
Reference-Guided Draft Genome Assembly, Annotation and SSR Mining Data of the Peruvian Creole Cattle (Bos taurus)

 ,
,  , , , ,
, , , , 
Abstract
1. Summary
2. Data Description
2.1. Genomic Survey
2.2. Assembly De Novo, Reference-Assisted Scaffolding, and Validation
2.3. Genomic Annotation
2.4. SSR Data Mining
2.5. Concluding Comments
3. Methods
3.1. Sample Collection and DNA Extraction
3.2. Genome Sequencing and Genomic Survey
3.3. De Novo Assembly and Validation
3.4. Genome Annotation
3.5. Identification of Simple Sequence Repeats
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Scheu, A.; Powell, A.; Bollongino, R.; Vigne, J.D.; Tresset, A.; Çakirlar, C.; Benecke, N.; Burger, J. The Genetic Prehistory of Domesticated Cattle from Their Origin to the Spread across Europe. BMC Genet. 2015, 16, 54. [Google Scholar] [CrossRef]
- Upadhyay, M.R.; Chen, W.; Lenstra, J.A.; Goderie, C.R.J.; Machugh, D.E.; Park, S.D.E.; Magee, D.A.; Matassino, D.; Ciani, F.; Megens, H.J.; et al. Genetic Origin, Admixture and Population History of Aurochs (Bos primigenius) and Primitive European Cattle. Heredity 2016, 118, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Garrick, D.J.; Ruvinsky, A. The Genetics of Cattle; CABI: London, UK, 2014; ISBN 9781119130536. [Google Scholar]
- Hiendleder, S.; Lewalski, H.; Janke, A. Complete Mitochondrial Genomes of Bos taurus and Bos indicus Provide New Insights into Intra-Species Variation, Taxonomy and Domestication. Cytogenet. Genome Res. 2008, 120, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Bovine Genome Sequencing and Analysis Consortium; Elsik, C.G.; Tellam, R.L.; Worley, K.C.; Gibbs, R.A.; Muzny, D.M.; Weinstock, G.M.; Adelson, D.L.; Eichler, E.E.; Einitski, L.; et al. The Genome Sequence of Taurine Cattle: A Window to Ruminant Biology and Evolution. Science 2009, 324, 522–528. [Google Scholar] [CrossRef] [PubMed]
- Delgado, J.V.; Martínez, A.M.; Acosta, A.; Álvarez, L.A.; Armstrong, E.; Camacho, E.; Cañón, J.; Cortés, O.; Dunner, S.; Landi, V.; et al. Genetic Characterization of Latin-American Creole Cattle Using Microsatellite Markers. Anim. Genet. 2012, 43, 2–10. [Google Scholar] [CrossRef]
- Giovambattista, G.; Takeshima, S.-N.; Ripoli, M.V.; Matsumoto, Y.; Angela, L.; Franco, A.; Saito, H.; Onuma, M.; Aida, Y. Characterization of Bovine MHC DRB3 Diversity in Latin American Creole Cattle Breeds. Gene 2013, 519, 150–158. [Google Scholar] [CrossRef]
- Ginja, C.; Gama, L.T.; Cortés, O.; Burriel, I.M.; Vega-Pla, J.L.; Penedo, C.; Sponenberg, P.; Cañón, J.; Sanz, A.; do Egito, A.A.; et al. The Genetic Ancestry of American Creole Cattle Inferred from Uniparental and Autosomal Genetic Markers. Sci. Rep. 2019, 9, 11486. [Google Scholar] [CrossRef]
- Raschia, M.A.; Poli, M. Phylogenetic Relationships of Argentinean Creole with Other Latin American Creole Cattle as Revealed by a Medium Density Single Nucleotide Polymorphism Microarray. Arch. Latinoam. Prod. Anim. 2021, 29, 91–100. [Google Scholar] [CrossRef]
- Liu, S.J.; Lv, J.Z.; Tan, Z.Y.; Ge, X.Y. The Complete Mitochondrial Genome of Uruguayan Native Cattle (Bos taurus). Mitochondrial DNA Part B Resour. 2020, 5, 443–444. [Google Scholar] [CrossRef]
- Aguirre Riofrio, L.; Apolo, G.; Chalco, L.; Martínez, A. Caracterización Genética de La Población Bovina Criolla de La Región Sur Del Ecuador y Su Relación Genética Con Otras Razas Bovinas. Anim. Genet. Resour. Génétiques Anim. Genéticos Anim. 2014, 54, 93–101. [Google Scholar] [CrossRef]
- Aracena, M.; Mujica, F. Caracterización Del Bovino Criollo Patagónico Chileno: Un Estudio de Caso. Agro Sur 2011, 39, 106–115. [Google Scholar] [CrossRef]
- Shieh, Y.-K.; Liu, S.-C.; Lung, L.C. Scaffolding Contigs Using Multiple Reference Genomes. In Computational Biology and Chemistry; Behzadi, P., Bernabò, N., Eds.; IntechOpen: London, UK, 2020. [Google Scholar] [CrossRef]
- Fertin, G.; Labarre, A.; Rusu, I.; Vialette, S.; Tannier, E. Combinatorics of Genome Rearrangements; MIT Press: London, UK, 2009. [Google Scholar]
- Kolmogorov, M.; Raney, B.; Paten, B.; Pham, S. Ragout—A Reference-Assisted Assembly Tool for Bacterial Genomes. Bioinformatics 2014, 30, i302–i309. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Yang, L.; Han, X.; Han, J.; Hu, Y.; Li, F.; Xia, H.; Peng, L.; Boschiero, C.; Rosen, B.D.; et al. Assembly of a Pangenome for Global Cattle Reveals Missing Sequences and Novel Structural Variations, Providing New Insights into Their Diversity and Evolutionary History. Genome Res. 2022, 32, 1585–1601. [Google Scholar] [CrossRef] [PubMed]
- Leonard, A.S.; Crysnanto, D.; Fang, Z.-H.; Heaton, M.P.; Ley, B.L.V.; Herrera, C.; Bollwein, H.; Bickhart, D.M.; Kuhn, K.L.; Smith, T.P.L.; et al. Structural Variant-Based Pangenome Construction Has Low Sensitivity to Variability of Haplotype-Resolved Bovine Assemblies. Nat. Commun. 2022, 13, 3012. [Google Scholar] [CrossRef]
- Rosen, B.D.; Bickhart, D.M.; Schnabel, R.D.; Koren, S.; Elsik, C.G.; Tseng, E.; Rowan, T.N.; Low, W.Y.; Zimin, A.; Couldrey, C.; et al. De Novo Assembly of the Cattle Reference Genome with Single-Molecule Sequencing. Gigascience 2020, 9, giaa021. [Google Scholar] [CrossRef]
- Yalta-Macedo, C.E.; Veli, E.A.; Díaz, G.R.; Vallejo-Trujillo, A. Paternal Ancestry of Peruvian Creole Cattle Inferred from Y-Chromosome Analysis. Livest. Sci. 2021, 244, 104376. [Google Scholar] [CrossRef]
- Arbizu, C.I.; Ferro-Mauricio, R.D.; Chávez-Galarza, J.C.; Vásquez, H.V.; Maicelo, J.L.; Poemape, C.; Gonzales, J.; Quilcate, C.; Corredor, F.-A. The Complete Mitochondrial Genome of a Neglected Breed, the Peruvian Creole Cattle (Bos taurus), and Its Phylogenetic Analysis. Data 2022, 7, 76. [Google Scholar] [CrossRef]
- Instituto Nacional de Estadística e Informática IV Censo Nacional Agropecuario. 2012. Available online: http://censos.inei.gob.pe/Cenagro/redatam/# (accessed on 14 March 2022).
- Mapiye, C.; Chikwanha, O.C.; Chimonyo, M.; Dzama, K. Strategies for Sustainable Use of Indigenous Cattle Genetic Resources in Southern Africa. Diversity 2019, 11, 214. [Google Scholar] [CrossRef]
- Ruiz, R.E.; Saucedo-uriarte, J.A.; Portocarrero-villegas, S.M.; Quispe-ccasa, H.A.; Cayo-colca, I.S. Zoometric Characterization of Creole Cows from the Southern Amazon Region of Peru. Diversity 2021, 13, 510. [Google Scholar] [CrossRef]
- Espinoza, R.; Urviola, G. Biometría y Constantes Clínicas Del Bovino Criollo En El Centro de Investigación y Producción Chuquibambilla de Puno (Perú). Arch. Zootec. 2005, 54, 233–236. [Google Scholar]
- Dipas Vargas, E.S. Zoometría e Índices Corporales Del Vacuno Criollo En El Matadero de Quicapata de La Provincia de Huamanga, A 2720 Msnm Ayacucho—2014; Universidad Nacional San Cristóbal de Huamanga: Ayacucho, Peru, 2015. [Google Scholar]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y.; et al. SOAPdenovo2: An Empirically Improved Memory-Efficient Short-Read de Novo Assembler. Gigascience 2012, 1, 2047-217X-1-18. [Google Scholar] [CrossRef] [PubMed]
- Zimin, A.V.; Marçais, G.; Puiu, D.; Roberts, M.; Salzberg, S.L.; Yorke, J.A. The MaSuRCA Genome Assembler. Bioinformatics 2013, 29, 2669–2677. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC A Quality Control Tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 5 August 2022).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Krueger Trim Galore! Babraham Bioinformatics. Available online: https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ (accessed on 5 August 2022).
- Marçais, G.; Kingsford, C. A Fast, Lock-Free Approach for Efficient Parallel Counting of Occurrences of k-Mers. Bioinformatics 2011, 27, 764–770. [Google Scholar] [CrossRef] [PubMed]
- Vurture, G.W.; Sedlazeck, F.J.; Nattestad, M.; Underwood, C.J.; Fang, H.; Gurtowski, J.; Schatz, M.C. GenomeScope: Fast Reference-Free Genome Profiling from Short Reads. Bioinformatics 2017, 33, 2202–2204. [Google Scholar] [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality Assessment Tool for Genome Assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Zimin, A.V.; Salzberg, S.L. The SAMBA Tool Uses Long Reads to Improve the Contiguity of Genome Assemblies. PLoS Comput. Biol. 2022, 18, e1009860. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing Genome Assembly and Annotation Completeness with Single-Copy Orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef]
- Wu, T.D.; Watanabe, C.K. GMAP: A Genomic Mapping and Alignment Program for MRNA and EST Sequences. Bioinformatics 2005, 21, 1859–1875. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Bao, Z.; Eddy, S. Automated de Novo Identification of Repeat Sequence Families in Sequenced Genomes. Genome Res. 2002, 12, 1269–1276. [Google Scholar] [CrossRef] [PubMed]
- Tarailo-Graovac, M.; Chen, N. Using Repeat Masker to Identify Repetitive Elements in Genomic Sequences. Curr. Protoc. Bioinforma. 2009, 25, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Jurka, J.; Kapitonov, V.V.; Pavlicek, A.; Klonowski, P.; Kohany, O.; Walichiewicz, J. Repbase Update, a Database of Eukaryotic Repetitive Elements. Cytogenet. Genome Res. 2005, 110, 462–467. [Google Scholar] [CrossRef]
- Bedell, J.A.; Korf, I.; Gish, W. MaskerAid: A Performance Enhancement to RepeatMasker. Bioinformatics 2000, 16, 1040–1041. [Google Scholar] [CrossRef]
- Campbell, M.S.; Holt, C.; Moore, B.; Yandell, M. Genome Annotation and Curation Using MAKER and MAKER-P. Curr. Protoc. Bioinforma. 2014, 48, 4.11.1–4.11.39. [Google Scholar] [CrossRef]
- Korf, I. Gene Finding in Novel Genomes. BMC Bioinform. 2004, 5, 59. [Google Scholar] [CrossRef]
- Stanke, M.; Diekhans, M.; Baertsch, R.; Haussler, D. Using Native and Syntenically Mapped CDNA Alignments to Improve de Novo Gene Finding. Bioinformatics 2008, 24, 637–644. [Google Scholar] [CrossRef]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-Web: A Web Server for Microsatellite Prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Statistic | Contigs | Scaffolds |
|---|---|---|
| N50 | 12,843 | 108,727,214 |
| N75 | 7242 | 74,944,637 |
| L50 | 63,921 | 11 |
| L75 | 133,082 | 19 |
| Largest contig | 109,017 | 164,677,788 |
| Total length | 2,679,899,159 | 2,814,362,078 |
| GC (%) | 41.92 | 41.87 |
| # contigs (≥1000 bp) | 307,114 | 10,953 |
| # contigs (≥5000 bp) | 179,627 | 1848 |
| # contigs (≥10,000 bp) | 92,431 | 777 |
| # contigs (≥25,000 bp) | 14,279 | 210 |
| # contigs (≥50,000 bp) | 726 | 75 |
| # N’s per 100 kbp | 0.0 | 5710.08 |
| Terms | Contigs | Scaffold |
|---|---|---|
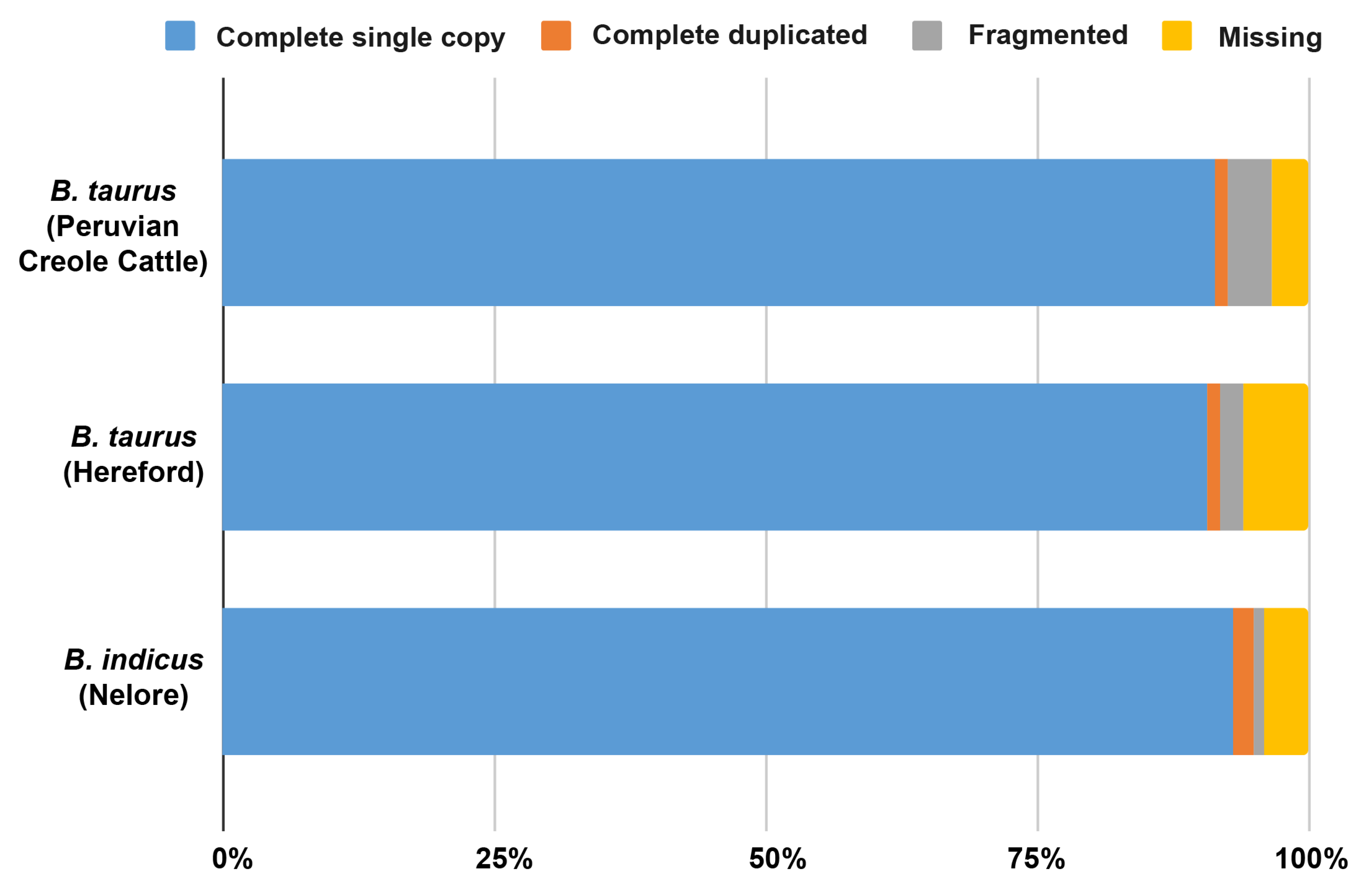
| Complete BUSCOs | 1620 | 3800 |
| Complete and single-copy BUSCOs | 1580 | 3744 |
| Complete and duplicated BUSCOs | 40 | 56 |
| Fragmented BUSCOs | 1573 | 165 |
| Missing BUSCOs | 911 | 139 |
| B. taurus | B. indicus | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Breed | PCC | Hereford | Jersey | Holstein | Brown Swiss | N’Dama | Nelore | Gyr | Ankole |
| Level Assembly | Scaffold | Chromosome | Chromosome | Chromosome | Scaffold | Chromosome | Chromosome | Chromosome | Chromosome |
| Total sequence length | 2,814,362,078 | 2,711,209,831 | 2,641,777,256 | 2,665,549,695 | 2,658,221,619 | 2,766,829,411 | 2,673,965,444 | 2,740,330,345 | 2,921,040,163 |
| Total ungapped length | 2,653,670,481 | 2,711,181,669 | 2,641,709,256 | 2,665,138,195 | 2,635,427,799 | 2,708,415,641 | 2,475,828,999 | 2,695,917,733 | 2,834,561,153 |
| Gaps between scaffolds | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Number of scaffolds | 12,639 | 1957 | 229 | 2306 | 14,725 | 1210 | 32 | 216,409 | 7581 |
| Scaffold N50 | 110,880,623 | 103,308,737 | 104,068,235 | 100,964,413 | 26,027,505 | 104,847,410 | 106,310,653 | 104,295,553 | 84,476,814 |
| Scaffold L50 | 11 | 12 | 11 | 12 | 29 | 12 | 11 | 12 | 13 |
| Number of contigs | 761,086 | 2343 | 365 | 3129 | 34,351 | 3601 | 253,770 | 337,292 | 8473 |
| Contig N50 | 6381 | 25,896,116 | 50,551,513 | 8,737,306 | 268,406 | 11,058,985 | 28,375 | 64,498 | 18,716,610 |
| Contig L50 | 126,374 | 32 | 17 | 90 | 2856 | 71 | 25,227 | 11,998 | 49 |
| Total number of chromosomes and plasmids | 0 | 31 | 32 | 32 | 0 | 32 | 32 | 31 | 30 |
| Number of component sequences (WGS or clone) | 12,639 | 1957 | 229 | 2306 | 14,725 | 1210 | 253,770 | 216,409 | 7581 |
| Repetitive DNA | Number of Elements | Length Occupied | Percentage of Sequence | |
|---|---|---|---|---|
| Retroelements | 3,484,900 | 897,585,367 bp | 32.39% | |
| SINEs | 256.918 | 28,733,533 bp | 104% | |
| LINEs | 2,890,366 | 791,282,631 bp | 28.55% | |
| L2/CR1/REX | 173.451 | 19,529,331 bp | 0.70% | |
| RTE/Bov-B | 1,426,552 | 452,420,074 bp | 16.32% | |
| L1/CIN4 | 1,111,156 | 291,303,229 bp | 10.51% | |
| LTR elements | 33.7616 | 77,569,203 bp | 2.80% | |
| Retroviral | 337.127 | 77,499,189 bp | 2.80% | |
| DNA transposons | 245.87 | 41,992,077 bp | 1.52% | |
| hobo-Activator | 84.758 | 27,282,703 bp | 0.98% | |
| Tc1-IS630-Pogo | 60.623 | 14,480,775 bp | 0.52% | |
| Unclassified | 665.577 | 96,490,946 bp | 3.48% | |
| Total interspersed repeats | 1,036,068,390 bp | 37.38% | ||
| Small RNA | 161.025 | 17,146,359 bp | 0.62% | |
| Satellites | 700 | 416,318 bp | 0.02% | |
| Simple repeats | 499.594 | 20,282,441 bp | 0.73 % | |
| B. taurus | B. indicus | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Breed | PCC | Hereford | Jersey | Holstein | Brown Swiss | N’Dama | Nelore | Gyr | Ankole |
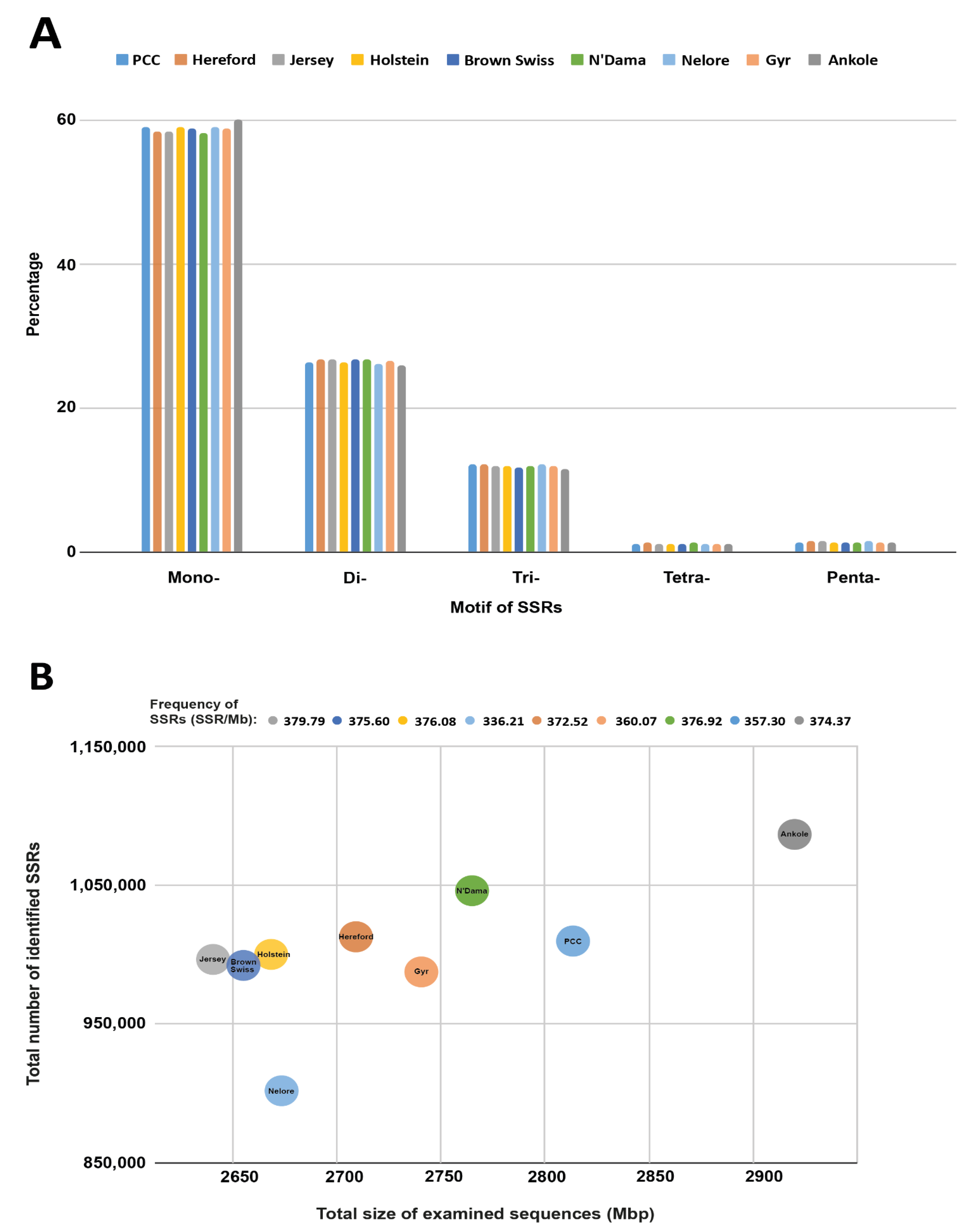
| Total size of examined sequences (Gbp, with gaps) | 2.8144 | 2.7112 | 2.6418 | 2.666 | 2.6582 | 2.7668 | 2.67380 | 2.7403 | 2.9210 |
| Total number of identified SSRs | 1,005,564 | 1,009,980 | 1,003,327 | 1,002,450 | 998,430 | 1,042,868 | 899,003 | 986,718 | 1,093,552 |
| Frequency (SSR/Mb) | 357.30 | 372.52 | 379.79 | 376.08 | 375.60 | 376.92 | 336.21 | 360.07 | 374.37 |
| Number of SSRs present in compound formation | 92,354 | 104,313 | 104,005 | 104,288 | 98,838 | 116,622 | 82,308 | 93,628 | 132,842 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Estrada, R.; Corredor, F.-A.; Figueroa, D.; Salazar, W.; Quilcate, C.; Vásquez, H.V.; Maicelo, J.L.; Gonzales, J.; Arbizu, C.I. Reference-Guided Draft Genome Assembly, Annotation and SSR Mining Data of the Peruvian Creole Cattle (Bos taurus). Data 2022, 7, 155. https://doi.org/10.3390/data7110155
Estrada R, Corredor F-A, Figueroa D, Salazar W, Quilcate C, Vásquez HV, Maicelo JL, Gonzales J, Arbizu CI. Reference-Guided Draft Genome Assembly, Annotation and SSR Mining Data of the Peruvian Creole Cattle (Bos taurus). Data. 2022; 7(11):155. https://doi.org/10.3390/data7110155
Chicago/Turabian StyleEstrada, Richard, Flor-Anita Corredor, Deyanira Figueroa, Wilian Salazar, Carlos Quilcate, Héctor V. Vásquez, Jorge L. Maicelo, Jhony Gonzales, and Carlos I. Arbizu. 2022. "Reference-Guided Draft Genome Assembly, Annotation and SSR Mining Data of the Peruvian Creole Cattle (Bos taurus)" Data 7, no. 11: 155. https://doi.org/10.3390/data7110155
APA StyleEstrada, R., Corredor, F.-A., Figueroa, D., Salazar, W., Quilcate, C., Vásquez, H. V., Maicelo, J. L., Gonzales, J., & Arbizu, C. I. (2022). Reference-Guided Draft Genome Assembly, Annotation and SSR Mining Data of the Peruvian Creole Cattle (Bos taurus). Data, 7(11), 155. https://doi.org/10.3390/data7110155