engl. J. Med. 2016, 375, 1925–1936. [Google Scholar] [CrossRef]
- Pfizer. Phase 1/2a Dose Escalation and Expansion Study Evaluating Safety, Tolerability, Pharmacokinetic, Pharmacody-namics and Anti-Tumor Activity of Pf-06873600 as a Single Agent and in Combination with Endocrine Therapy; Springer: Berlin/Heidelberg, Germany, 2022. [Google Scholar]
- Rosenblum, D.; Gutkin, A.; Dammes, N.; Peer, D. Progress and Challenges towards CRISPR/Cas Clinical Translation. Adv. Drug Deliv. Rev. 2020, 154–155, 176–186. [Google Scholar] [CrossRef]
- Hanker, A.B.; Chatterjee, S.; Wang, Y.; Ye, D.; Sudhan, D.R.; Larsen, B.M.; Smith, L.C.; Zhang, Y.; Kandagatla, V.; Majmudar, K.; et al. Abstract PD2-01: A Platform of CDK4/6 Inhibitor-Resistant Patient-Derived Breast Cancer Organoids Illuminates Mechanisms of Resistance and Therapeutic Vulnerabilities. Cancer Res. 2022, 82, PD2-01. [Google Scholar] [CrossRef]
- Xavier, P.L.P.; Müller, S.; Fukumasu, H. Epigenetic Mechanisms in Canine Cancer. Front. Oncol. 2020, 10, 591843. [Google Scholar] [CrossRef]
- da Costa Ferreira, V.; Pinheiro, D.d.R.; de Sousa, R.M.; de Aguirra, L.R.V.M.; Pereira, W.L.A.; Burbano, R.M.R.; Borges, B.d.N. Methylation Pattern and Mutational Status of BRCA1 in Canine Mammary Tumors in a Brazilian Population. Comp. Clin. Path 2019, 28, 63–67. [Google Scholar] [CrossRef]
- Qiu, H.; Lin, D. Roles of DNA Mutation in the Coding Region and DNA Methylation in the 5′ Flanking Region of BRCA1 in Canine Mammary Tumors. J. Vet. Med. Sci. 2016, 78, 943–949. [Google Scholar] [CrossRef]
- Brandão, Y.d.O.; Toledo, M.B.; Chequin, A.; Cristo, T.G.; Sousa, R.S.; Ramos, E.A.S.; Klassen, G. DNA Methylation Status of the Estrogen Receptor α Gene in Canine Mammary Tumors. Vet. Pathol. 2018, 55, 510–516. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.-J.; Lee, K.-H.; Nam, A.-R.; Cho, J.-Y. Genome-Wide Methylation Profiling in Canine Mammary Tumor Reveals MiRNA Candidates Associated with Human Breast Cancer. Cancers 2019, 11, 1466. [Google Scholar] [CrossRef]
- Choi, H.J.; Jang, S.; Ryu, J.E.; Lee, H.J.; Lee, H.B.; Ahn, W.S.; Kim, H.J.; Lee, H.J.; Lee, H.J.; Gong, G.Y.; et al. Significance of EZH2 Expression in Canine Mammary Tumors. BMC Vet. Res. 2016, 12, 164. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Xiong, H.; Ellis, A.E.; Northrup, N.C.; Rodriguez, C.O.; O’Regan, R.M.; Dalton, S.; Zhao, S. Molecular Homology and Difference between Spontaneous Canine Mammary Cancer and Human Breast Cancer. Cancer Res. 2014, 74, 5045–5056. [Google Scholar] [CrossRef] [PubMed]
- Boggs, R.M.; Wright, Z.M.; Stickney, M.J.; Porter, W.W.; Murphy, K.E. MicroRNA Expression in Canine Mammary Cancer. Mamm. Genome 2008, 19, 561–569. [Google Scholar] [CrossRef]
- Bulkowska, M.; Rybicka, A.; Senses, K.M.; Ulewicz, K.; Witt, K.; Szymanska, J.; Taciak, B.; Klopfleisch, R.; Hellmén, E.; Dolka, I.; et al. MicroRNA Expression Patterns in Canine Mammary Cancer Show Significant Differences between Metastatic and Non-Metastatic Tumours. BMC Cancer 2017, 17, 728. [Google Scholar] [CrossRef]
- Fish, E.J.; Irizarry, K.J.; DeInnocentes, P.; Ellis, C.J.; Prasad, N.; Moss, A.G.; Curt Bird, R. Malignant Canine Mammary Epithelial Cells Shed Exosomes Containing Differentially Expressed MicroRNA That Regulate Oncogenic Networks. BMC Cancer 2018, 18, 1–20. [Google Scholar] [CrossRef]
- Morgan, R.G.; Chambers, A.C.; Legge, D.N.; Coles, S.J.; Greenhough, A.; Williams, A.C. Optimized Delivery of SiRNA into 3D Tumor Spheroid Cultures in Situ. Sci. Rep. 2018, 8, 7952. [Google Scholar] [CrossRef]
- Nadauld, L.D.; Garcia, S.; Natsoulis, G.; Bell, J.M.; Miotke, L.; Hopmans, E.S.; Xu, H.; Pai, R.K.; Palm, C.; Regan, J.F.; et al. Metastatic Tumor Evolution and Organoid Modeling Implicate TGFBR2as a Cancer Driver in Diffuse Gastric Cancer. Genome Biol. 2014, 15, 428. [Google Scholar] [CrossRef]
- Menche, C.; Farin, H.F. Strategies for Genetic Manipulation of Adult Stem Cell-Derived Organoids. Exp. Mol. Med. 2021, 53, 1483–1494. [Google Scholar] [CrossRef]
- Sun, D.; Evans, L.; Perrone, F.; Sokleva, V.; Lim, K.; Rezakhani, S.; Lutolf, M.; Zilbauer, M.; Rawlins, E.L. A Functional Genetic Toolbox for Human Tissue-Derived Organoids. Elife 2021, 10, e67886. [Google Scholar] [CrossRef]
- Andersson-Rolf, A.; Mustata, R.C.; Merenda, A.; Kim, J.; Perera, S.; Grego, T.; Andrews, K.; Tremble, K.; Silva, J.C.R.; Fink, J.; et al. One-Step Generation of Conditional and Reversible Gene Knockouts. Nat. Methods 2017, 14, 287–289. [Google Scholar] [CrossRef]
- Natsume, T.; Kiyomitsu, T.; Saga, Y.; Kanemaki, M.T. Rapid Protein Depletion in Human Cells by Auxin-Inducible Degron Tagging with Short Homology Donors. Cell Rep. 2016, 15, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, K.; Fukagawa, T.; Takisawa, H.; Kakimoto, T.; Kanemaki, M. An Auxin-Based Degron System for the Rapid Depletion of Proteins in Nonplant Cells. Nat. Methods 2009, 6, 917–922. [Google Scholar] [CrossRef]
- Sachs, N.; de Ligt, J.; Kopper, O.; Gogola, E.; Bounova, G.; Weeber, F.; Balgobind, A.V.; Wind, K.; Gracanin, A.; Begthel, H.; et al. A Living Biobank of Breast Cancer Organoids Captures Disease Heterogeneity. Cell 2018, 172, 373–386.e10. [Google Scholar] [CrossRef] [PubMed]
- Marx, V. Method of the Year: Spatially Resolved Transcriptomics. Nat. Methods 2021, 18, 9–14. [Google Scholar] [CrossRef]
- Chen, H.; Zhang, Z.; Jiang, S.; Li, R.; Li, W.; Zhao, C.; Hong, H.; Huang, X.; Li, H.; Bo, X. New Insights on Human Essential Genes Based on Integrated Analysis and the Construction of the HEGIAP Web-Based Platform. Brief. Bioinform. 2019, 21, 1397–1410. [Google Scholar] [CrossRef]
- Hu, M.; Fu, X.; Si, Z.; Li, C.; Sun, J.; Du, X.; Zhang, H. Identification of Differently Expressed Genes Associated With Prognosis and Growth in Colon Adenocarcinoma Based on Integrated Bioinformatics Analysis. Front. Genet. 2019, 10, 1245. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Program—NCI. Available online: https://www.cancer.gov/tcga (accessed on 22 September 2022).
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A Web Server for Cancer and Normal Gene Expression Profiling and Interactive Analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef]
| Gene | Gene Description | p-Value_N | lfc_N | p-Value_C | lfc_C |
|---|---|---|---|---|---|
| HSP90AA1 | Heat shock protein HSP 90-alpha | 0.70337 | 0.18138 | 0.02346 | −1.547 |
| FANCD2 | Fanconi anemia group D2 protein | 0.603 | −0.22887 | 0.04947 | −1.1426 |
| CDK2 | Cyclin-dependent kinase 2 | 0.53595 | −0.27009 | 0.039806 | −1.4028 |
| TOP2B | DNA topoisomerase 2-beta | 0.39481 | −0.0917 | 0.0087646 | −1.5298 |
| RPTOR | Regulatory-associated protein of mTOR | 0.39167 | −0.45626 | 0.048055 | −1.0535 |
| BCL2L1 | Bcl-2-like protein 1 | 0.31849 | −0.26447 | 0.013289 | −1.2187 |
| MYBL1 | Myb proto-oncogene like 1 | 0.31416 | −0.5715 | 0.024879 | −1.6852 |
| TTK | Serine/threonine-protein kinase ttk/mps1 | 0.28795 | −0.61574 | 0.013661 | −1.584 |
| FANCC | Fanconi anemia group C protein | 0.24945 | −0.54887 | 0.0073124 | −1.9841 |
| POLE | DNA polymerase ε catalytic subunit A | 0.24504 | −0.27922 | 0.013083 | −1.3574 |
| CDK12 | Cyclin-dependent kinase 12 | 0.23879 | −0.16167 | 0.008662 | −1.9851 |
| GMPS | GMP synthase [glutamine-hydrolyzing] | 0.23684 | −0.25904 | 0.035056 | −1.4074 |
| CCND3 | G1/S-specific cyclin-D3 | 0.18021 | −0.86676 | 0.017937 | −1.3585 |
| USP5 | Ubiquitin carboxyl-terminal hydrolase 5 | 0.16569 | −0.87191 | 0.023271 | −1.2658 |
| NFKBIA | NF-kappa-B inhibitor alpha | 0.14329 | −0.99465 | 0.0078846 | −2.0144 |
| PI4KB | Phosphatidylinositol 4-kinase beta | 0.10617 | −0.86682 | 0.036475 | −1.4132 |
| FANCE | Fanconi anemia group E protein | 0.09315 | −0.67103 | 0.026213 | −1.027 |
| Library | Gene | p-Value_N | lfc_N | p-Value_C | lfc_C |
|---|---|---|---|---|---|
| CP1736 | CDK2 | 0.53595 | −0.27009 | 0.039806 | −1.4028 |
| CDK4 | 0.92691 | 0.28251 | 0.16707 | −0.84773 | |
| CDK6 | 0.10149 | −1.1315 | 0.0024321 | −1.6764 | |
| CP1737 | CDK2 | 0.31995 | −0.74951 | 0.024024 | −1.3541 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Inglebert, M.; Dettwiler, M.; He, C.; Markkanen, E.; Opitz, L.; Naguleswaran, A.; Rottenberg, S. Individualized Pooled CRISPR/Cas9 Screenings Identify CDK2 as a Druggable Vulnerability in a Canine Mammary Carcinoma Patient. Vet. Sci. 2025, 12, 183. https://doi.org/10.3390/vetsci12020183
Inglebert M, Dettwiler M, He C, Markkanen E, Opitz L, Naguleswaran A, Rottenberg S. Individualized Pooled CRISPR/Cas9 Screenings Identify CDK2 as a Druggable Vulnerability in a Canine Mammary Carcinoma Patient. Veterinary Sciences. 2025; 12(2):183. https://doi.org/10.3390/vetsci12020183
Chicago/Turabian StyleInglebert, Marine, Martina Dettwiler, Chang He, Enni Markkanen, Lennart Opitz, Arunasalam Naguleswaran, and Sven Rottenberg. 2025. "Individualized Pooled CRISPR/Cas9 Screenings Identify CDK2 as a Druggable Vulnerability in a Canine Mammary Carcinoma Patient" Veterinary Sciences 12, no. 2: 183. https://doi.org/10.3390/vetsci12020183
APA StyleInglebert, M., Dettwiler, M., He, C., Markkanen, E., Opitz, L., Naguleswaran, A., & Rottenberg, S. (2025). Individualized Pooled CRISPR/Cas9 Screenings Identify CDK2 as a Druggable Vulnerability in a Canine Mammary Carcinoma Patient. Veterinary Sciences, 12(2), 183. https://doi.org/10.3390/vetsci12020183