Comparative Aspects of Human, Canine, and Feline Obesity and Factors Predicting Progression to Diabetes

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
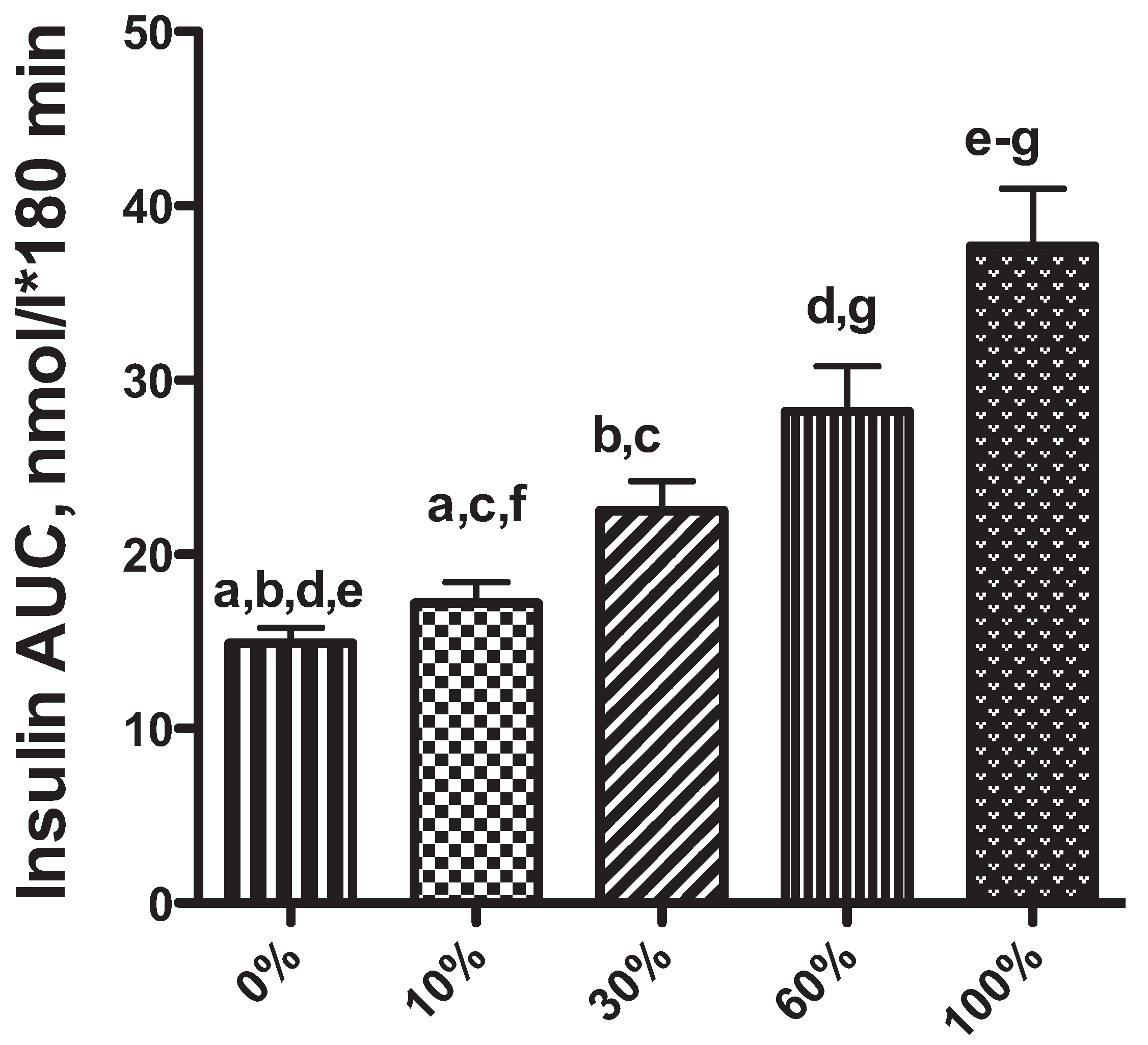
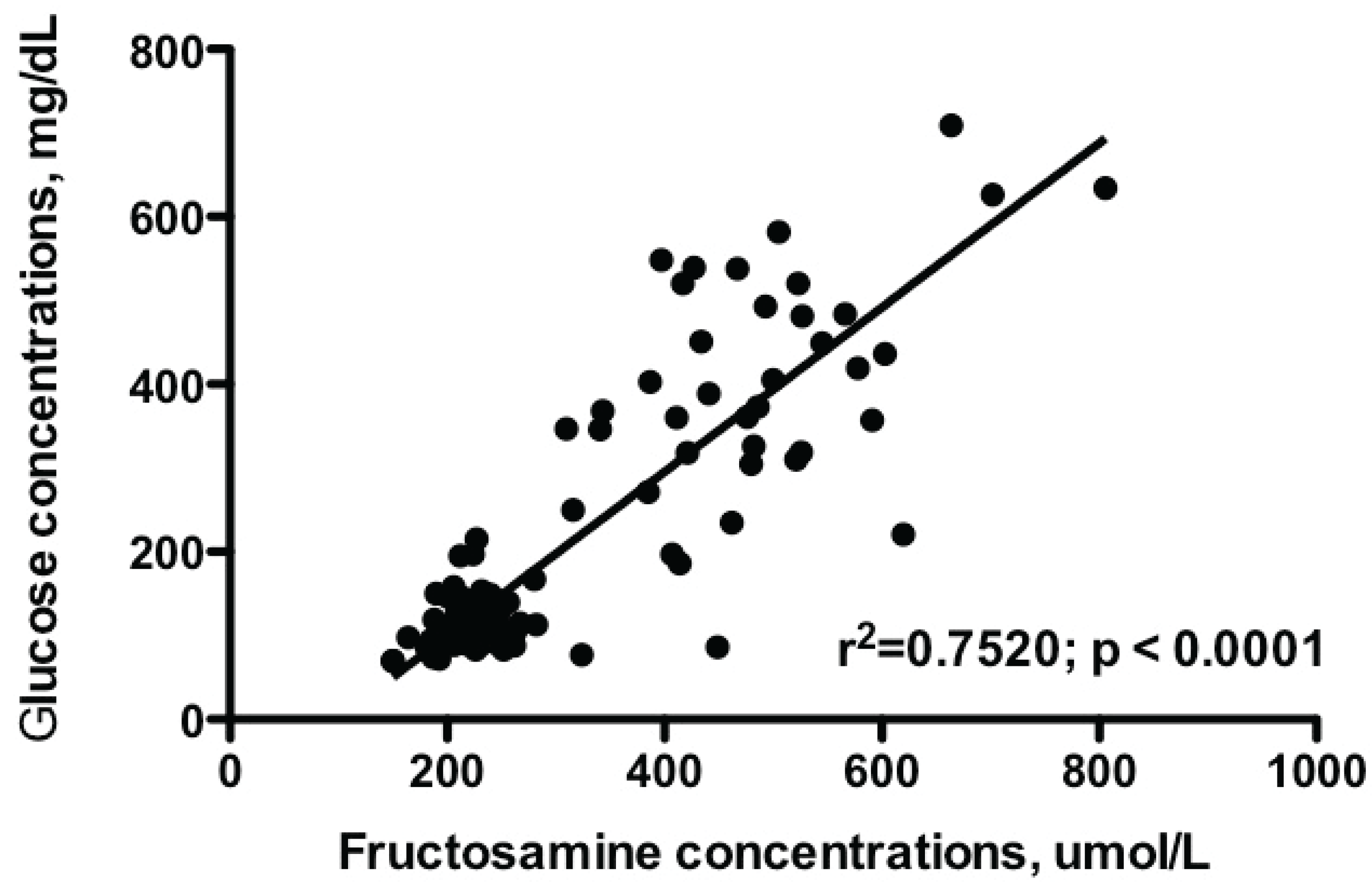
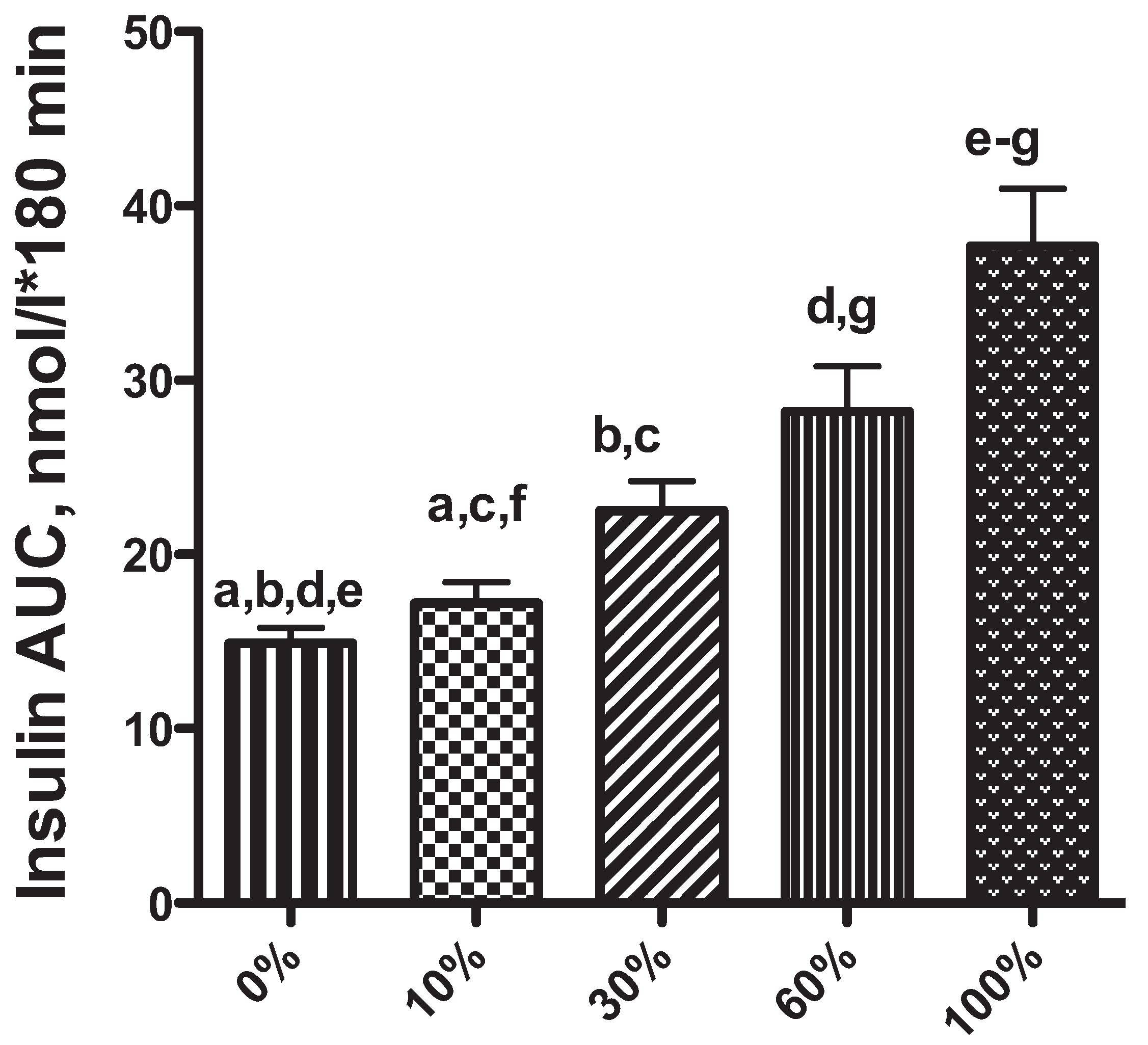
2. Obesity-Associated Changes in Insulin Secretion: A Risk Factor for Diabetes Mellitus in Dogs and Cats?
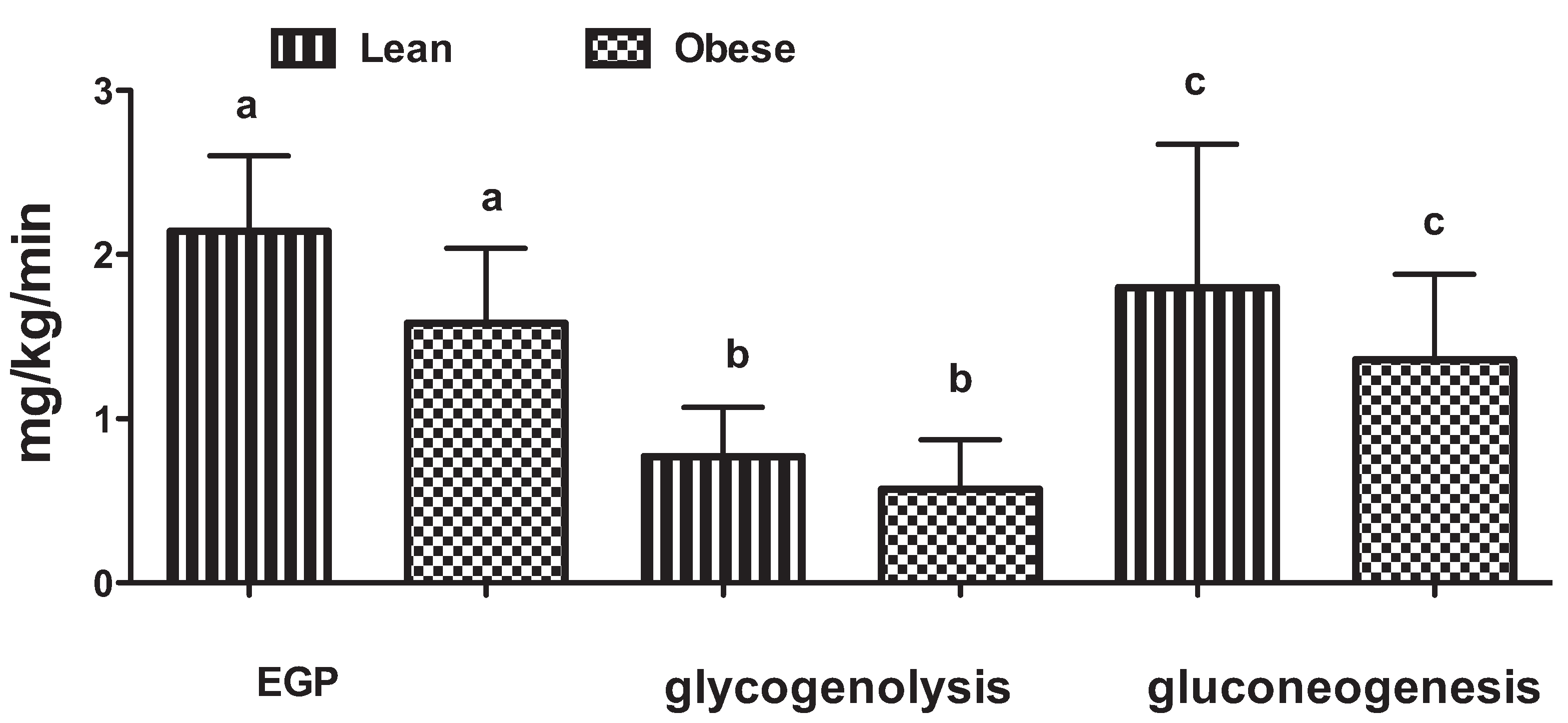
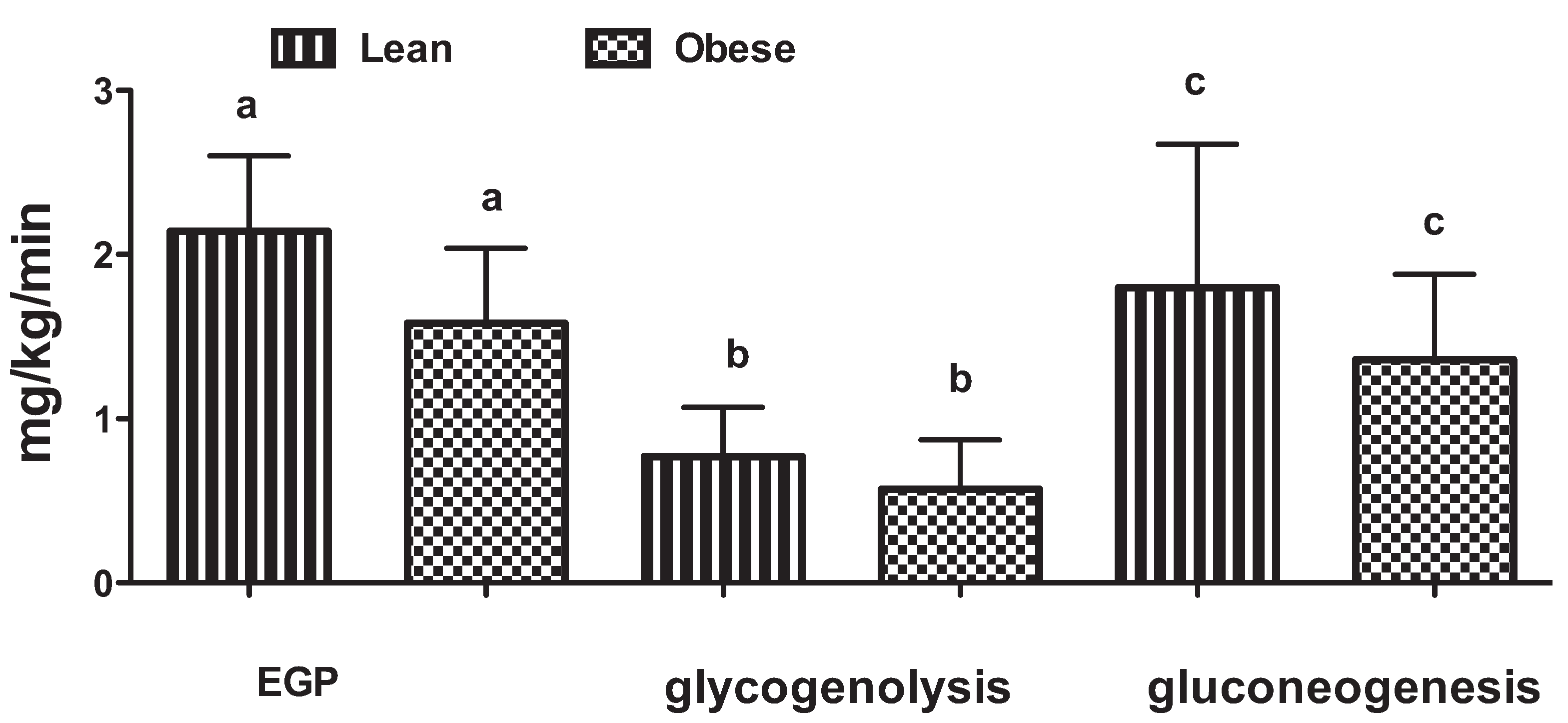
3. The Role of the Liver in Glucose Control

4. The Role of Inflammation in Obesity and Diabetes
5. Conclusions
Conflicts of Interest
References
- Howard, B. Banfield sees bump in fat pets. DVM Newsmag. Vet. Med. 2012, 43, 1. [Google Scholar]
- Kealy, R.D.; Lawler, D.F.; Ballam, J.M.; Mantz, S.L.; Biery, D.N.; Greeley, E.H.; Lust, G.; Segre, M.; Smith, G.K.; Stowe, H.D. Effects of diet restriction on life span and age-related changes in dogs. J. Am. Vet. Med. Assoc. 2002, 220, 1315–1320. [Google Scholar]
- Fall, T.; Hamlin, H.H.; Hedhammar, A.; Kampe, O.; Egenvall, A. Diabetes mellitus in a population of 180,000 insured dogs: Incidence, survival, and breed distribution. J. Vet. Intern. Med. 2007, 21, 1209–1216. [Google Scholar] [CrossRef]
- Kaneko, J.J.; Mattheeuws, D.; Rottiers, R.P.; Vermeulen, A. Glucose tolerance and insulin response in diabetes mellitus of dogs. J. Small Anim. Pract. 1978, 19, 85–94. [Google Scholar] [CrossRef]
- Verkest, K.R.; Fleeman, L.M.; Rand, J.S.; Morton, J.M. Evaluation of beta-cell sensitivity to glucose and first-phase insulin secretion in obese dogs. Am. J. Vet. Res. 2011, 72, 357–366. [Google Scholar] [CrossRef]
- Karam, J.H.; Grodsky, G.M.; Forsham, P.H. Excessive insulin response to glucose in obese subjects as measured by immunochemical assay. Diabetes 1963, 12, 197–204. [Google Scholar]
- Polonsky, K.S.; Given, B.D.; Hirsch, L.; Shapiro, E.T.; Tillil, H.; Beebe, C.; Galloway, J.A.; Frank, B.H.; Karrison, T.; van Cauter, E. Quantitative study of insulin secretion and clearance in normal and obese subjects. J. Clin. Invest. 1988, 81, 435–441. [Google Scholar] [CrossRef]
- Seino, S.; Shibasaki, T.; Minami, K. Dynamics of insulin secretion and the clinical implications for obesity and diabetes. J. Clin. Investig. 2011, 121, 2118–2125. [Google Scholar] [CrossRef]
- Stancakova, A.; Javorsky, M.; Kuulasmaa, T.; Haffner, S.M.; Kuusisto, J.; Laakso, M. Changes in insulin sensitivity and insulin release in relation to glycemia and glucose tolerance in 6414 Finnish men. Diabetes 2009, 58, 1212–1221. [Google Scholar]
- Weir, G.C.; Bonner-Weir, S. Five stages of evolving beta-cell dysfunction during progression to diabetes. Diabetes 2004, 53, S16–S21. [Google Scholar] [CrossRef]
- Maclean, N.; Ogilvie, R.F. Quantitative estimation of the pancreatic islet tissue in diabetic subjects. Diabetes 1955, 4, 367–376. [Google Scholar]
- Butler, A.E.; Janson, J.; Bonner-Weir, S.; Ritzel, R.; Rizza, R.A.; Butler, P.C. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 2003, 52, 102–110. [Google Scholar] [CrossRef]
- Chan, J.M.; Rimm, E.B.; Colditz, G.A.; Stampfer, M.J.; Willett, W.C. Obesity, fat distribution, and weight gain as risk factors for clinical diabetes in men. Diabetes Care 1994, 17, 961–969. [Google Scholar]
- Ionut, V.; Liu, H.; Mooradian, V.; Castro, A.V.; Kabir, M.; Stefanovski, D.; Zheng, D.; Kirkman, E.L.; Bergman, R.N. Novel canine models of obese prediabetes and mild type 2 diabetes. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E38–E48. [Google Scholar] [CrossRef]
- Ader, M.; Stefanovski, D.; Kim, S.P.; Richey, J.M.; Ionut, V.; Catalano, K.J.; Hucking, K.; Ellmerer, M.; van Citters, G.; Hsu, I.R.; et al. Hepatic insulin clearance is the primary determinant of insulin sensitivity in the normal dog. Obesity (Silver Spring) 2014, 22, 1238–1245. [Google Scholar] [CrossRef]
- Hoenig, M.; Thomaseth, K.; Brandao, J.; Waldron, M.; Ferguson, D.C. Assessment and mathematical modeling of glucose turnover and insulin sensitivity in lean and obese cats. Domest. Anim. Endocrinol. 2006, 31, 373–389. [Google Scholar] [CrossRef]
- Hoenig, M.; Thomaseth, K.; Waldron, M.; Ferguson, D.C. Insulin sensitivity, fat distribution, and adipocytokine response to different diets in lean and obese cats before and after weight loss. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 292, R227–R234. [Google Scholar]
- Hoenig, M.; Alexander, S.; Holson, J.; Ferguson, D.C. Influence of glucose dosage on interpretation of intravenous glucose tolerance tests in lean and obese cats. J. Vet. Intern. Med. 2002, 16, 529–532. [Google Scholar] [CrossRef]
- Hoenig, M.; Pach, N.; Thomaseth, K.; Devries, F.; Ferguson, D.C. Evaluation of long-term glucose homeostasis in lean and obese cats by use of continuous glucose monitoring. Am. J. Vet. Res. 2012, 73, 1100–1106. [Google Scholar] [CrossRef]
- Hoenig, M.; Traas, A.M.; Schaeffer, D.J. Evaluation of routine hematology profile results and fructosamine, thyroxine, insulin, and proinsulin concentrations in lean, overweight, obese, and diabetic cats. J. Am. Vet. Med. Assoc. 2013, 243, 1302–1309. [Google Scholar]
- Hoenig, M.; Pach, N.; Thomaseth, K.; Le, A.; Schaeffer, D.; Ferguson, D.C. Cats differ from other species in their cytokine and antioxidant enzyme response when developing obesity. Obesity (Silver Spring) 2013, 21, E407–E414. [Google Scholar]
- Matsumoto, K.; Miyake, S.; Yano, M.; Ueki, Y.; Yamaguchi, Y.; Akazawa, S.; Tominaga, Y. Glucose tolerance, insulin secretion, and insulin sensitivity in nonobese and obese Japanese subjects. Diabetes Care 1997, 20, 1562–1568. [Google Scholar]
- Dankner, R.; Abdul-Ghani, M.A.; Gerber, Y.; Chetrit, A.; Wainstein, J.; Raz, I. Predicting the 20-year diabetes incidence rate. Diabetes Metab. Res. Rev. 2007, 23, 551–558. [Google Scholar]
- Appleton, D.J.; Rand, J.S.; Priest, J.; Sunvold, G.D. Determination of reference values for glucose tolerance, insulin tolerance, and insulin sensitivity tests in clinically normal cats. Am. J. Vet. Res. 2001, 62, 630–636. [Google Scholar] [CrossRef]
- Link, K.R.; Rand, J.S.; Hendrikz, J.K. Evaluation of a simplified intravenous glucose tolerance test and a reflectance glucose meter for use in cats. Vet. Rec. 1997, 140, 253–256. [Google Scholar] [CrossRef]
- Link, K.R.; Rand, J.S. Reference values for glucose tolerance and glucose tolerance status in cats. J. Am. Vet. Med. Assoc. 1998, 213, 492–496. [Google Scholar]
- Hoenig, M.; Hall, G.; Ferguson, D.; Jordan, K.; Henson, M.; Johnson, K.; O’Brien, T. A feline model of experimentally induced islet amyloidosis. Am. J. Pathol. 2000, 157, 2143–2150. [Google Scholar] [CrossRef]
- Muller, C.; Assimacopoulos-Jeannet, F.; Mosimann, F.; Schneiter, P.; Riou, J.P.; Pachiaudi, C.; Felber, J.P.; Jequier, E.; Jeanrenaud, B.; Tappy, L. Endogenous glucose production, gluconeogenesisand liver glycogen concentration in obese non-diabetic patients. Diabetologia 1997, 40, 463–468. [Google Scholar] [CrossRef]
- Gastaldelli, A.; Baldi, S.; Pettiti, M.; Toschi, E.; Camastra, S.; Natali, A.; Landau, B.R.; Ferrannini, E. Influence of obesity and type 2 diabetes on gluconeogenesis and glucose output in humans: A quantitative study. Diabetes 2000, 49, 1367–1373. [Google Scholar] [CrossRef]
- Basu, R.; Chandramouli, V.; Dicke, B.; Landau, B.; Rizza, R. Obesity and type 2 diabetes impair insulin-induced suppression of glycogenolysis as well as gluconeogenesis. Diabetes 2005, 54, 1942–1948. [Google Scholar] [CrossRef]
- Weyer, C.; Bogardus, C.; Mott, D.M.; Pratley, R.E. The natural history of insulin secretory dysfunction and insulin resistance in the pathogenesis of type 2 diabetes mellitus. J. Clin. Investig. 1999, 104, 787–794. [Google Scholar] [CrossRef]
- Kley, S.; Hoenig, M.; Glushka, J.; Jin, E.S.; Burgess, S.C.; Waldron, M.; Jordan, E.T.; Prestegard, J.H.; Ferguson, D.C.; Wu, S.; et al. The impact of obesity, sex, and diet on hepatic glucose production in cats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 296, R936–R943. [Google Scholar]
- Hoenig, M.; Jordan, E.T.; Glushka, J.; Kley, S.; Patil, A.; Waldron, M.; Prestegard, J.H.; Ferguson, D.C.; Wu, S.; Olson, D.E. Effect of macronutrients, age, and obesity on 6- and 24-h postprandial glucose metabolism in cats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 301, R1798–R1807. [Google Scholar]
- Edgerton, D.S.; Cardin, S.; Emshwiller, M.; Neal, D.; Chandramouli, V.; Schumann, W.C.; Landau, B.R.; Rossetti, L.; Cherrington, A.D. Small increases in insulin inhibit hepatic glucose production solely caused by an effect on glycogen metabolism. Diabetes 2001, 50, 1872–1882. [Google Scholar] [CrossRef]
- Rothman, D.L.; Magnusson, I.; Katz, L.D.; Shulman, R.G.; Shulman, G.I. Quantitation of hepatic glycogenolysis and gluconeogenesis in fasting humans with 13C NMR. Science 1991, 254, 573–576. [Google Scholar]
- Chiasson, J.L.; Liljenquist, J.E.; Finger, F.E.; Lacy, W.W. Differential sensitivity of glycogenolysis and gluconeogenesis to insulin infusions in dogs. Diabetes 1976, 25, 283–291. [Google Scholar] [CrossRef]
- Sokal, J.E.; Gerszi, K.E. Human liver glycogen levels. J. Lab. Clin. Med. 1959, 53, 876–881. [Google Scholar]
- Fittschen, C.; Bellamy, J.E. Prednisone-induced morphologic and chemical changes in the liver of dogs. Vet. Pathol. 1984, 21, 399–406. [Google Scholar]
- Korenblat, K.M.; Fabbrini, E.; Mohammed, B.S.; Klein, S. Liver, muscle, and adipose tissue insulin action is directly related to intrahepatic triglyceride content in obese subjects. Gastroenterology 2008, 134, 1369–1375. [Google Scholar]
- Fabbrini, E.; Sullivan, S.; Klein, S. Obesity and nonalcoholic fatty liver disease: Biochemical, metabolic, and clinical implications. Hepatology 2010, 51, 679–689. [Google Scholar] [CrossRef]
- Hijona, E.; Hijona, L.; Arenas, J.I.; Bujanda, L. Inflammatory mediators of hepatic steatosis. Mediat. Inflamm. 2010. [Google Scholar] [CrossRef]
- Kleiner, D.E.; Brunt, E.M.; van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.C.; Torbenson, M.S.; Unalp-Arida, A.; et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar]
- Clark, M.H.; Larsen, R.; Lu, W.; Hoenig, M. Investigation of 1H MRS for quantification of hepatic triglyceride in lean and obese cats. Res. Vet. Sci. 2013, 95, 678–680. [Google Scholar] [CrossRef]
- Hall, J.A.; Barstad, L.A.; Connor, W.E. Lipid composition of hepatic and adipose tissues from normal cats and from cats with idiopathic hepatic lipidosis. J. Vet. Intern. Med. 1997, 11, 238–242. [Google Scholar]
- Ibrahim, W.H.; Bailey, N.; Sunvold, G.D.; Bruckner, G.G. Effects of carnitine and taurine on fatty acid metabolism and lipid accumulation in the liver of cats during weight gain and weight loss. Am. J. Vet. Res. 2003, 64, 1265–1277. [Google Scholar]
- Lehrke, M.; Lazar, M.A. The many faces of PPARgamma. Cell 2005, 123, 993–999. [Google Scholar]
- Walczak, R.; Tontonoz, P. PPARadigms and PPARadoxes: Expanding roles for PPARgamma in the control of lipid metabolism. J. Lipid Res. 2002, 43, 177–186. [Google Scholar]
- Hoenig, M.; Ferguson, D.C. Effect of darglitazone on glucose clearance and lipid metabolism in obese cats. Am. J. Vet. Res. 2003, 64, 1409–1413. [Google Scholar] [CrossRef]
- Ikeda, H.; Taketomi, S.; Sugiyama, Y.; Shimura, Y.; Sohda, T.; Meguro, K.; Fujita, T. Effects of pioglitazone on glucose and lipid metabolism in normal and insulin resistant animals. Arzneimittelforschung 1990, 40, 156–162. [Google Scholar]
- Yki-Jarvinen, H. Thiazolidinediones. N. Engl. J. Med. 2004, 351, 1106–1118. [Google Scholar] [CrossRef]
- Bastard, J.P.; Maachi, M.; Lagathu, C.; Kim, M.J.; Caron, M.; Vidal, H.; Capeau, J.; Feve, B. Recent advances in the relationship between obesity, inflammation, and insulin resistance. Eur. Cytokine Netw. 2006, 17, 4–12. [Google Scholar]
- Dandona, P.; Aljada, A.; Bandyopadhyay, A. Inflammation: The link between insulin resistance, obesity and diabetes. Trends Immunol. 2004, 25, 4–7. [Google Scholar] [CrossRef]
- Lumeng, C.N.; Saltiel, A.R. Inflammatory links between obesity and metabolic disease. J. Clin. Investig. 2011, 121, 2111–2117. [Google Scholar] [CrossRef]
- Shoelson, S.E.; Herrero, L.; Naaz, A. Obesity, inflammation, and insulin resistance. Gastroenterology 2007, 132, 2169–2180. [Google Scholar] [CrossRef]
- Houstis, N.; Rosen, E.D.; Lander, E.S. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature 2006, 440, 944–948. [Google Scholar] [CrossRef]
- Jeusette, I.; Torre, C.; Salas, A.; Iraculis, N.; Compagnucci, M.; Romano, V.; Kirschvink, N. Effects of consuming diets containing various fats or citrus flavanones on plasma lipid and urinary F2-isoprostane concentrations in overweight cats. Am. J. Vet. Res. 2010, 71, 1039–1044. [Google Scholar]
- Tvarijonaviciute, A.; Ceron, J.J.; Holden, S.L.; Morris, P.J.; Biourge, V.; German, A.J. Effects of weight loss in obese cats on biochemical analytes related to inflammation and glucose homeostasis. Domest. Anim. Endocrinol. 2012, 42, 129–141. [Google Scholar] [CrossRef]
- Jaso-Friedmann, L.; Leary, J.H., 3rd; Praveen, K.; Waldron, M.; Hoenig, M. The effects of obesity and fatty acids on the feline immune system. Vet. Immunol. Immunopathol. 2008, 122, 146–152. [Google Scholar] [CrossRef]
- Tvarijonaviciute, A.; Tecles, F.; Martinez-Subiela, S.; Ceron, J.J. Effect of weight loss on inflammatory biomarkers in obese dogs. Vet. J. 2012, 193, 570–572. [Google Scholar] [CrossRef]
- German, A.J.; Hervera, M.; Hunter, L.; Holden, S.L.; Morris, P.J.; Biourge, V.; Trayhurn, P. Improvement in insulin resistance and reduction in plasma inflammatory adipokines after weight loss in obese dogs. Domest. Anim. Endocrinol. 2009, 37, 214–226. [Google Scholar] [CrossRef]
- Clark, M.; Thomaseth, K.; Dirikolu, L.; Ferguson, D.C.; Hoenig, M. Effects of pioglitazone on insulin sensitivity and serum lipids in obese cats. J. Vet. Intern. Med. 2014, 28, 166–174. [Google Scholar] [CrossRef]
- Gayet, C.; Leray, V.; Saito, M.; Siliart, B.; Nguyen, P. The effects of obesity-associated insulin resistance on mRNA expression of peroxisome proliferator-activated receptor-gamma target genes, in dogs. Br. J. Nutr. 2007, 98, 497–503. [Google Scholar] [CrossRef]
- Kim, K.Y.; Kim, J.K.; Han, S.H.; Lim, J.S.; Kim, K.I.; Cho, D.H.; Lee, M.S.; Lee, J.H.; Yoon, D.Y.; Yoon, S.R.; et al. Adiponectin is a negative regulator of NK cell cytotoxicity. J. Immunol. 2006, 176, 5958–5964. [Google Scholar] [CrossRef]
- Holland, W.L.; Miller, R.A.; Wang, Z.V.; Sun, K.; Barth, B.M.; Bui, H.H.; Davis, K.E.; Bikman, B.T.; Halberg, N.; Rutkowski, J.M.; et al. Receptor-mediated activation of ceramidase activity initiates the pleiotropic actions of adiponectin. Nat. Med. 2011, 17, 55–63. [Google Scholar] [CrossRef]
- Wijesekara, N.; Krishnamurthy, M.; Bhattacharjee, A.; Suhail, A.; Sweeney, G.; Wheeler, M.B. Adiponectin-induced ERK and Akt phosphorylation protects against pancreatic beta cell apoptosis and increases insulin gene expression and secretion. J. Biol. Chem. 2010, 285, 33623–33631. [Google Scholar]
- Weyer, C.; Funahashi, T.; Tanaka, S.; Hotta, K.; Matsuzawa, Y.; Pratley, R.E.; Tataranni, P.A. Hypoadiponectinemia in obesity and type 2 diabetes: Close association with insulin resistance and hyperinsulinemia. J. Clin. Endocrinol. Metab. 2001, 86, 1930–1935. [Google Scholar]
- Ishioka, K.; Omachi, A.; Sagawa, M.; Shibata, H.; Honjoh, T.; Kimura, K.; Saito, M. Canine adiponectin: cDNA structure, mRNA expression in adipose tissues and reduced plasma levels in obesity. Res. Vet. Sci. 2006, 80, 127–132. [Google Scholar] [CrossRef]
- Verkest, K.R.; Rand, J.S.; Fleeman, L.M.; Morton, J.M.; Richards, A.A.; Rose, F.J.; Whitehead, J.P. Distinct adiponectin profiles might contribute to differences in susceptibility to type 2 diabetes in dogs and humans. Domest. Anim. Endocrinol. 2011, 41, 67–73. [Google Scholar] [CrossRef]
- Tvarijonaviciute, A.; Ceron, J.J.; Holden, S.L.; Cuthbertson, D.J.; Biourge, V.; Morris, P.J.; German, A.J. Obesity-related metabolic dysfunction in dogs: A comparison with human metabolic syndrome. BMC Vet. Res. 2012, 8. [Google Scholar] [CrossRef]
- Considine, R.V.; Sinha, M.K.; Heiman, M.L.; Kriauciunas, A.; Stephens, T.W.; Nyce, M.R.; Ohannesian, J.P.; Marco, C.C.; McKee, L.J.; Bauer, T.L.; et al. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N. Engl. J. Med. 1996, 334, 292–295. [Google Scholar] [CrossRef]
- Backus, R.C.; Havel, P.J.; Gingerich, R.L.; Rogers, Q.R. Relationship between serum leptin immunoreactivity and body fat mass as estimated by use of a novel gas-phase Fourier transform infrared spectroscopy deuterium dilution method in cats. Am. J. Vet. Res. 2000, 61, 796–801. [Google Scholar] [CrossRef]
- Jeusette, I.C.; Lhoest, E.T.; Istasse, L.P.; Diez, M.O. Influence of obesity on plasma lipid and lipoprotein concentrations in dogs. Am. J. Vet. Res. 2005, 66, 81–86. [Google Scholar] [CrossRef]
- Tian, Z.; Sun, R.; Wei, H.; Gao, B. Impaired natural killer (NK) cell activity in leptin receptor deficient mice: Leptin as a critical regulator in NK cell development and activation. Biochem. Biophys. Res. Commun. 2002, 298, 297–302. [Google Scholar] [CrossRef]
- Sanchez-Margalet, V.; Martin-Romero, C.; Santos-Alvarez, J.; Goberna, R.; Najib, S.; Gonzalez-Yanes, C. Role of leptin as an immunomodulator of blood mononuclear cells: Mechanisms of action. Clin. Exp. Immunol. 2003, 133, 11–19. [Google Scholar]
- Soderberg, S.; Ahren, B.; Jansson, J.H.; Johnson, O.; Hallmans, G.; Asplund, K.; Olsson, T. Leptin is associated with increased risk of myocardial infarction. J. Intern. Med. 1999, 246, 409–418. [Google Scholar] [CrossRef]
- Wolk, R.; Berger, P.; Lennon, R.J.; Brilakis, E.S.; Johnson, B.D.; Somers, V.K. Plasma leptin and prognosis in patients with established coronary atherosclerosis. J. Am. Coll. Cardiol. 2004, 44, 1819–1824. [Google Scholar] [CrossRef]
- Zhang, F.; Basinski, M.B.; Beals, J.M.; Briggs, S.L.; Churgay, L.M.; Clawson, D.K.; DiMarchi, R.D.; Furman, T.C.; Hale, J.E.; Hsiung, H.M.; et al. Crystal structure of the obese protein leptin-E100. Nature 1997, 387, 206–209. [Google Scholar] [CrossRef]
- Bodey, A.R.; Michell, A.R. Epidemiological study of blood pressure in domestic dogs. J. Small Anim. Pract. 1996, 37, 116–125. [Google Scholar]
- Mehlman, E.; Bright, J.M.; Jeckel, K.; Porsche, C.; Veeramachaneni, D.N.; Frye, M. Echocardiographic evidence of left ventricular hypertrophy in obese dogs. J. Vet. Intern. Med. 2013, 27, 62–68. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hoenig, M. Comparative Aspects of Human, Canine, and Feline Obesity and Factors Predicting Progression to Diabetes. Vet. Sci. 2014, 1, 121-135. https://doi.org/10.3390/vetsci1020121
Hoenig M. Comparative Aspects of Human, Canine, and Feline Obesity and Factors Predicting Progression to Diabetes. Veterinary Sciences. 2014; 1(2):121-135. https://doi.org/10.3390/vetsci1020121
Chicago/Turabian StyleHoenig, Margarethe. 2014. "Comparative Aspects of Human, Canine, and Feline Obesity and Factors Predicting Progression to Diabetes" Veterinary Sciences 1, no. 2: 121-135. https://doi.org/10.3390/vetsci1020121
APA StyleHoenig, M. (2014). Comparative Aspects of Human, Canine, and Feline Obesity and Factors Predicting Progression to Diabetes. Veterinary Sciences, 1(2), 121-135. https://doi.org/10.3390/vetsci1020121