Free Radical and Viral Infection: A Review from the Perspective of Ferroptosis

 ,
,  ,
, {kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
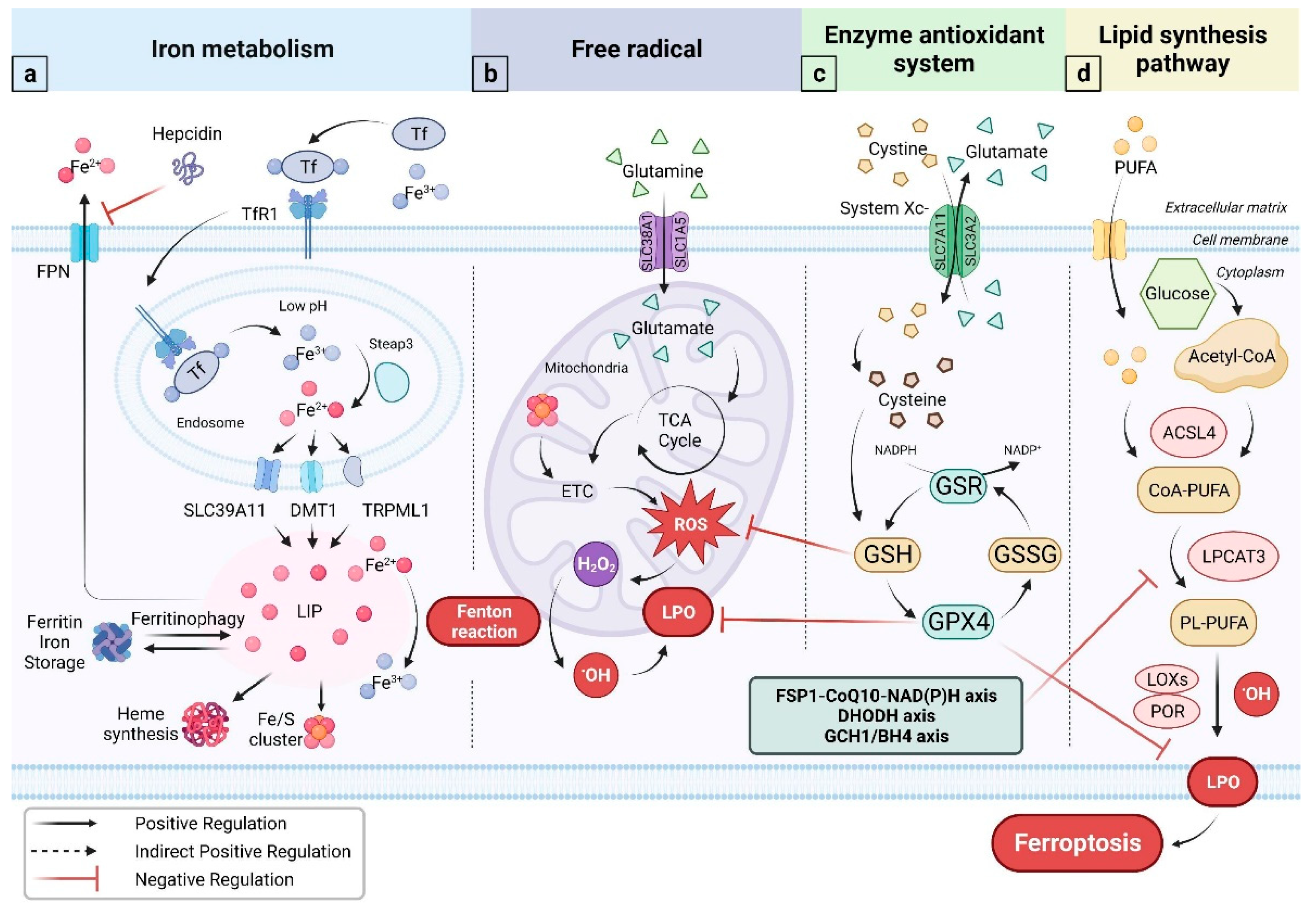
2. Free Radical, Iron Metabolism and Viral Infections
2.1. Free Radical
2.2. Iron Metabolism
3. Ferroptosis
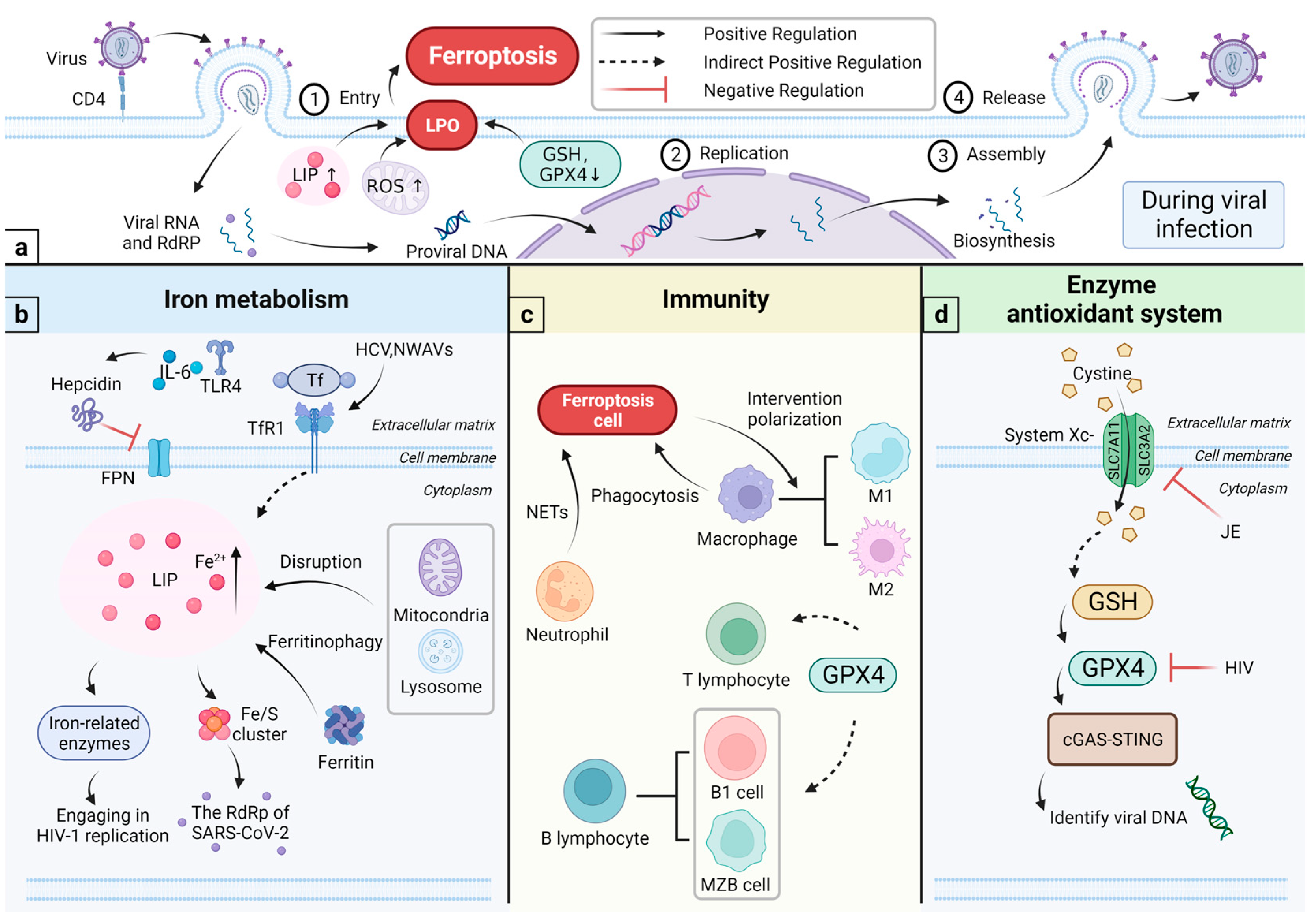
4. Ferroptosis and Viral Infections
4.1. Relationship among Viral Infections, Iron Metabolism, and Immunity
4.1.1. Viral Infections and Iron Metabolism
4.1.2. Iron and Immunity
4.2. Relationship among Viral Infections, Enzyme Antioxidant System, and Immunity
4.2.1. Viral Infections and Enzyme Antioxidant System
4.2.2. Enzyme Antioxidant System and the Immunity
5. Intervention of Viral Infections through Ferroptosis
5.1. Ferroptosis Inducers and Viral Infections
5.2. Ferroptosis Inhibitors and Viral Infections
6. Conclusions and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dixon, L.K.; Stahl, K.; Jori, F.; Vial, L.; Pfeiffer, D.U. African Swine Fever Epidemiology and Control. Annu. Rev. Anim. Biosci. 2020, 8, 221–246. [Google Scholar] [CrossRef]
- Lake, M.A. What we know so far: COVID-19 current clinical knowledge and research. Clin. Med. 2020, 20, 124–127. [Google Scholar] [CrossRef] [PubMed]
- Kaye, A.D.; Okeagu, C.N.; Pham, A.D.; Silva, R.A.; Hurley, J.J.; Arron, B.L.; Sarfraz, N.; Lee, H.N.; Ghali, G.E.; Gamble, J.W.; et al. Economic impact of COVID-19 pandemic on healthcare facilities and systems: International perspectives. Best. Pract. Res. Clin. Anaesthesiol. 2021, 35, 293–306. [Google Scholar] [CrossRef]
- Lindert, J.; Jakubauskiene, M.; Bilsen, J. The COVID-19 disaster and mental health-assessing, responding and recovering. Eur. J. Public. Health 2021, 31, iv31–iv35. [Google Scholar] [CrossRef] [PubMed]
- Troyer, E.A.; Kohn, J.N.; Hong, S. Are we facing a crashing wave of neuropsychiatric sequelae of COVID-19? Neuropsychiatric symptoms and potential immunologic mechanisms. Brain Behav. Immun. 2020, 87, 34–39. [Google Scholar] [CrossRef]
- Foo, J.; Bellot, G.; Pervaiz, S.; Alonso, S. Mitochondria-mediated oxidative stress during viral infection. Trends Microbiol. 2022, 30, 679–692. [Google Scholar] [CrossRef]
- Pham-Huy, L.A.; He, H.; Pham-Huy, C. Free radicals, antioxidants in disease and health. Int. J. Biomed. Sci. 2008, 4, 89–96. [Google Scholar]
- Silva, J.P.; Coutinho, O.P. Free radicals in the regulation of damage and cell death-basic mechanisms and prevention. Drug Discov. Ther. 2010, 4, 144–167. [Google Scholar]
- Ashida, H.; Mimuro, H.; Ogawa, M.; Kobayashi, T.; Sanada, T.; Kim, M.; Sasakawa, C. Cell death and infection: A double-edged sword for host and pathogen survival. J. Cell Biol. 2011, 195, 931–942. [Google Scholar] [CrossRef]
- Jorgensen, I.; Rayamajhi, M.; Miao, E.A. Programmed cell death as a defence against infection. Nat. Rev. Immunol. 2017, 17, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Checconi, P.; De Angelis, M.; Marcocci, M.E.; Fraternale, A.; Magnani, M.; Palamara, A.T.; Nencioni, L. Redox-Modulating Agents in the Treatment of Viral Infections. Int. J. Mol. Sci. 2020, 21, 4084. [Google Scholar] [CrossRef]
- Guillin, O.M.; Vindry, C.; Ohlmann, T.; Chavatte, L. Selenium, Selenoproteins and Viral Infection. Nutrients 2019, 11, 2101. [Google Scholar] [CrossRef]
- Silwal, P.; Kim, J.K.; Kim, Y.J.; Jo, E.-K. Mitochondrial Reactive Oxygen Species: Double-Edged Weapon in Host Defense and Pathological Inflammation During Infection. Front. Immunol. 2020, 11, 1649. [Google Scholar] [CrossRef]
- Zolini, G.P.; Lima, G.K.; Lucinda, N.; Silva, M.A.; Dias, M.F.; Pessoa, N.L.; Coura, B.P.; Cartelle, C.T.; Arantes, R.M.; Kroon, E.G.; et al. Defense against HSV-1 in a murine model is mediated by iNOS and orchestrated by the activation of TLR2 and TLR9 in trigeminal ganglia. J. NeuroInflamm. 2014, 11, 20. [Google Scholar] [CrossRef] [PubMed]
- Croen, K.D. Evidence for antiviral effect of nitric oxide. Inhibition of herpes simplex virus type 1 replication. J. Clin. Investig. 1993, 91, 2446–2452. [Google Scholar] [CrossRef]
- de Souza, K.P.R.; Silva, E.G.; de Oliveira Rocha, E.S.; Figueiredo, L.B.; de Almeida-Leite, C.M.; Arantes, R.M.E.; de Assis Silva Gomes, J.; Ferreira, G.P.; de Oliveira, J.G.; Kroon, E.G.; et al. Nitric oxide synthase expression correlates with death in an experimental mouse model of dengue with CNS involvement. Virol. J. 2013, 10, 267. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.-J.; Chien, H.-L.; Lin, S.-Y.; Chang, B.-L.; Yu, H.-P.; Tang, W.-C.; Lin, Y.-L. MCPIP1 ribonuclease exhibits broad-spectrum antiviral effects through viral RNA binding and degradation. Nucleic Acids Res. 2013, 41, 3314–3326. [Google Scholar] [CrossRef] [PubMed]
- Damle, V.G.; Wu, K.; Arouri, D.J.; Schirhagl, R. Detecting free radicals post viral infections. Free Radic. Biol. Med. 2022, 191, 8–23. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Wang, M.-p.; Joshua, B.; Jin, N.-y.; Du, S.-w.; Li, C. Ferroptosis in viral infection: The unexplored possibility. Acta Pharmacol. Sin. 2022, 43, 1905–1915. [Google Scholar] [CrossRef]
- Drakesmith, H.; Prentice, A. Viral infection and iron metabolism. Nat. Rev. Microbiol. 2008, 6, 541–552. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Guo, P.; Xie, X.; Wang, Y.; Chen, G. Ferroptosis, a new form of cell death, and its relationships with tumourous diseases. J. Cell Mol. Med. 2017, 21, 648–657. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Li, X.; Liu, L.; Yu, B.; Xue, Y.; Liu, Y. Erastin sensitizes glioblastoma cells to temozolomide by restraining xCT and cystathionine-γ-lyase function. Oncol. Rep. 2015, 33, 1465–1474. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Hsu, J.L.; Chen, C.T.; Wang, Y.N.; Hsu, M.C.; Chang, S.S.; Du, Y.; Ko, H.W.; Herbst, R.; Hung, M.C. Caspase-independent cell death is involved in the negative effect of EGF receptor inhibitors on cisplatin in non-small cell lung cancer cells. Clin. Cancer Res. 2013, 19, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Zhou, X.; Xie, F.; Zhang, L.; Yan, H.; Huang, J.; Zhang, C.; Zhou, F.; Chen, J.; Zhang, L. Ferroptosis in cancer and cancer immunotherapy. Cancer Commun. 2022, 42, 88–116. [Google Scholar] [CrossRef]
- Stockwell, B.R. Ferroptosis turns 10: Emerging mechanisms, physiological functions, and therapeutic applications. Cell 2022, 185, 2401–2421. [Google Scholar] [CrossRef]
- Forman, H.J.; Zhang, H. Targeting oxidative stress in disease: Promise and limitations of antioxidant therapy. Nat. Rev. Drug Discov. 2021, 20, 689–709. [Google Scholar] [CrossRef]
- Phaniendra, A.; Jestadi, D.B.; Periyasamy, L. Free radicals: Properties, sources, targets, and their implication in various diseases. Indian. J. Clin. Biochem. 2015, 30, 11–26. [Google Scholar] [CrossRef]
- Sirokmány, G.; Donkó, Á.; Geiszt, M. Nox/Duox Family of NADPH Oxidases: Lessons from Knockout Mouse Models. Trends Pharmacol. Sci. 2016, 37, 318–327. [Google Scholar] [CrossRef]
- Yang, L.; Cao, L.-m.; Zhang, X.-j.; Chu, B. Targeting ferroptosis as a vulnerability in pulmonary diseases. Cell Death Dis. 2022, 13, 649. [Google Scholar] [CrossRef]
- Gong, Y.; Tang, N.; Liu, P.; Sun, Y.; Lu, S.; Liu, W.; Tan, L.; Song, C.; Qiu, X.; Liao, Y.; et al. Newcastle disease virus degrades SIRT3 via PINK1-PRKN-dependent mitophagy to reprogram energy metabolism in infected cells. Autophagy 2022, 18, 1503–1521. [Google Scholar] [CrossRef] [PubMed]
- Meynard, D.; Babitt, J.L.; Lin, H.Y. The liver: Conductor of systemic iron balance. Blood 2014, 123, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, F.; Rocha, S.; Fernandes, R. Iron metabolism: From health to disease. J. Clin. Lab. Anal. 2014, 28, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Hentze, M.W.; Muckenthaler, M.U.; Andrews, N.C. Balancing acts: Molecular control of mammalian iron metabolism. Cell 2004, 117, 285–297. [Google Scholar] [CrossRef] [PubMed]
- Ganz, T.; Nemeth, E. The hepcidin-ferroportin system as a therapeutic target in anemias and iron overload disorders. Hematol. Am. Soc. Hematol. Educ. Program. 2011, 2011, 538–542. [Google Scholar] [CrossRef] [PubMed]
- Vashisht, A.A.; Zumbrennen, K.B.; Huang, X.; Powers, D.N.; Durazo, A.; Sun, D.; Bhaskaran, N.; Persson, A.; Uhlen, M.; Sangfelt, O.; et al. Control of iron homeostasis by an iron-regulated ubiquitin ligase. Science 2009, 326, 718–721. [Google Scholar] [CrossRef]
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The molecular machinery of regulated cell death. Cell Res. 2019, 29, 347–364. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef]
- Cheng, X.; Zhang, J.; Xiao, Y.; Wang, Z.; He, J.; Ke, M.; Liu, S.; Wang, Q.; Zhang, L. Mitochondrial Regulation of Ferroptosis in Cancer Therapy. Int. J. Mol. Sci. 2023, 24, 10037. [Google Scholar] [CrossRef]
- Zheng, J.; Conrad, M. The Metabolic Underpinnings of Ferroptosis. Cell Metab. 2020, 32, 920–937. [Google Scholar] [CrossRef]
- Lu, C.; Deng, K.; Hu, C.; Lyu, L. Dual-reaction-center catalytic process continues Fenton’s story. Front. Environ. Sci. Eng. 2020, 14, 82. [Google Scholar] [CrossRef]
- Lillo-Moya, J.; Rojas-Solé, C.; Muñoz-Salamanca, D.; Panieri, E.; Saso, L.; Rodrigo, R. Targeting Ferroptosis against Ischemia/Reperfusion Cardiac Injury. Antioxidants 2021, 10, 667. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Monian, P.; Pan, Q.; Zhang, W.; Xiang, J.; Jiang, X. Ferroptosis is an autophagic cell death process. Cell Res. 2016, 26, 1021–1032. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Stockwell, B.R. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem. Biol. 2008, 15, 234–245. [Google Scholar] [CrossRef] [PubMed]
- Ju, X.; Yan, Y.; Liu, Q.; Li, N.; Sheng, M.; Zhang, L.; Li, X.; Liang, Z.; Huang, F.; Liu, K.; et al. Neuraminidase of Influenza A Virus Binds Lysosome-Associated Membrane Proteins Directly and Induces Lysosome Rupture. J. Virol. 2015, 89, 10347–10358. [Google Scholar] [CrossRef]
- Gao, X.; Qian, M.; Campian, J.L.; Marshall, J.; Zhou, Z.; Roberts, A.M.; Kang, Y.J.; Prabhu, S.D.; Sun, X.F.; Eaton, J.W. Mitochondrial dysfunction may explain the cardiomyopathy of chronic iron overload. Free Radic. Biol. Med. 2010, 49, 401–407. [Google Scholar] [CrossRef]
- Brigelius-Flohé, R.; Maiorino, M. Glutathione peroxidases. Biochim. Biophys. Acta 2013, 1830, 3289–3303. [Google Scholar] [CrossRef]
- Maiorino, M.; Conrad, M.; Ursini, F. GPx4, Lipid Peroxidation, and Cell Death: Discoveries, Rediscoveries, and Open Issues. Antioxid. Redox Signal 2018, 29, 61–74. [Google Scholar] [CrossRef]
- Mao, C.; Liu, X.; Zhang, Y.; Lei, G.; Yan, Y.; Lee, H.; Koppula, P.; Wu, S.; Zhuang, L.; Fang, B.; et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature 2021, 593, 586–590. [Google Scholar] [CrossRef]
- Stockwell, B.R. A powerful cell-protection system prevents cell death by ferroptosis. Nature 2019, 575, 597–598. [Google Scholar] [CrossRef]
- Ding, C.C.; Rose, J.; Sun, T.; Wu, J.; Chen, P.H.; Lin, C.C.; Yang, W.H.; Chen, K.Y.; Lee, H.; Xu, E.; et al. MESH1 is a cytosolic NADPH phosphatase that regulates ferroptosis. Nat. Metab. 2020, 2, 270–277. [Google Scholar] [CrossRef]
- Kraft, V.A.N.; Bezjian, C.T.; Pfeiffer, S.; Ringelstetter, L.; Müller, C.; Zandkarimi, F.; Merl-Pham, J.; Bao, X.; Anastasov, N.; Kössl, J.; et al. GTP Cyclohydrolase 1/Tetrahydrobiopterin Counteract Ferroptosis through Lipid Remodeling. ACS Cent. Sci. 2020, 6, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Soula, M.; Weber, R.A.; Zilka, O.; Alwaseem, H.; La, K.; Yen, F.; Molina, H.; Garcia-Bermudez, J.; Pratt, D.A.; Birsoy, K. Metabolic determinants of cancer cell sensitivity to canonical ferroptosis inducers. Nat. Chem. Biol. 2020, 16, 1351–1360. [Google Scholar] [CrossRef] [PubMed]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017, 13, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Lei, P.; Bai, T.; Sun, Y. Mechanisms of Ferroptosis and Relations With Regulated Cell Death: A Review. Front. Physiol. 2019, 10, 139. [Google Scholar] [CrossRef]
- Chen, X.; Kang, R.; Kroemer, G.; Tang, D. Ferroptosis in infection, inflammation, and immunity. J. Exp. Med. 2021, 218, e20210518. [Google Scholar] [CrossRef] [PubMed]
- Yan, B.; Ai, Y.; Sun, Q.; Ma, Y.; Cao, Y.; Wang, J.; Zhang, Z.; Wang, X. Membrane Damage during Ferroptosis Is Caused by Oxidation of Phospholipids Catalyzed by the Oxidoreductases POR and CYB5R1. Mol. Cell 2021, 81, 355–369. [Google Scholar] [CrossRef]
- Shen, Q.; Wu, C.; Freniere, C.; Tripler, T.N.; Xiong, Y. Nuclear Import of HIV-1. Viruses 2021, 13, 2242. [Google Scholar] [CrossRef]
- Martin, D.N.; Uprichard, S.L. Identification of transferrin receptor 1 as a hepatitis C virus entry factor. Proc. Natl. Acad. Sci. USA 2013, 110, 10777–10782. [Google Scholar] [CrossRef]
- Zhang, S.; Hu, W.; Yuan, L.; Yang, Q. Transferrin receptor 1 is a supplementary receptor that assists transmissible gastroenteritis virus entry into porcine intestinal epithelium. Cell Commun. Signal 2018, 16, 69. [Google Scholar] [CrossRef] [PubMed]
- Radoshitzky, S.R.; Abraham, J.; Spiropoulou, C.F.; Kuhn, J.H.; Nguyen, D.; Li, W.; Nagel, J.; Schmidt, P.J.; Nunberg, J.H.; Andrews, N.C.; et al. Transferrin receptor 1 is a cellular receptor for New World haemorrhagic fever arenaviruses. Nature 2007, 446, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Zong, M.; Fofana, I.; Choe, H. Human and host species transferrin receptor 1 use by North American arenaviruses. J. Virol. 2014, 88, 9418–9428. [Google Scholar] [CrossRef] [PubMed]
- Ilbäck, N.G.; Frisk, P.; Mohamed, N.; Gadhasson, I.L.; Blomberg, J.; Friman, G. Virus induces metal-binding proteins and changed trace element balance in the brain during the course of a common human infection (coxsackievirus B3) in mice. Sci. Total Environ. 2007, 381, 88–98. [Google Scholar] [CrossRef]
- Armitage, A.E.; Stacey, A.R.; Giannoulatou, E.; Marshall, E.; Sturges, P.; Chatha, K.; Smith, N.M.; Huang, X.; Xu, X.; Pasricha, S.R.; et al. Distinct patterns of hepcidin and iron regulation during HIV-1, HBV, and HCV infections. Proc. Natl. Acad. Sci. USA 2014, 111, 12187–12192. [Google Scholar] [CrossRef]
- Cavezzi, A.; Troiani, E.; Corrao, S. COVID-19: Hemoglobin, Iron, and Hypoxia beyond Inflammation. A Narrative Review. Clin. Pract. 2020, 10, 1271. [Google Scholar] [CrossRef]
- Nabel, G.; Baltimore, D. An inducible transcription factor activates expression of human immunodeficiency virus in T cells. Nature 1987, 326, 711–713. [Google Scholar] [CrossRef]
- Xiong, S.; She, H.; Takeuchi, H.; Han, B.; Engelhardt, J.F.; Barton, C.H.; Zandi, E.; Giulivi, C.; Tsukamoto, H. Signaling role of intracellular iron in NF-kappaB activation. J. Biol. Chem. 2003, 278, 17646–17654. [Google Scholar] [CrossRef]
- Nekhai, S.; Jeang, K.T. Transcriptional and post-transcriptional regulation of HIV-1 gene expression: Role of cellular factors for Tat and Rev. Future Microbiol. 2006, 1, 417–426. [Google Scholar] [CrossRef]
- Yang, X.; Gold, M.O.; Tang, D.N.; Lewis, D.E.; Aguilar-Cordova, E.; Rice, A.P.; Herrmann, C.H. TAK, an HIV Tat-associated kinase, is a member of the cyclin-dependent family of protein kinases and is induced by activation of peripheral blood lymphocytes and differentiation of promonocytic cell lines. Proc. Natl. Acad. Sci. USA 1997, 94, 12331–12336. [Google Scholar] [CrossRef]
- Kim, Y.S.; Kang, K.R.; Wolff, E.C.; Bell, J.K.; McPhie, P.; Park, M.H. Deoxyhypusine hydroxylase is a Fe(II)-dependent, HEAT-repeat enzyme. Identification of amino acid residues critical for Fe(II) binding and catalysis [corrected]. J. Biol. Chem. 2006, 281, 13217–13225. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, C.; Klein, K.C.; Kiser, P.K.; Singh, A.R.; Firestein, B.L.; Riba, S.C.; Lingappa, J.R. Identification of a host protein essential for assembly of immature HIV-1 capsids. Nature 2002, 415, 88–92. [Google Scholar] [CrossRef] [PubMed]
- Maio, N.; Lafont, B.A.P.; Sil, D.; Li, Y.; Bollinger, J.M., Jr.; Krebs, C.; Pierson, T.C.; Linehan, W.M.; Rouault, T.A. Fe-S cofactors in the SARS-CoV-2 RNA-dependent RNA polymerase are potential antiviral targets. Science 2021, 373, 236–241. [Google Scholar] [CrossRef] [PubMed]
- Honarmand Ebrahimi, K.; Ciofi-Baffoni, S.; Hagedoorn, P.L.; Nicolet, Y.; Le Brun, N.E.; Hagen, W.R.; Armstrong, F.A. Iron-sulfur clusters as inhibitors and catalysts of viral replication. Nat. Chem. 2022, 14, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Li, Y. The interaction between ferroptosis and lipid metabolism in cancer. Signal Transduct. Target. Ther. 2020, 5, 108. [Google Scholar] [CrossRef]
- Yamane, D.; Hayashi, Y.; Matsumoto, M.; Nakanishi, H.; Imagawa, H.; Kohara, M.; Lemon, S.M.; Ichi, I. FADS2-dependent fatty acid desaturation dictates cellular sensitivity to ferroptosis and permissiveness for hepatitis C virus replication. Cell Chem. Biol. 2022, 29, 799–810. [Google Scholar] [CrossRef]
- Pasricha, S.R.; Tye-Din, J.; Muckenthaler, M.U.; Swinkels, D.W. Iron deficiency. Lancet 2021, 397, 233–248. [Google Scholar] [CrossRef]
- Nemeth, E.; Rivera, S.; Gabayan, V.; Keller, C.; Taudorf, S.; Pedersen, B.K.; Ganz, T. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J. Clin. Investig. 2004, 113, 1271–1276. [Google Scholar] [CrossRef]
- Peyssonnaux, C.; Zinkernagel, A.S.; Datta, V.; Lauth, X.; Johnson, R.S.; Nizet, V. TLR4-dependent hepcidin expression by myeloid cells in response to bacterial pathogens. Blood 2006, 107, 3727–3732. [Google Scholar] [CrossRef]
- Camaschella, C.; Nai, A.; Silvestri, L. Iron metabolism and iron disorders revisited in the hepcidin era. Haematologica 2020, 105, 260–272. [Google Scholar] [CrossRef]
- Kowdley, K.V.; Gochanour, E.M.; Sundaram, V.; Shah, R.A.; Handa, P. Hepcidin Signaling in Health and Disease: Ironing Out the Details. Hepatol. Commun. 2021, 5, 723–735. [Google Scholar] [CrossRef]
- Luo, X.; Gong, H.B.; Gao, H.Y.; Wu, Y.P.; Sun, W.Y.; Li, Z.Q.; Wang, G.; Liu, B.; Liang, L.; Kurihara, H.; et al. Oxygenated phosphatidylethanolamine navigates phagocytosis of ferroptotic cells by interacting with TLR2. Cell Death Differ. 2021, 28, 1971–1989. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Xu, B.; Hu, N.; Guo, Z.; Bao, W.; Shao, B.; Yang, W. Targeting the Macrophage-Ferroptosis Crosstalk: A Novel Insight into Tumor Immunotherapy. Front. Biosci. Landmark 2022, 27, 203. [Google Scholar] [CrossRef] [PubMed]
- Kapralov, A.A.; Yang, Q.; Dar, H.H.; Tyurina, Y.Y.; Anthonymuthu, T.S.; Kim, R.; St Croix, C.M.; Mikulska-Ruminska, K.; Liu, B.; Shrivastava, I.H.; et al. Redox lipid reprogramming commands susceptibility of macrophages and microglia to ferroptotic death. Nat. Chem. Biol. 2020, 16, 278–290. [Google Scholar] [CrossRef]
- Sun, Q.; Hu, S.; Lou, Z.; Gao, J. The macrophage polarization in inflammatory dermatosis and its potential drug candidates. Biomed. Pharmacother. 2023, 161, 114469. [Google Scholar] [CrossRef]
- Jaeschke, H.; Hasegawa, T. Role of neutrophils in acute inflammatory liver injury. Liver Int. 2006, 26, 912–919. [Google Scholar] [CrossRef]
- Yotsumoto, S.; Muroi, Y.; Chiba, T.; Ohmura, R.; Yoneyama, M.; Magarisawa, M.; Dodo, K.; Terayama, N.; Sodeoka, M.; Aoyagi, R.; et al. Hyperoxidation of ether-linked phospholipids accelerates neutrophil extracellular trap formation. Sci. Rep. 2017, 7, 16026. [Google Scholar] [CrossRef] [PubMed]
- Fujita, N.; Takei, Y. Iron, hepatitis C virus, and hepatocellular carcinoma: Iron reduction preaches the gospel for chronic hepatitis C. J. Gastroenterol. 2007, 42, 923–926. [Google Scholar] [CrossRef]
- Gattoni, A.; Parlato, A.; Vangieri, B.; Bresciani, M.; Derna, R.; Baldassarre, R. Role of hemochromatosis genes in chronic hepatitis C. Clin. Ter. 2006, 157, 61–68. [Google Scholar]
- Nahon, P.; Sutton, A.; Rufat, P.; Ziol, M.; Thabut, G.; Schischmanoff, P.O.; Vidaud, D.; Charnaux, N.; Couvert, P.; Ganne-Carrie, N.; et al. Liver iron, HFE gene mutations, and hepatocellular carcinoma occurrence in patients with cirrhosis. Gastroenterology 2008, 134, 102–110. [Google Scholar] [CrossRef]
- Fujita, N.; Sugimoto, R.; Takeo, M.; Urawa, N.; Mifuji, R.; Tanaka, H.; Kobayashi, Y.; Iwasa, M.; Watanabe, S.; Adachi, Y.; et al. Hepcidin expression in the liver: Relatively low level in patients with chronic hepatitis C. Mol. Med. 2007, 13, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Fillebeen, C.; Muckenthaler, M.; Andriopoulos, B.; Bisaillon, M.; Mounir, Z.; Hentze, M.W.; Koromilas, A.E.; Pantopoulos, K. Expression of the subgenomic hepatitis C virus replicon alters iron homeostasis in Huh7 cells. J. Hepatol. 2007, 47, 12–22. [Google Scholar] [CrossRef]
- Sadiq, I.Z. Free Radicals and Oxidative Stress: Signaling Mechanisms, Redox Basis for Human Diseases, and Cell Cycle Regulation. Curr. Mol. Med. 2023, 23, 13–35. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.C. Glutathione synthesis. Biochim. Biophys. Acta 2013, 1830, 3143–3153. [Google Scholar] [CrossRef] [PubMed]
- Carr, M.; Gonzalez, G.; Martinelli, A.; Wastika, C.E.; Ito, K.; Orba, Y.; Sasaki, M.; Hall, W.W.; Sawa, H. Upregulated expression of the antioxidant sestrin 2 identified by transcriptomic analysis of Japanese encephalitis virus-infected SH-SY5Y neuroblastoma cells. Virus Genes 2019, 55, 630–642. [Google Scholar] [CrossRef]
- Li, F.J.; Long, H.Z.; Zhou, Z.W.; Luo, H.Y.; Xu, S.G.; Gao, L.C. System X(c) (-)/GSH/GPX4 axis: An important antioxidant system for the ferroptosis in drug-resistant solid tumor therapy. Front. Pharmacol. 2022, 13, 910292. [Google Scholar] [CrossRef]
- Matsushita, M.; Freigang, S.; Schneider, C.; Conrad, M.; Bornkamm, G.W.; Kopf, M. T cell lipid peroxidation induces ferroptosis and prevents immunity to infection. J. Exp. Med. 2015, 212, 555–568. [Google Scholar] [CrossRef]
- Pruett, S.B.; Obiri, N.; Kiel, J.L. Involvement and relative importance of at least two distinct mechanisms in the effects of 2-mercaptoethanol on murine lymphocytes in culture. J. Cell Physiol. 1989, 141, 40–45. [Google Scholar] [CrossRef]
- Kang, R.; Zeng, L.; Zhu, S.; Xie, Y.; Liu, J.; Wen, Q.; Cao, L.; Xie, M.; Ran, Q.; Kroemer, G.; et al. Lipid Peroxidation Drives Gasdermin D-Mediated Pyroptosis in Lethal Polymicrobial Sepsis. Cell Host Microbe 2018, 24, 97–108. [Google Scholar] [CrossRef]
- Jia, M.; Qin, D.; Zhao, C.; Chai, L.; Yu, Z.; Wang, W.; Tong, L.; Lv, L.; Wang, Y.; Rehwinkel, J.; et al. Redox homeostasis maintained by GPX4 facilitates STING activation. Nat. Immunol. 2020, 21, 727–735. [Google Scholar] [CrossRef]
- Ma, R.; Ortiz Serrano, T.P.; Davis, J.; Prigge, A.D.; Ridge, K.M. The cGAS-STING pathway: The role of self-DNA sensing in inflammatory lung disease. FASEB J. 2020, 34, 13156–13170. [Google Scholar] [CrossRef]
- Muri, J.; Thut, H.; Bornkamm, G.W.; Kopf, M. B1 and Marginal Zone B Cells but Not Follicular B2 Cells Require Gpx4 to Prevent Lipid Peroxidation and Ferroptosis. Cell Rep. 2019, 29, 2731–2744.e2734. [Google Scholar] [CrossRef]
- Hao, S.; Yu, J.; He, W.; Huang, Q.; Zhao, Y.; Liang, B.; Zhang, S.; Wen, Z.; Dong, S.; Rao, J.; et al. Cysteine Dioxygenase 1 Mediates Erastin-Induced Ferroptosis in Human Gastric Cancer Cells. Neoplasia 2017, 19, 1022–1032. [Google Scholar] [CrossRef] [PubMed]
- Huo, H.; Zhou, Z.; Qin, J.; Liu, W.; Wang, B.; Gu, Y. Erastin Disrupts Mitochondrial Permeability Transition Pore (mPTP) and Induces Apoptotic Death of Colorectal Cancer Cells. PLoS ONE 2016, 11, e0154605. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Zhu, S.; Song, X.; Sun, X.; Fan, Y.; Liu, J.; Zhong, M.; Yuan, H.; Zhang, L.; Billiar, T.R.; et al. The Tumor Suppressor p53 Limits Ferroptosis by Blocking DPP4 Activity. Cell Rep. 2017, 20, 1692–1704. [Google Scholar] [CrossRef]
- Eling, N.; Reuter, L.; Hazin, J.; Hamacher-Brady, A.; Brady, N.R. Identification of artesunate as a specific activator of ferroptosis in pancreatic cancer cells. Oncoscience 2015, 2, 517–532. [Google Scholar] [CrossRef] [PubMed]
- Greenshields, A.L.; Shepherd, T.G.; Hoskin, D.W. Contribution of reactive oxygen species to ovarian cancer cell growth arrest and killing by the anti-malarial drug artesunate. Mol. Carcinog. 2017, 56, 75–93. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Qu, Y.; Zhang, Q.; Li, F.; Li, B.; Li, Z.; Dong, Y.; Lu, L.; Cai, X. Exosomes derived from hepatitis B virus-infected hepatocytes promote liver fibrosis via miR-222/TFRC axis. Cell Biol. Toxicol. 2022, 39, 467–481. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.F.; Xiang, Y.Q.; Fang, F.; Gao, R.; Zhang, L.F.; Xie, S.H.; Liu, Z.; Du, J.L.; Chen, S.H.; Hong, M.H.; et al. Hepatitis B virus infection and risk of nasopharyngeal carcinoma in southern China. Cancer Epidemiol. Biomark. Prev. 2015, 24, 1766–1773. [Google Scholar] [CrossRef]
- Li, T.Y.; Yang, Y.; Zhou, G.; Tu, Z.K. Immune suppression in chronic hepatitis B infection associated liver disease: A review. World J. Gastroenterol. 2019, 25, 3527–3537. [Google Scholar] [CrossRef]
- Yuan, S.; Wei, C.; Liu, G.; Zhang, L.; Li, J.; Li, L.; Cai, S.; Fang, L. Sorafenib attenuates liver fibrosis by triggering hepatic stellate cell ferroptosis via HIF-1α/SLC7A11 pathway. Cell Prolif. 2022, 55, e13158. [Google Scholar] [CrossRef] [PubMed]
- Hodgson, N.C. Merkel cell carcinoma: Changing incidence trends. J. Surg. Oncol. 2005, 89, 1–4. [Google Scholar] [CrossRef]
- Feng, H.; Shuda, M.; Chang, Y.; Moore, P.S. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science 2008, 319, 1096–1100. [Google Scholar] [CrossRef] [PubMed]
- Efferth, T. From ancient herb to modern drug: Artemisia annua and artemisinin for cancer therapy. Semin. Cancer Biol. 2017, 46, 65–83. [Google Scholar] [CrossRef]
- Wang, N.; Zeng, G.Z.; Yin, J.L.; Bian, Z.X. Artesunate activates the ATF4-CHOP-CHAC1 pathway and affects ferroptosis in Burkitt’s Lymphoma. Biochem. Biophys. Res. Commun. 2019, 519, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Sarma, B.; Willmes, C.; Angerer, L.; Adam, C.; Becker, J.C.; Kervarrec, T.; Schrama, D.; Houben, R. Artesunate Affects T Antigen Expression and Survival of Virus-Positive Merkel Cell Carcinoma. Cancers 2020, 12, 919. [Google Scholar] [CrossRef]
- Seibt, T.M.; Proneth, B.; Conrad, M. Role of GPX4 in ferroptosis and its pharmacological implication. Free Radic. Biol. Med. 2019, 133, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Sun, X.; Luo, X.; Ding, J.; Fan, F.; Li, P.; Shen, X.; Fang, Y. Encapsulation of selenium-containing peptides in xanthan gum-lysozyme nanoparticles as a powerful gastrointestinal delivery system. Food Res. Int. 2022, 156, 111351. [Google Scholar] [CrossRef]
- Rushworth, G.F.; Megson, I.L. Existing and potential therapeutic uses for N-acetylcysteine: The need for conversion to intracellular glutathione for antioxidant benefits. Pharmacol. Ther. 2014, 141, 150–159. [Google Scholar] [CrossRef]
- Anderson, M.E.; Meister, A. Glutathione monoesters. Anal. Biochem. 1989, 183, 16–20. [Google Scholar] [CrossRef]
- Levy, E.J.; Anderson, M.E.; Meister, A. Transport of glutathione diethyl ester into human cells. Proc. Natl. Acad. Sci. USA 1993, 90, 9171–9175. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Wang, M.; Wang, Y.; Guo, Y.; Tao, X.; Wang, X.; Cao, Y.; Tian, S.; Li, Q. p53-mediated ferroptosis is required for 1-methyl-4-phenylpyridinium-induced senescence of PC12 cells. Toxicol. Vitr. 2021, 73, 105146. [Google Scholar] [CrossRef]
- Ji, Q.; Fu, S.; Zuo, H.; Huang, Y.; Chu, L.; Zhu, Y.; Hu, J.; Wu, Y.; Chen, S.; Wang, Y.; et al. ACSL4 is essential for radiation-induced intestinal injury by initiating ferroptosis. Cell Death Discov. 2022, 8, 332. [Google Scholar] [CrossRef] [PubMed]
- Debebe, Z.; Ammosova, T.; Jerebtsova, M.; Kurantsin-Mills, J.; Niu, X.; Charles, S.; Richardson, D.R.; Ray, P.E.; Gordeuk, V.R.; Nekhai, S. Iron chelators ICL670 and 311 inhibit HIV-1 transcription. Virology 2007, 367, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Salhi, Y.; Costagliola, D.; Rebulla, P.; Dessi, C.; Karagiorga, M.; Lena-Russo, D.; de Montalembert, M.; Girot, R. Serum ferritin, desferrioxamine, and evolution of HIV-1 infection in thalassemic patients. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 1998, 18, 473–478. [Google Scholar] [CrossRef]
- Tabor, E.; Epstein, J.S.; Hewlett, I.K.; Lee, S.F. Inhibition by desferrioxamine of in-vitro replication of HIV-1. Lancet 1991, 337, 795. [Google Scholar] [CrossRef]
- Khadivjam, B.; Stegen, C.; Hogue-Racine, M.A.; El Bilali, N.; Döhner, K.; Sodeik, B.; Lippé, R. The ATP-Dependent RNA Helicase DDX3X Modulates Herpes Simplex Virus 1 Gene Expression. J. Virol. 2017, 91, 8. [Google Scholar] [CrossRef]
- Romeo, A.M.; Christen, L.; Niles, E.G.; Kosman, D.J. Intracellular chelation of iron by bipyridyl inhibits DNA virus replication: Ribonucleotide reductase maturation as a probe of intracellular iron pools. J. Biol. Chem. 2001, 276, 24301–24308. [Google Scholar] [CrossRef]
- Kuo, C.Y.; Chiu, V.; Hsieh, P.C.; Huang, C.Y.; Huang, S.J.; Tzeng, I.S.; Tsai, F.M.; Chen, M.L.; Liu, C.T.; Chen, Y.R. Chrysophanol attenuates hepatitis B virus X protein-induced hepatic stellate cell fibrosis by regulating endoplasmic reticulum stress and ferroptosis. J. Pharmacol. Sci. 2020, 144, 172–182. [Google Scholar] [CrossRef]
- Xiao, Q.; Yan, L.; Han, J.; Yang, S.; Tang, Y.; Li, Q.; Lao, X.; Chen, Z.; Xiao, J.; Zhao, H.; et al. Metabolism-dependent ferroptosis promotes mitochondrial dysfunction and inflammation in CD4(+) T lymphocytes in HIV-infected immune non-responders. EBioMedicine 2022, 86, 104382. [Google Scholar] [CrossRef]
- Battaglia, A.M.; Chirillo, R.; Aversa, I.; Sacco, A.; Costanzo, F.; Biamonte, F. Ferroptosis and Cancer: Mitochondria Meet the “Iron Maiden” Cell Death. Cells 2020, 9, 1505. [Google Scholar] [CrossRef] [PubMed]
- Long, H.Z.; Cheng, Y.; Zhou, Z.W.; Luo, H.Y.; Wen, D.D.; Gao, L.C. The key roles of organelles and ferroptosis in Alzheimer’s disease. J. Neurosci. Res. 2022, 100, 1257–1280. [Google Scholar] [CrossRef]
- Toyokuni, S.; Ito, F.; Yamashita, K.; Okazaki, Y.; Akatsuka, S. Iron and thiol redox signaling in cancer: An exquisite balance to escape ferroptosis. Free Radic. Biol. Med. 2017, 108, 610–626. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Dielschneider, R.F.; Henson, E.S.; Xiao, W.; Choquette, T.R.; Blankstein, A.R.; Chen, Y.; Gibson, S.B. Ferroptosis and autophagy induced cell death occur independently after siramesine and lapatinib treatment in breast cancer cells. PLoS ONE 2017, 12, e0182921. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wei, X.; Tao, F.; Deng, C.; Lv, C.; Chen, C.; Cheng, Y. The potential application of nanomaterials for ferroptosis-based cancer therapy. Biomed. Mater. 2021, 16, 042013. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Chen, Z.; Zhang, H.; Chen, C.; Zeng, M.; Yunis, J.; Wei, Y.; Wan, Y.; Wang, N.; Zhou, M.; et al. Selenium–GPX4 axis protects follicular helper T cells from ferroptosis. Nat. Immunol. 2021, 22, 1127–1139. [Google Scholar] [CrossRef]
- Upadhyayula, P.S.; Higgins, D.M.; Mela, A.; Banu, M.; Dovas, A.; Zandkarimi, F.; Patel, P.; Mahajan, A.; Humala, N.; Nguyen, T.T.T.; et al. Dietary restriction of cysteine and methionine sensitizes gliomas to ferroptosis and induces alterations in energetic metabolism. Nat. Commun. 2023, 14, 1187. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, J.; Fu, J.; Zhao, S.; Zhang, X.; Chao, Y.; Pan, Q.; Sun, H.; Zhang, J.; Li, B.; Xue, T.; et al. Free Radical and Viral Infection: A Review from the Perspective of Ferroptosis. Vet. Sci. 2023, 10, 456. https://doi.org/10.3390/vetsci10070456
Chen J, Fu J, Zhao S, Zhang X, Chao Y, Pan Q, Sun H, Zhang J, Li B, Xue T, et al. Free Radical and Viral Infection: A Review from the Perspective of Ferroptosis. Veterinary Sciences. 2023; 10(7):456. https://doi.org/10.3390/vetsci10070456
Chicago/Turabian StyleChen, Jun, Jinping Fu, Sha Zhao, Xiaoxi Zhang, Yuyang Chao, Qunxing Pan, Huawei Sun, Jingfeng Zhang, Bin Li, Tao Xue, and et al. 2023. "Free Radical and Viral Infection: A Review from the Perspective of Ferroptosis" Veterinary Sciences 10, no. 7: 456. https://doi.org/10.3390/vetsci10070456
APA StyleChen, J., Fu, J., Zhao, S., Zhang, X., Chao, Y., Pan, Q., Sun, H., Zhang, J., Li, B., Xue, T., Li, J., & Liu, C. (2023). Free Radical and Viral Infection: A Review from the Perspective of Ferroptosis. Veterinary Sciences, 10(7), 456. https://doi.org/10.3390/vetsci10070456