

Genetic and Phylogenetic Analysis of Feline Coronavirus in Guangxi Province of China from 2021 to 2024

 , ,
, ,
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Primers Design
2.3. Test for FCoV in Clinical Samples
2.4. S, M, and N Gene Amplification and Sequencing
2.5. Sequence Analysis
2.6. Phylogenetic Analysis
2.7. Homologous Recombination Analysis
2.8. System Dynamics Analysis
2.9. Selection Pressure and Site Mutation Analysis
3. Results
3.1. Detection and Sequencing Results
3.2. Similarity Analysis
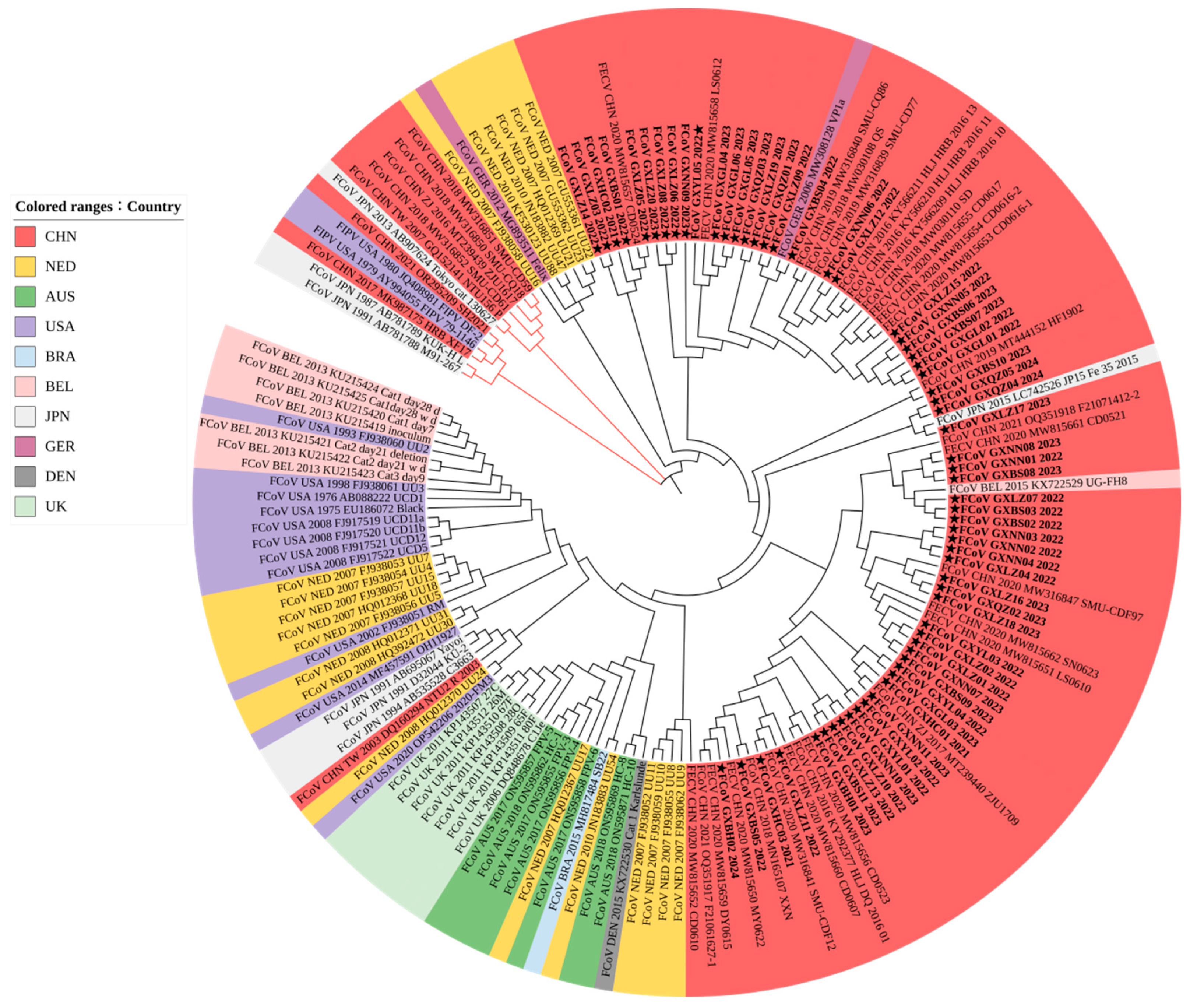
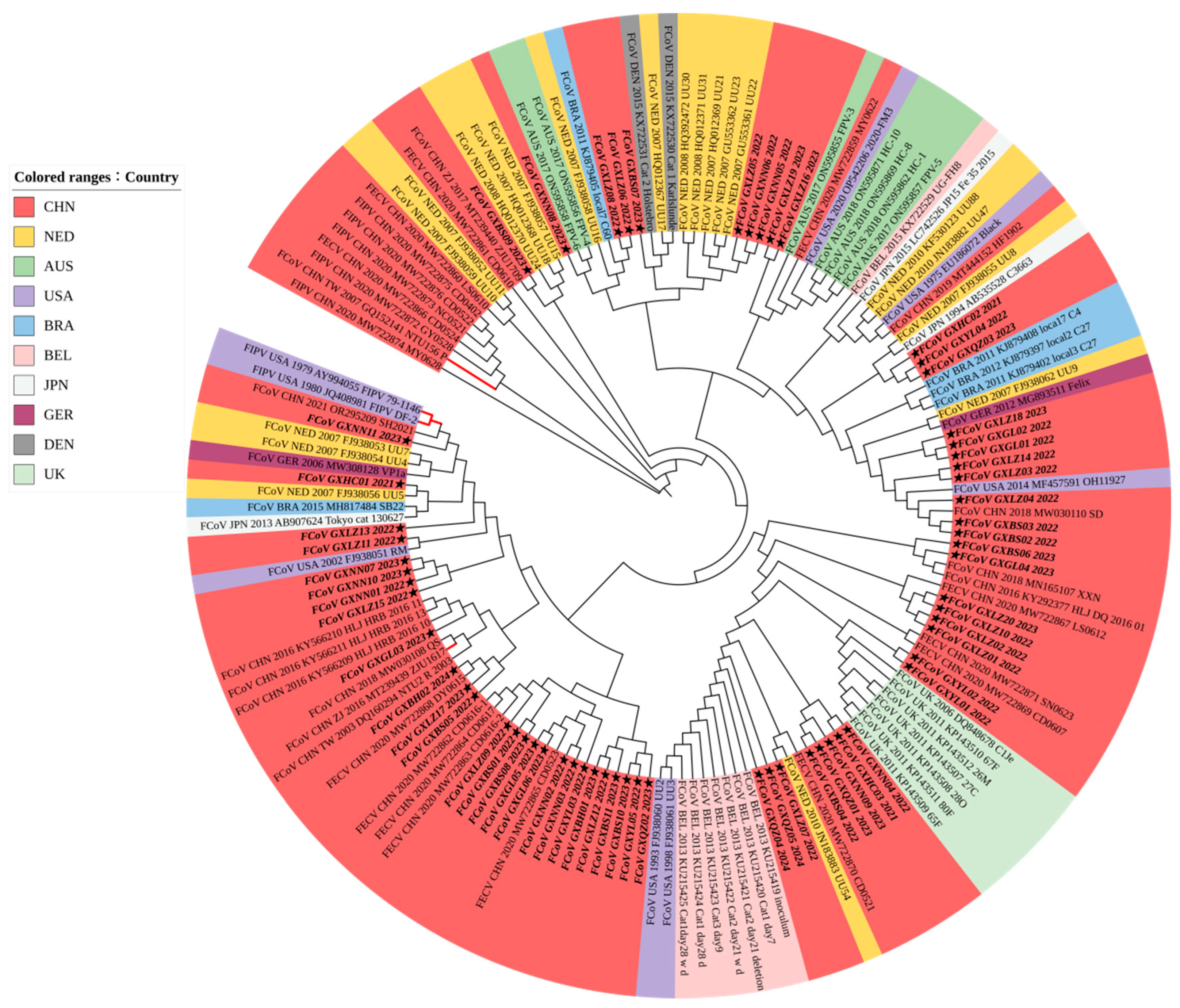
3.3. Phylogenetic Analysis
3.3.1. Phylogenetic Analysis Based on S Gene Sequences
3.3.2. Phylogenetic Analysis Based on M Gene Sequences
3.3.3. Phylogenetic Analysis Based on N Gene Sequences
3.4. Genetic Evolution Rate
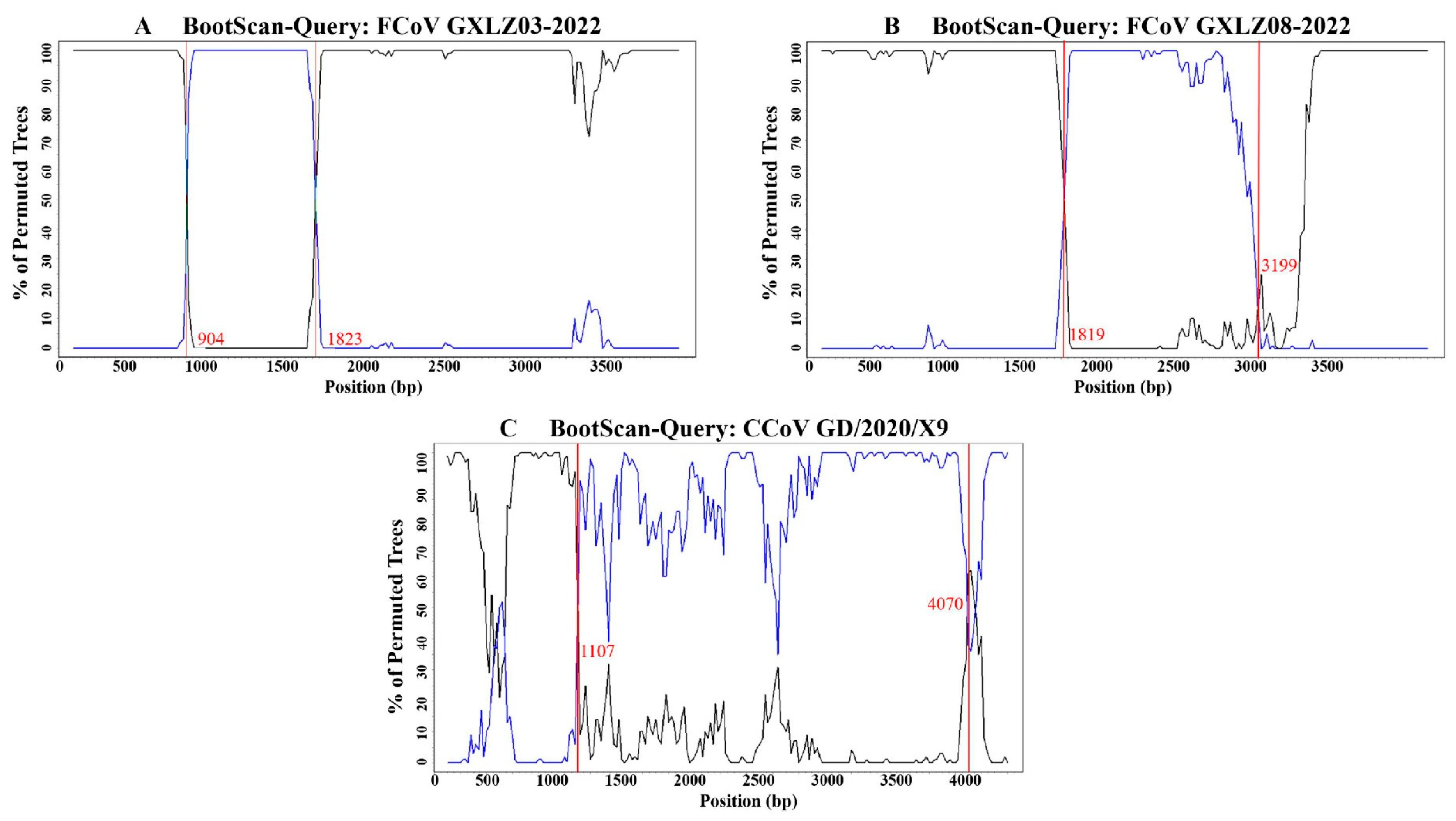
3.5. Analysis of Homologous Recombination of the S Gene
3.6. Positive Selection Pressure and Comparison of Mutation Sites of the S Gene
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Woo, P.C.Y.; de Groot, R.J.; Haagmans, B.; Lau, S.K.P.; Neuman, B.W.; Perlman, S.; Sola, I.; van der Hoek, L.; Wong, A.C.P.; Yeh, S.H. ICTV virus taxonomy profile: Coronaviridae 2023. J. Gen. Virol. 2023, 104, 001843. [Google Scholar] [CrossRef] [PubMed]
- Tekes, G.; Thiel, H.J. Feline coronaviruses: Pathogenesis of feline infectious peritonitis. Adv. Virus Res. 2016, 96, 193–218. [Google Scholar] [PubMed]
- Jaimes, J.A.; Millet, J.K.; Stout, A.E.; André, N.M.; Whittaker, G.R. A tale of two viruses: The distinct spike glycoproteins of feline coronaviruses. Viruses 2020, 12, 83. [Google Scholar] [CrossRef] [PubMed]
- Belouzard, S.; Millet, J.K.; Licitra, B.N.; Whittaker, G.R. Mechanisms of coronavirus cell entry mediated by the viral spike protein. Viruses 2012, 4, 1011–1033. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Peng, Q.; Lang, Y.; Du, S.; Cao, S.; Wu, R.; Zhao, Q.; Huang, X.; Wen, Y.; Lin, J.; et al. Phylogeny, evolution, and transmission dynamics of canine and feline coronaviruses: A retro-prospective study. Front. Microbiol. 2022, 13, 850516. [Google Scholar] [CrossRef]
- Wong, N.A.; Saier, M.H. The SARS-coronavirus infection cycle: A survey of viral membrane proteins, their functional interactions and pathogenesis. Int. J. Mol. Sci. 2021, 22, 1308. [Google Scholar] [CrossRef]
- McBride, R.; van Zyl, M.; Fielding, B.C. The coronavirus nucleocapsid is a multifunctional protein. Viruses 2014, 6, 2991–3018. [Google Scholar] [CrossRef]
- Li, B.; Gao, Y.; Ma, Y.; Shi, K.; Shi, Y.; Feng, S.; Yin, Y.; Long, F.; Sun, W. Genetic and evolutionary analysis of porcine deltacoronavirus in Guangxi province, southern China, from 2020 to 2023. Microorganisms 2024, 12, 416. [Google Scholar] [CrossRef]
- Shi, K.; Li, B.; Shi, Y.; Feng, S.; Yin, Y.; Long, F.; Pan, Y.; Wei, Y. Phylogenetic and evolutionary analysis of porcine epidemic diarrhea virus in Guangxi province, China, during 2020 and 2024. Viruses 2024, 16, 1126. [Google Scholar] [CrossRef]
- Gao, Y.Y.; Wang, Q.; Liang, X.Y.; Zhang, S.; Bao, D.; Zhao, H.; Li, S.B.; Wang, K.; Hu, G.X.; Gao, F.S. An updated review of feline coronavirus: Mind the two biotypes. Virus Res. 2023, 326, 199059. [Google Scholar] [CrossRef]
- Pedersen, N.C. An update on feline infectious peritonitis: Virology and immunopathogenesis. Vet. J. 2014, 201, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Herrewegh, A.A.; Smeenk, I.; Horzinek, M.C.; Rottier, P.J.; de Groot, R.J. Feline coronavirus type II strains 79-1683 and 79-1146 originate from a double recombination between feline coronavirus type I and canine coronavirus. J. Virol. 1998, 72, 4508–4514. [Google Scholar] [CrossRef] [PubMed]
- Amer, A.; Siti Suri, A.; Abdul Rahman, O.; Mohd, H.B.; Faruku, B.; Saeed, S.; Tengku Azmi, T.I. Isolation and molecular characterization of type I and type II feline coronavirus in Malaysia. Virol. J. 2012, 9, 278. [Google Scholar] [CrossRef] [PubMed]
- An, D.J.; Jeoung, H.Y.; Jeong, W.; Park, J.Y.; Lee, M.H.; Park, B.K. Prevalence of Korean cats with natural feline coronavirus infections. Virol. J. 2011, 8, 455. [Google Scholar] [CrossRef]
- Doki, T.; Yabe, M.; Takano, T.; Hohdatsu, T. Differential induction of type I interferon by type I and type II feline coronaviruses in vitro. Res. Vet. Sci. 2018, 120, 57–62. [Google Scholar] [CrossRef]
- Tasker, S. Diagnosis of feline infectious peritonitis: Update on evidence supporting available tests. J. Feline Med. Surg. 2018, 20, 228–243. [Google Scholar] [CrossRef]
- Ehmann, R.; Kristen-Burmann, C.; Bank-Wolf, B.; König, M.; Herden, C.; Hain, T.; Thiel, H.J.; Ziebuhr, J.; Tekes, G. Reverse genetics for type I feline coronavirus field isolate to study the molecular pathogenesis of feline infectious peritonitis. Mbio 2018, 9, e01422-18. [Google Scholar] [CrossRef]
- Licitra, B.N.; Millet, J.K.; Regan, A.D.; Hamilton, B.S.; Rinaldi, V.D.; Duhamel, G.E.; Whittaker, G.R. Mutation in spike protein cleavage site and pathogenesis of feline coronavirus. Emerg. Infect. Dis. 2013, 19, 1066–1073. [Google Scholar] [CrossRef]
- Porter, E.; Tasker, S.; Day, M.J.; Harley, R.; Kipar, A.; Siddell, S.G.; Helps, C.R. Amino acid changes in the spike protein of feline coronavirus correlate with systemic spread of virus from the intestine and not with feline infectious peritonitis. Vet. Res. 2014, 45, 49. [Google Scholar] [CrossRef]
- Chang, H.W.; de Groot, R.J.; Egberink, H.F.; Rottier, P.J. Feline infectious peritonitis: Insights into feline coronavirus pathobiogenesis and epidemiology based on genetic analysis of the viral 3c gene. J. Gen. Virol. 2010, 91, 415–420. [Google Scholar] [CrossRef]
- Lednicky, J.A.; Tagliamonte, M.S.; White, S.K.; Blohm, G.M.; Alam, M.M.; Iovine, N.M.; Salemi, M.; Mavian, C.; Morris, J.G. Isolation of a novel recombinant canine coronavirus from a visitor to Haiti: Further evidence of transmission of coronaviruses of zoonotic origin to humans. Clin. Infect. Dis. 2022, 75, e1184–e1187. [Google Scholar] [CrossRef] [PubMed]
- He, W.T.; Ji, X.; He, W.; Dellicour, S.; Wang, S.; Li, G.; Zhang, L.; Gilbert, M.; Zhu, H.; Xing, G.; et al. Genomic epidemiology, evolution, and transmission dynamics of porcine deltacoronavirus. Mol. Biol. Evol. 2020, 37, 2641–2654. [Google Scholar] [CrossRef] [PubMed]
- Bahoussi, A.N.; Wang, P.H.; Shah, P.T.; Bu, H.; Wu, C.; Xing, L. Evolutionary plasticity of zoonotic porcine deltacoronavirus (PDCoV): Genetic characteristics and geographic distribution. BMC Vet. Res. 2022, 18, 444. [Google Scholar] [CrossRef] [PubMed]
- Vlasova, A.N.; Diaz, A.; Damtie, D.; Xiu, L.; Toh, T.H.; Lee, J.S.; Saif, L.J.; Gray, G.C. Novel canine coronavirus isolated from a hospitalized patient with pneumonia in east Malaysia. Clin. Infect. Dis. 2022, 74, 446–454. [Google Scholar] [CrossRef]
- Ouyang, H.; Liu, J.; Yin, Y.; Cao, S.; Yan, R.; Ren, Y.; Zhou, D.; Li, Q.; Li, J.; Liao, X.; et al. Epidemiology and comparative analyses of the S gene on feline coronavirus in central China. Pathogens 2022, 11, 460. [Google Scholar] [CrossRef]
- Song, X.L.; Li, W.F.; Shan, H.; Yang, H.Y.; Zhang, C.M. Prevalence and genetic variation of the M, N, and S2 genes of feline coronavirus in Shandong province, China. Arch. Virol. 2023, 168, 227. [Google Scholar] [CrossRef]
- Zhou, Q.; Li, Y.; Huang, J.; Fu, N.; Song, X.; Sha, X.; Zhang, B. Prevalence and molecular characteristics of feline coronavirus in southwest China from 2017 to 2020. J. Gen. Virol. 2021, 102, 001654. [Google Scholar] [CrossRef]
- He, M.; Feng, S.; Shi, K.; Shi, Y.; Long, F.; Yin, Y.; Li, Z. One-step triplex TaqMan quantitative reverse transcription polymerase chain reaction for the detection of feline coronavirus, feline panleukopenia virus, and feline leukemia virus. Vet. World 2024, 17, 946–955. [Google Scholar] [CrossRef]
- Martin, D.P.; Varsani, A.; Roumagnac, P.; Botha, G.; Maslamoney, S.; Schwab, T.; Kelz, Z.; Kumar, V.; Murrell, B. RDP5: A computer program for analyzing recombination in, and removing signals of recombination from, nucleotide sequence datasets. Virus Evol. 2021, 7, veaa087. [Google Scholar] [CrossRef]
- Chang, H.W.; Egberink, H.F.; Halpin, R.; Spiro, D.J.; Rottier, P.J. Spike protein fusion peptide and feline coronavirus virulence. Emerg. Infect. Dis. 2012, 18, 1089–1095. [Google Scholar] [CrossRef]
- Vijaykrishna, D.; Smith, G.J.; Zhang, J.X.; Peiris, J.S.; Chen, H.; Guan, Y. Evolutionary insights into the ecology of coronaviruses. J. Virol. 2007, 81, 4012–4020. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Lu, Q.; Jin, Q.; Li, P.; Xing, G.; Zhang, G. Phylogenetically evolutionary analysis provides insights into the genetic diversity and adaptive evolution of porcine deltacoronavirus. BMC Vet. Res. 2024, 20, 22. [Google Scholar] [CrossRef] [PubMed]
- Callaway, E. Why a highly mutated coronavirus variant has scientists on alert. Nature 2023, 620, 934. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Liu, Q.; Kong, F.; Guo, D.; Zhai, J.; Su, M.; Sun, D. Circulation and genetic diversity of feline coronavirus type I and II from clinically healthy and FIP-suspected cats in China. Transbound. Emerg. Dis. 2019, 66, 763–775. [Google Scholar] [CrossRef]
- Dong, B.; Zhang, X.; Zhong, X.; Hu, W.; Lin, Z.; Zhang, S.; Deng, H.; Lin, W. Prevalence of natural feline coronavirus infection in domestic cats in Fujian, China. Virol. J. 2024, 21, 2. [Google Scholar] [CrossRef]
- Kipar, A.; Baptiste, K.; Barth, A.; Reinacher, M. Natural FCoV infection: Cats with FIP exhibit significantly higher viral loads than healthy infected cats. J. Feline Med. Surg. 2006, 8, 69–72. [Google Scholar] [CrossRef]
- Tanaka, Y.; Sasaki, T.; Matsuda, R.; Uematsu, Y.; Yamaguchi, T. Molecular epidemiological study of feline coronavirus strains in Japan using RT-PCR targeting nsp14 gene. BMC Vet. Res. 2015, 11, 57. [Google Scholar] [CrossRef]
- Myrrha, L.W.; Silva, F.M.F.; Vidigal, P.M.P.; Resende, M.; Bressan, G.C.; Fietto, J.L.R.; Santos, M.R.; Silva, L.M.N.; Assao, V.S.; Silva-Jú Nior, A.; et al. Feline coronavirus isolates from a part of Brazil: Insights into molecular epidemiology and phylogeny inferred from the 7b gene. J. Vet. Med. Sci. 2019, 81, 1455–1460. [Google Scholar] [CrossRef]
- Lauzi, S.; Stranieri, A.; Giordano, A.; Luzzago, C.; Zehender, G.; Paltrinieri, S.; Ebranati, E. Origin and transmission of feline coronavirus type I in domestic cats from northern Italy: A phylogeographic approach. Vet. Microbiol. 2020, 244, 108667. [Google Scholar] [CrossRef]
- Guarnieri, C.; Bertola, L.; Ferrari, L.; Quintavalla, C.; Corradi, A.; Di Lecce, R. Myocarditis in an FIP-diseased cat with FCoV M1058L mutation: Clinical and pathological changes. Animals 2024, 14, 1673. [Google Scholar] [CrossRef]
- Wells, H.L.; Bonavita, C.M.; Navarrete-Macias, I.; Vilchez, B.; Rasmussen, A.L.; Anthony, S.J. The coronavirus recombination pathway. Cell Host Microbe 2023, 31, 874–889. [Google Scholar] [CrossRef] [PubMed]
- Jaimes, J.A.; Whittaker, G.R. Feline coronavirus: Insights into viral pathogenesis based on the spike protein structure and function. Virology 2018, 517, 108–121. [Google Scholar] [CrossRef] [PubMed]
- Decaro, N.; Mari, V.; Lanave, G.; Lorusso, E.; Lucente, M.S.; Desario, C.; Colaianni, M.L.; Elia, G.; Ferringo, F.; Alfano, F.; et al. Mutation analysis of the spike protein in Italian feline infectious peritonitis virus and feline enteric coronavirus sequences. Res. Vet. Sci. 2021, 135, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Paltrinieri, S.; Giordano, A.; Stranieri, A.; Lauzi, S. Feline infectious peritonitis (FIP) and coronavirus disease 19 (COVID-19): Are they similar? Transbound. Emerg. Dis. 2021, 68, 1786–1799. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Deng, S.; Mou, D.; Zhang, G.; Fu, Y.; Huang, W.; Zhang, Y.; Lyu, Y. Analysis of spike and accessory 3c genes mutations of less virulent and FIP-associated feline coronaviruses in Beijing, China. Virology 2024, 589, 109919. [Google Scholar] [CrossRef]
- Takano, T.; Yamada, S.; Doki, T.; Hohdatsu, T. Pathogenesis of oral type I feline infectious peritonitis virus (FIPV) infection: Antibody-dependent enhancement infection of cats with type I FIPV via the oral route. J. Vet. Med. Sci. 2019, 81, 911–915. [Google Scholar] [CrossRef]
- Olsen, C.W.; Corapi, W.V.; Ngichabe, C.K.; Baines, J.D.; Scott, F.W. Monoclonal antibodies to the spike protein of feline infectious peritonitis virus mediate antibody-dependent enhancement of infection of feline macrophages. J. Virol. 1992, 66, 956–965. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Sequence (5′→3′) | Position | Product/bp |
|---|---|---|---|
| FCoV-M-F | CCCGACGAAGCATTYTTGGTTTGAACTA | 26,192–26,219 | 880 |
| FCoV-M-R | GGAAGGTTCATCTCCCCAGTTGACG | 27,047–27,071 | |
| FCoV-N-F | GCACGTACTGAYAATTTGAGTGAAC | 26,964–26,988 | 1202 |
| FCoV-N-R | TGCGTTTAGTTCGTAACCTC | 28,146–28,165 | |
| FCoV-S1-F | GATATGGTYGTTGGATTRCTAAGG | 20,376–20,399 | 960 |
| FCoV-S1-R | TGCCRTCARTTGGCACAAAG | 21,316–21,335 | |
| FCoV-S2-F | GTGCCAATTGATGGCAAGATAC | 21,320–21,341 | 918 |
| FCoV-S2-R | TCCATTCGCCTGTGCTATTT | 22,218–22,237 | |
| FCoV-S3-F | ACTCTCACTTGCTGAYATACAC | 22,192–22,213 | 982 |
| FCoV-S3-R | TCTGTTACCATTGCAGACATACT | 23,151–23,173 | |
| FCoV-S4-F | AGGCCGAATACATTCAGATTCA | 23,100–23,121 | 1032 |
| FCoV-S4-R | TGCCACAGAAACCATACCTATC | 24,110–24,131 | |
| FCoV-S5-F | GTATGCTGAAGTCAAGGCTAGT | 24,040–24,061 | 895 |
| FCoV-S5-R | CAAGTACAGCGTCAACAGAGA | 24,914–24,934 |
| Gene | Sequence Similarity among Obtained Strains | Sequence Similarity between Obtained Strains and Reference Strains | ||
|---|---|---|---|---|
| nt | aa | nt | aa | |
| S | 81.2–99.6% | 70.2–99.5% | 68.9–90% | 58.1–82.8% |
| M | 89.9–100% | 91.6–100% | 79.8–95.9% | 80.8–98.5% |
| N | 90.1–100% | 91.5–100% | 88.7–95.5% | 89.3–97.9% |
| Recombinant Strain | Accession No. | Major Parent | Similarity | Minor Parent | Similarity |
|---|---|---|---|---|---|
| FCoV GXLZ03-2022 | PP448818 | FCoV GXLZ14-2022 | 99.2% | FCoV GXLZ05-2022 | 97.2% |
| FCoV GXLZ08-2022 | PP448823 | FCoV GXLZ06-2022 | 99.2% | FCoV GXBS01-2022 | 99.3% |
| CCoV GD/2020/X9 | MZ320954 | FCoV ZJU1617 | 97.3% | CCoV 341/05 | 99.6% |
| Site | FEL | SLAC | FUBAR | MEME |
|---|---|---|---|---|
| H275S | + | + | + | + |
| I290T | + | + | + | + |
| N374K | + | + | + | + |
| F378L | + | + | + | + |
| V830T | + | + | + | + |
| T854I | + | + | + | + |
| K980T | + | + | + | + |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, K.; He, M.; Shi, Y.; Long, F.; Shi, Y.; Yin, Y.; Pan, Y.; Li, Z.; Feng, S. Genetic and Phylogenetic Analysis of Feline Coronavirus in Guangxi Province of China from 2021 to 2024. Vet. Sci. 2024, 11, 455. https://doi.org/10.3390/vetsci11100455
Shi K, He M, Shi Y, Long F, Shi Y, Yin Y, Pan Y, Li Z, Feng S. Genetic and Phylogenetic Analysis of Feline Coronavirus in Guangxi Province of China from 2021 to 2024. Veterinary Sciences. 2024; 11(10):455. https://doi.org/10.3390/vetsci11100455
Chicago/Turabian StyleShi, Kaichuang, Mengyi He, Yuwen Shi, Feng Long, Yandi Shi, Yanwen Yin, Yi Pan, Zongqiang Li, and Shuping Feng. 2024. "Genetic and Phylogenetic Analysis of Feline Coronavirus in Guangxi Province of China from 2021 to 2024" Veterinary Sciences 11, no. 10: 455. https://doi.org/10.3390/vetsci11100455
APA StyleShi, K., He, M., Shi, Y., Long, F., Shi, Y., Yin, Y., Pan, Y., Li, Z., & Feng, S. (2024). Genetic and Phylogenetic Analysis of Feline Coronavirus in Guangxi Province of China from 2021 to 2024. Veterinary Sciences, 11(10), 455. https://doi.org/10.3390/vetsci11100455