
An Improved Duplex Real-Time Quantitative RT-PCR Assay with a Canine Endogenous Internal Positive Control for More Sensitive and Reliable Detection of Canine Parainfluenza Virus 5
 , ,
, ,
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Samples and Nucleic Acid Extraction
2.2. Reference qRT-PCR Assays
2.3. Reference Gene Construction for dqRT-PCR Assay
2.4. Primers and Probe for dqRT-PCR Assay
2.5. Optimization of dqRT-PCR Conditions
2.6. Sensitivity of dqRT-PCR Assays
2.7. Specificity of dqRT-PCR Assays
2.8. Precision of dqRT-PCR Assays
2.9. Clinical Evaluation of dqRT-PCR Assay
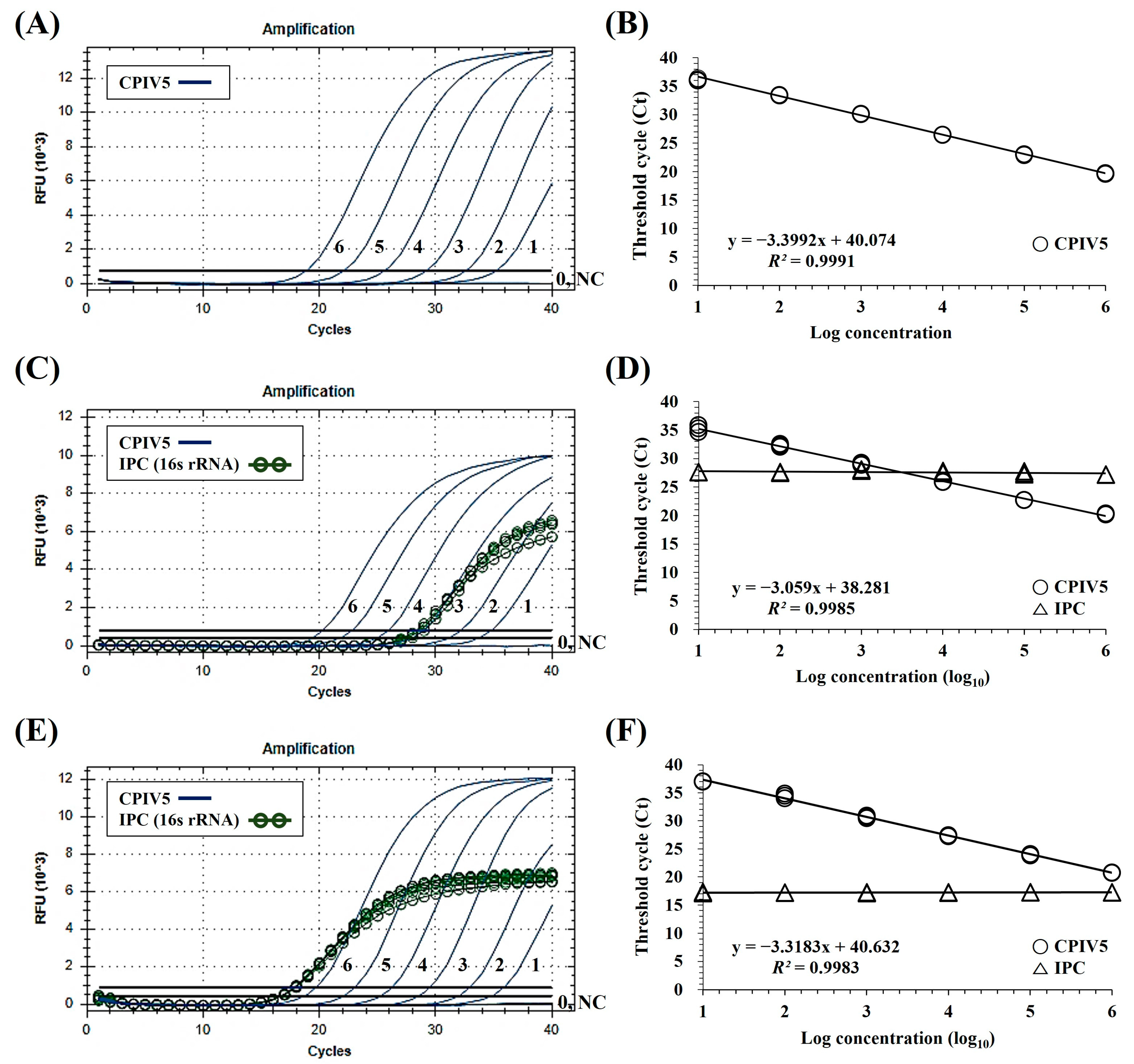
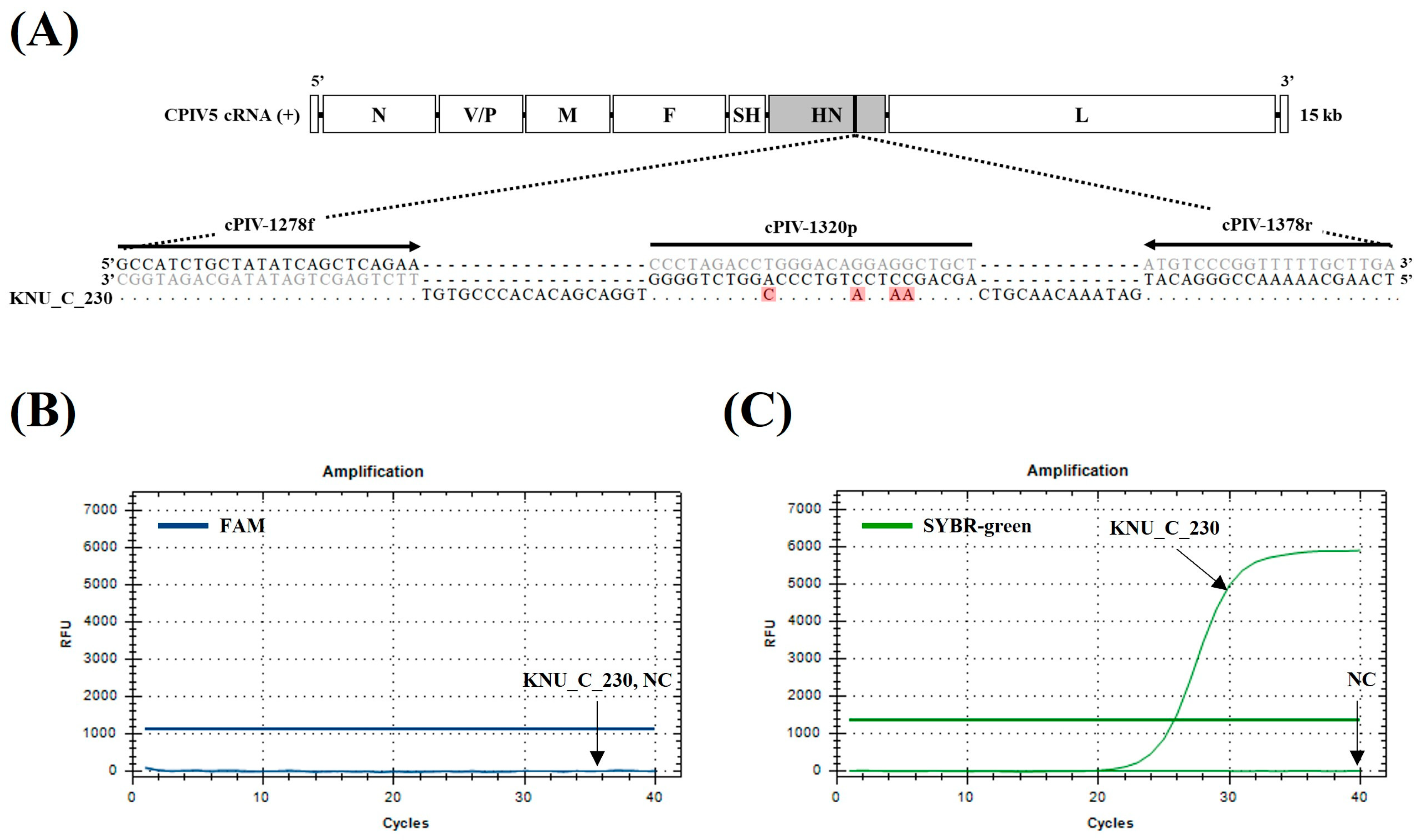
3. Results
3.1. Interpretation of the dqRT-PCR Assay
3.2. Specificity and Sensitivity of the dqRT-PCR Assay
3.3. Precision of the dqRT-PCR Assay
3.4. Comparative Clinical Evaluation of the dqRT-PCR Assay
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rima, B.; Balkema-Buschmann, A.; Dundon, W.G.; Duprex, P.; Easton, A.; Fouchier, R.; Kurath, G.; Lamb, R.; Lee, B.; Rota, P.; et al. ICTV Report Consortium. ICTV Virus Taxonomy Profile: Paramyxoviridae. J. Gen. Virol. 2019, 100, 1593–1594. [Google Scholar] [CrossRef]
- Hsiung, G.D. Parainfluenza-5 virus. Infection of man and animal. Prog. Med. Virol. 1972, 14, 241–274. [Google Scholar]
- Chatziandreou, N.; Stock, N.; Young, D.; Andrejeva, J.; Hagmaier, K.; McGeoch, D.J.; Randall, R.E. Relationships and host range of human, canine, simian and porcine isolates of simian virus 5 (parainfluenza virus 5). J. Gen. Virol. 2004, 85, 3007–3016. [Google Scholar] [CrossRef]
- Jiang, N.; Wang, E.; Guo, D.; Wang, X.; Su, M.; Kong, F.; Yuan, D.; Zhai, J.; Sun, D. Isolation and molecular characterization of parainfluenza virus 5 in diarrhea-affected piglets in China. J. Vet. Med. Sci. 2018, 80, 590–593. [Google Scholar] [CrossRef]
- Lee, Y.N.; Park, C.K.; Kim, S.H.; Lee, S.; Shin, J.H.; Lee, C. Characterization in vitro and in vivo of a novel porcine parainfluenza virus 5 isolate in Korea. Virus Res. 2013, 178, 423–429. [Google Scholar] [CrossRef]
- Oem, J.K.; Kim, S.H.; Kim, Y.H.; Lee, M.H.; Lee, K.K. Molecular characteristics of canine parainfluenza viruses type 5 (CPIV-5) isolated in Korea. Can. J. Vet. Res. 2015, 79, 64–67. [Google Scholar] [PubMed]
- Xie, J.; Tong, P.; Zhang, A.; Zhang, L.; Song, X.; Kuang, L. Identification and characterization of the first equine parainfluenza Virus 5. Virol. Sin. 2020, 35, 245–247. [Google Scholar] [CrossRef] [PubMed]
- Zhai, J.Q.; Zhai, S.L.; Lin, T.; Liu, J.K.; Wang, H.X.; Li, B.; Zhang, H.; Zou, S.Z.; Zhou, X.; Wu, M.F.; et al. First complete genome sequence of parainfluenza virus 5 isolated from lesser panda. Arch. Virol. 2017, 162, 1413–1418. [Google Scholar] [CrossRef]
- Binn, L.N.; Eddy, G.A.; Lazar, E.C.; Helms, J.; Murnane, T. Viruses recovered from laboratory dogs with respiratory disease. Proc. Soc. Exp. Biol. Med. 1967, 126, 140–145. [Google Scholar] [CrossRef] [PubMed]
- Day, M.J.; Carey, S.; Clercx, C.; Kohn, B.; MarsilIo, F.; Thiry, E.; Freyburger, L.; Schulz, B.; Walker, D.J. Aetiology of canine infectious respiratory disease complex and prevalence of its pathogens in Europe. J. Comp. Pathol. 2020, 176, 86–108. [Google Scholar] [CrossRef]
- Ellis, J.A.; Krakowka, G.S. A review of canine parainfluenza virus infection in dogs. J. Am. Vet. Med. Assoc. 2012, 240, 273–284. [Google Scholar] [CrossRef]
- Joffe, D.J.; Lelewski, R.; Weese, J.S.; Mcgill-Worsley, J.; Shankel, C.; Mendonca, S.; Sager, T.; Smith, M.; Poljak, Z. Factors associated with development of canine infectious respiratory disease complex (CIRDC) in dogs in 5 Canadian small animal clinics. Can. Vet. J. 2016, 57, 46–51. [Google Scholar]
- Liu, C.; Li, X.; Zhang, J.; Yang, L.; Li, F.; Deng, J.; Tan, F.; Sun, M.; Liu, Y.; Tian, K. Isolation and genomic characterization of a canine parainfluenza virus type 5 strain in China. Arch. Virol. 2017, 162, 2337–2344. [Google Scholar] [CrossRef]
- Charoenkul, K.; Nasamran, C.; Janetanakit, T.; Chaiyawong, S.; Bunpapong, N.; Boonyapisitsopa, S.; Tangwangvivat, R.; Amonsin, A. Molecular detection and whole genome characterization of Canine Parainfluenza type 5 in Thailand. Sci. Rep. 2021, 85, 3007–3016. [Google Scholar] [CrossRef]
- Erles, K.; Dubovi, E.J.; Brooks, H.W.; Brownlie, J. Longitudinal study of viruses associated with canine infectious respiratory disease. J. Clin. Microbiol. 2004, 42, 4524–4529. [Google Scholar] [CrossRef]
- Posuwan, N.; Payungporn, S.; Thontiravong, A.; Kitikoon, P.; Amonsin, A.; Poovorawan, Y. Prevalence of respiratory viruses isolated from dogs in Thailand during 2008–2009. Asian Biomed. 2010, 4, 563–569. [Google Scholar] [CrossRef]
- Decaro, N.; Mari, V.; Larocca, V.; Losurdo, M.; Lanave, G.; Lucente, M.S.; Corrente, M.; Catella, C.; Bo, S.; Elia, G.; et al. Molecular surveillance of traditional and emerging pathogens associated with canine infectious respiratory disease. Vet. Microbiol. 2016, 192, 21–25. [Google Scholar] [CrossRef]
- Hiebl, A.; Auer, A.; Bagrinovschi, G.; Stejskal, M.; Hirt, R.; Rümenapf, H.T.; Tichy, A.; Künzel, F. Detection of selected viral pathogens in dogs with canine infectious respiratory disease in Austria. J. Small. Anim. Pract. 2019, 60, 594–600. [Google Scholar] [CrossRef]
- Piewbang, C.; Rungsipipat, A.; Poovorawan, Y.; Techangamsuwan, S. Development and application of multiplex PCR assays for detection of virus-induced respiratory disease complex in dogs. J. Vet. Med. Sci. 2017, 78, 1847–1854. [Google Scholar] [CrossRef]
- Hao, X.; Liu, R.; He, Y.; Xiao, X.; Xiao, W.; Zheng, Q.; Lin, X.; Tao, P.; Zhou, P.; Li, S. Multiplex PCR methods for detection of several viruses associated with canine respiratory and enteric diseases. PLoS ONE 2019, 14, e0213295. [Google Scholar] [CrossRef]
- Mochizuki, M.; Yachi, A.; Ohshima, T.; Ohuchi, A.; Ishida, T. Etiologic study of upper respiratory infections of household dogs. J. Vet. Med. Sci. 2008, 70, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Arya, M.; Shergill, I.S.; Williamson, M.; Gommersall, L.; Arya, N.; Patel, H.R. Basic principles of real-time quantitative PCR. Expert Rev. Mol. Diagn. 2005, 5, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Kralik, P.; Ricchi, M. A basic guide to real time PCR in microbial diagnostics: Definitions, parameters, and everything. Front. Microbiol. 2017, 8, 108. [Google Scholar] [CrossRef]
- Matsuu, A.; Yabuki, M.; Aoki, E.; Iwahana, M. Molecular detection of canine respiratory pathogens between 2017 and 2018 in Japan. J. Vet. Med. Sci. 2018, 82, 690–694. [Google Scholar] [CrossRef]
- More, G.D.; Cave, N.J.; Biggs, P.J.; Acke, E.; Dunowska, M. A molecular survey of canine respiratory viruses in New Zealand. N. Z. Vet. J. 2021, 69, 224–233. [Google Scholar] [CrossRef]
- Dong, J.; Tsui, W.N.T.; Leng, X.; Fu, J.; Lohman, M.; Anderson, J.; Hamill, V.; Lu, N.; Porter, E.P.; Gray, M.; et al. Development of a three-panel multiplex real-time PCR assay for simultaneous detection of nine canine respiratory pathogens. J. Microbiol. Methods 2022, 199, 106528. [Google Scholar] [CrossRef]
- Windsor, R.C.; Johnson, L.R.; Sykes, J.E.; Drazenovich, T.L.; Leutenegger, C.M.; De Cock, H.E. Molecular detection of microbes in nasal tissue of dogs with idiopathic lymphoplasmacytic rhinitis. J. Vet. Intern. Med. 2006, 20, 250–256. [Google Scholar] [CrossRef]
- Bustin, S.; Huggett, J. qPCR primer design revisited. Biomol. Detect. Quantif. 2017, 14, 19–28. [Google Scholar] [CrossRef]
- Cordey, S.; Thomas, Y.; Cherpillod, P.; van Belle, S.; Tapparel, C.; Kaiser, L. Simultaneous detection of parainfluenza viruses 1 and 3 by real-time reverse transcription-polymerase chain reaction. J. Virol. Methods. 2009, 156, 166–168. [Google Scholar] [CrossRef]
- Tong, S.; Chern, S.W.; Li, Y.; Pallansch, M.A.; Anderson, L.J. Sensitive and broadly reactive reverse transcription-PCR assays to detect novel paramyxoviruses. J. Clin. Microbiol. 2008, 46, 2652–2658. [Google Scholar] [CrossRef]
- Thellin, O.; Zorzi, W.; Lakaye, B.; De Borman, B.; Coumans, B.; Hennen, G.; Grisar, T.; Igout, A.; Heinen, E.D. Housekeeping genes as internal standards: Use and limits. J. Biotechnol. 1999, 75, 291–295. [Google Scholar] [CrossRef]
- Brinkhof, B.; Spee, B.; Rothuizen, J.; Penning, L.C. Development and evaluation of canine reference genes for accurate quantification of gene expression. Anal. Biochem. 2006, 356, 36–43. [Google Scholar] [CrossRef]
- Modarelli, J.J.; Ferro, P.J.; Esteve-Gasent, M.D. Development and application of a canine endogenous internal positive control for use in real-time PCR assays. J. Vet. Diagn. Investig. 2018, 30, 789–792. [Google Scholar] [CrossRef]
- Rima, B.K.; Gatherer, D.; Young, D.F.; Norsted, H.; Randall, R.E.; Davison, A.J. Stability of the parainfluenza virus 5 genome revealed by deep sequencing of strains isolated from different hosts and following passage in cell culture. J. Virol. 2014, 88, 3826–3836. [Google Scholar] [CrossRef]
- Navarro, E.; Serrano-Heras, G.; Castañoa, M.J.; Solera, J. Real-time PCR detection chemistry. Clin. Chim. Acta. 2015, 439, 231–250. [Google Scholar] [CrossRef]
- Kwiecien, R.; Kopp-Schneider, A.; Blettner, M. Concordance analysis: Part 16 of a series on evaluation of scientific publications. Dtsch. Ärztebl. Int. 2011, 108, 515–521. [Google Scholar] [CrossRef]
- Durchfeld, B.; Baumgärtner, W.; Krakowka, S. Intranasal infection of ferrets (Mustela putorius furo) with canine parainfluenza virus. Zentralbl Veterinarmed B. 1991, 38, 505–512. [Google Scholar] [CrossRef]
- Randall, R.E.; Young, D.F.; Goswami, K.K.; Russell, W.C. Isolation and characterization of monoclonal antibodies to simian virus 5 and their use in revealing antigenic diferences between human, canine and simian isolates. J. Gen. Virol. 1987, 68, 2769–2780. [Google Scholar] [CrossRef]
- Klungthong, C.; Chinnawirotpisan, P.; Hussem, K.; Phonpakobsin, T.; Manasatienkij, W.; Ajariyakhajorn, C.; Rungrojcharoenkit, K.; Gibbons, R.V.; Jarman, R.G. The impact of primer and probe-template mismatches on the sensitivity of pandemic influenza A/H1N1/2009 virus detection by real-time RT-PCR. J. Clin. Virol. 2010, 48, 91–95. [Google Scholar] [CrossRef]
- Lengerova, M.; Racil, Z.; Volfova, P.; Lochmanova, J.; Berkovcova, J.; Dvorakova, D.; Vorlicek, J.; Mayer, J. Real-time PCR diagnostics failure caused by nucleotide variability within exon 4 of the human cytomegalovirus major immediate-early gene. J. Clin. Microbiol. 2007, 45, 1042–1044. [Google Scholar] [CrossRef] [PubMed]
- Süß, B.; Flekna, G.; Wagner, M.; Hein, I. Studying the effect of single mismatches in primer and probe binding regions on amplification curves and quantification in real-time PCR. J. Microbiol. Methods. 2009, 76, 316–319. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.R.; Kuo, C.Y.; Huang, H.Y.; Wu, F.T.; Huang, Y.L.; Cheng, C.Y.; Su, Y.T.; Chang, F.Y.; Wu, H.S.; Liu, M.T. Newly emerging mutations in the matrix genes of the human influenza A(H1N1)pdm09 and A(H3N2) viruses reduce the detection sensitivity of real-time reverse transcription-PCR. J. Clin. Microbiol. 2014, 52, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.K.; Yoon, S.S.; Kim, B.H.; Byun, J.W.; Lee, K.W.; Kim, Y.H.; Choi, S.S.; Cha, H.J.; Son, S.W. Incidence of canine parainfluenza virus 2 in Korean stay dogs by immunohistochemistry and serology. Korean J. Vet. Public Health 2009, 33, 1–6. [Google Scholar]
- Koh, B.R.D.; Kim, H.N.; Kim, H.J.; Oh, A.R.; Jung, B.R.; Park, J.S.; Lee, J.G.; Na, H.M.; Kim, Y.H. A survey of respiratory pathogens in dogs for adoption in Gwangju metropolitan city animal shelter, South Korea. Korean J. Vet. Serv. 2020, 43, 67–77. [Google Scholar] [CrossRef]
- Na, H.M.; Bae, S.Y.; Lee, Y.E.; Park, J.S.; Park, S.D.; Kim, E.S.; Kim, Y.H. Prediction survey on the viral disease of companion animals in Gwangju area, Korea. Korean J. Vet. Serv. 2013, 36, 187–192. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Method | Target Gene | Primer and Probe | Primer/Probe Sequence (5′–3′) a | Genome Position b | Amplicon Size (bp) | Reference |
|---|---|---|---|---|---|---|
| dqRT-PCR | L | CPIV-LF | GCTTGGACAATGATATCTATCTC | 10649–10671 | 83 | This study |
| CPIV-LR | TCTCCCTGCACCATACTC | 10714–10731 | ||||
| CPIV-LP | FAM–CCACAGAGTCTGGCACACGAGTA–BHQ1 | 10689–10711 | ||||
| Canine 16S rRNA | EIPC-F | AGACGAGAAGACCCTATG | 2142–2159 | 116 | This study | |
| EIPC-R | GGTCACCCCAACCTAAAT | 2240–2257 | ||||
| EIPC-P | HEX–ACCTACAAGGCATAACATAACACCA–BHQ1 | 2199–2223 | ||||
| qRT-PCR | HN | cPIV-1278f | GCCATCTGCTATATCAGCTCAGAA | 7861–7884 | 101 | Windsor et al. [28] |
| cPIV-1378r | TCAAGCAAAAACCGGGACAT | 7942–7961 | ||||
| cPIV-1320p | FAM–AGCAGCCTCCTGTCCCAGGTCTAGGG–BHQ1 | 7903–7928 | ||||
| N | NF | GATCATTCCGCTTAATCCCC | 480–499 | 77 | Dong et al. [27] | |
| NR | TTCTGCAAGTGCAGCATAGG | 537–556 | ||||
| NP | FAM–TCGTTCAGGTATGAGCCGTGGA–BHQ1 | 505–526 |
| Pathogen | Strain | Source a | Amplification of Target Gene | |
|---|---|---|---|---|
| CPIV5 (FAM) | EIPC (HEX) | |||
| Canine parainfluenza virus 5 | D008 | CAVS | + | + |
| Canine parainfluenza virus 5 | Cornell | CAVS | + | + |
| Canine parainfluenza virus 5 | NL-CPI-5 | CAVS | + | + |
| Canine parainfluenza virus 5 | Korean isolate | ADIC | + | + |
| Canine respiratory coronavirus | K37 | APQA | − | − |
| Canine coronavirus | K378 | CAVS | − | − |
| Canine distemper virus | Onderstepoort | CAVS | − | + |
| Canine influenza virus | A/Canine/Republic of Korea/01/07(H3N2) | CAVS | − | + |
| Canine adenovirus 2 | Ditchfield | CAVS | − | + |
| Canine parvovirus | 7809 16-LP | CAVS | − | + |
| Bordetella bronchiseptica | S-55 | CAVS | − | + |
| Non-infected canine swab sample | - | APQA | − | + |
| Non-infected canine lung sample | - | APQA | − | + |
| MDCK cell | - | ADIC | − | + |
| ST cell | - | ADIC | − | − |
| Vero cell | - | ADIC | − | − |
| Dilutions (TCID50/mL) | Results of Different Assays a | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| L gene-Specific dqRT-PCR | HN Gene-Specific qRT-PCR | N Gene-Specific qRT-PCR | |||||||
| 1-Folds | 2-Folds | 3-Folds | 1-Folds | 2-Folds | 3-Folds | 1-Folds | 2-Folds | 3-Folds | |
| 104 | 21.49 | 21.53 | 21.52 | 22.10 | 22.74 | 22.49 | 21.79 | 21.87 | 21.86 |
| 103 | 24.75 | 24.97 | 24.94 | 25.82 | 26.08 | 26.19 | 25.14 | 25.37 | 25.29 |
| 102 | 28.03 | 28.10 | 28.16 | 29.59 | 29.85 | 30.03 | 28.50 | 28.28 | 28.35 |
| 101 | 31.26 | 31.46 | 34.35 | 32.99 | 33.53 | 33.17 | 31.39 | 31.33 | 31.47 |
| 100 | 34.95 | 33.97 | 34.11 | 37.75 | NC | 36.85 | 34.94 | 34.32 | 34.45 |
| 10−1 | NC | NC | NC | NC | NC | NC | NC | NC | NC |
| Concentration of RNA (Copies/Reaction) | Ct Values of the dqRT-PCR for CPIV5 RNAs | |||||
|---|---|---|---|---|---|---|
| Intra-Assay | Inter-Assay | |||||
| Mean | SD | CV (%) | Mean | SD | CV (%) | |
| High (106) | 20.73 | 0.03 | 0.1 | 20.33 | 0.29 | 1.4 |
| Medium (104) | 27.34 | 0.11 | 0.4 | 26.60 | 0.51 | 1.9 |
| Low (102) | 34.45 | 0.42 | 1.2 | 33.40 | 0.66 | 2.0 |
| Test Results of Different Assays | New dqRT-PCR | Detection Rate | Overall Agreement | |||
|---|---|---|---|---|---|---|
| Positive | Negative | Total | ||||
| HN gene-specific qRT-PCR | Positive | 15 | 0 | 15 | 5.6% | 99.6% |
| Negative | 1 | 253 | 254 | |||
| Total | 16 | 253 | 269 | |||
| N gene-specific qRT-PCR | Positive | 16 | 0 | 16 | 5.9% | 100.0% |
| Negative | 0 | 253 | 253 | |||
| Total | 16 | 253 | 269 | |||
| Detection rate | 5.9% | |||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeon, G.-T.; Kim, H.-R.; Shin, Y.-K.; Kwon, O.-K.; Kang, H.-E.; Kwon, O.-D.; Park, C.-K. An Improved Duplex Real-Time Quantitative RT-PCR Assay with a Canine Endogenous Internal Positive Control for More Sensitive and Reliable Detection of Canine Parainfluenza Virus 5. Vet. Sci. 2023, 10, 142. https://doi.org/10.3390/vetsci10020142
Jeon G-T, Kim H-R, Shin Y-K, Kwon O-K, Kang H-E, Kwon O-D, Park C-K. An Improved Duplex Real-Time Quantitative RT-PCR Assay with a Canine Endogenous Internal Positive Control for More Sensitive and Reliable Detection of Canine Parainfluenza Virus 5. Veterinary Sciences. 2023; 10(2):142. https://doi.org/10.3390/vetsci10020142
Chicago/Turabian StyleJeon, Gyu-Tae, Hye-Ryung Kim, Yeun-Kyung Shin, Oh-Kyu Kwon, Hae-Eun Kang, Oh-Deog Kwon, and Choi-Kyu Park. 2023. "An Improved Duplex Real-Time Quantitative RT-PCR Assay with a Canine Endogenous Internal Positive Control for More Sensitive and Reliable Detection of Canine Parainfluenza Virus 5" Veterinary Sciences 10, no. 2: 142. https://doi.org/10.3390/vetsci10020142
APA StyleJeon, G.-T., Kim, H.-R., Shin, Y.-K., Kwon, O.-K., Kang, H.-E., Kwon, O.-D., & Park, C.-K. (2023). An Improved Duplex Real-Time Quantitative RT-PCR Assay with a Canine Endogenous Internal Positive Control for More Sensitive and Reliable Detection of Canine Parainfluenza Virus 5. Veterinary Sciences, 10(2), 142. https://doi.org/10.3390/vetsci10020142