First Draft Genome Assembly of the Malaysian Stingless Bee, Heterotrigona itama (Apidae, Meliponinae)

 , , ,
, , ,
Abstract
1. Summary
2. Data Description
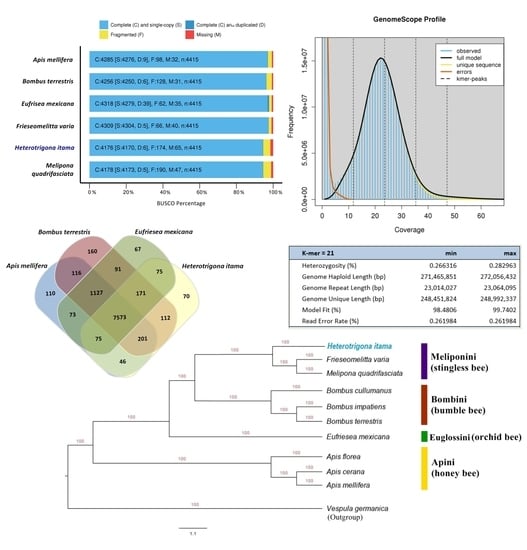
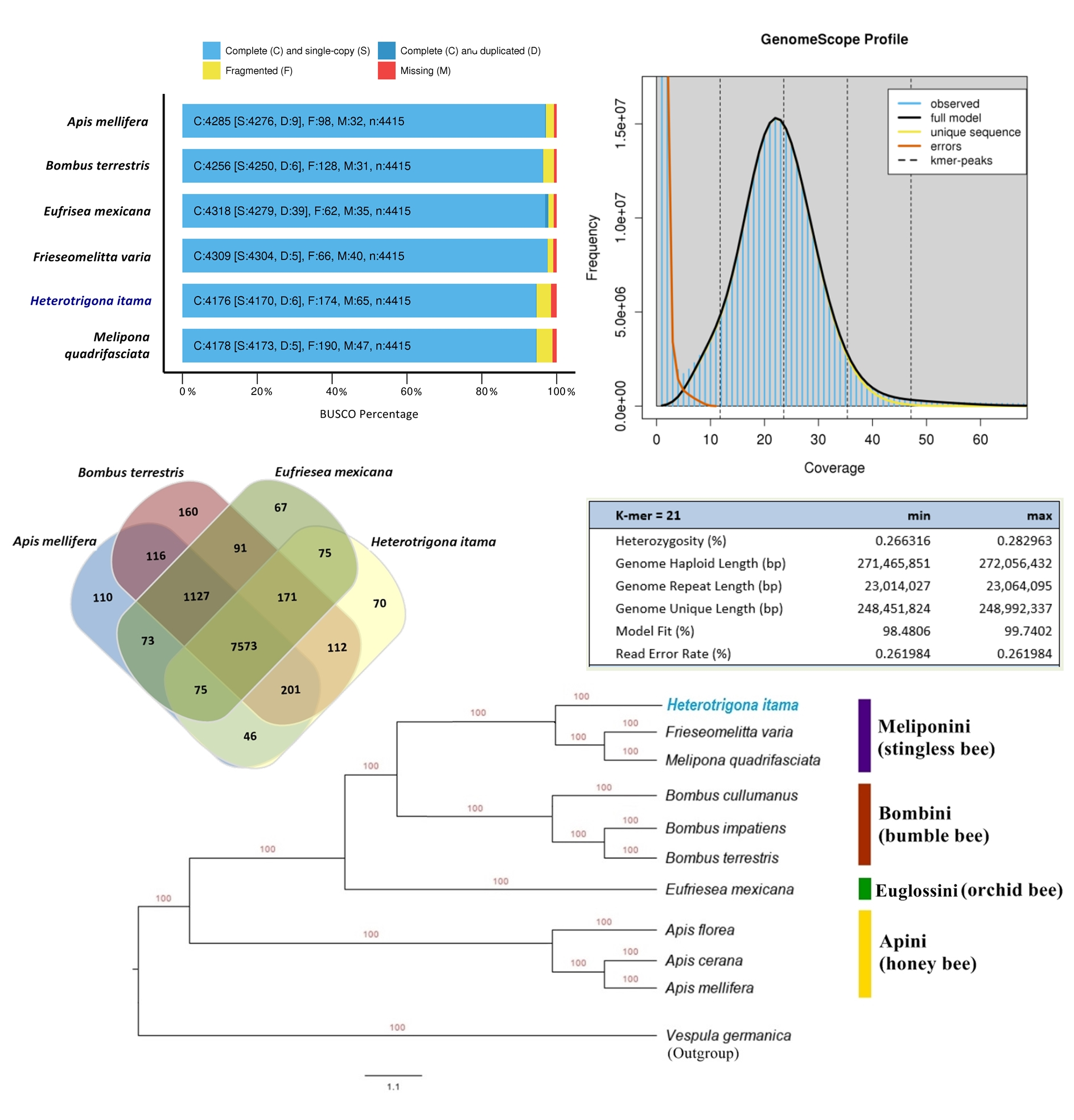
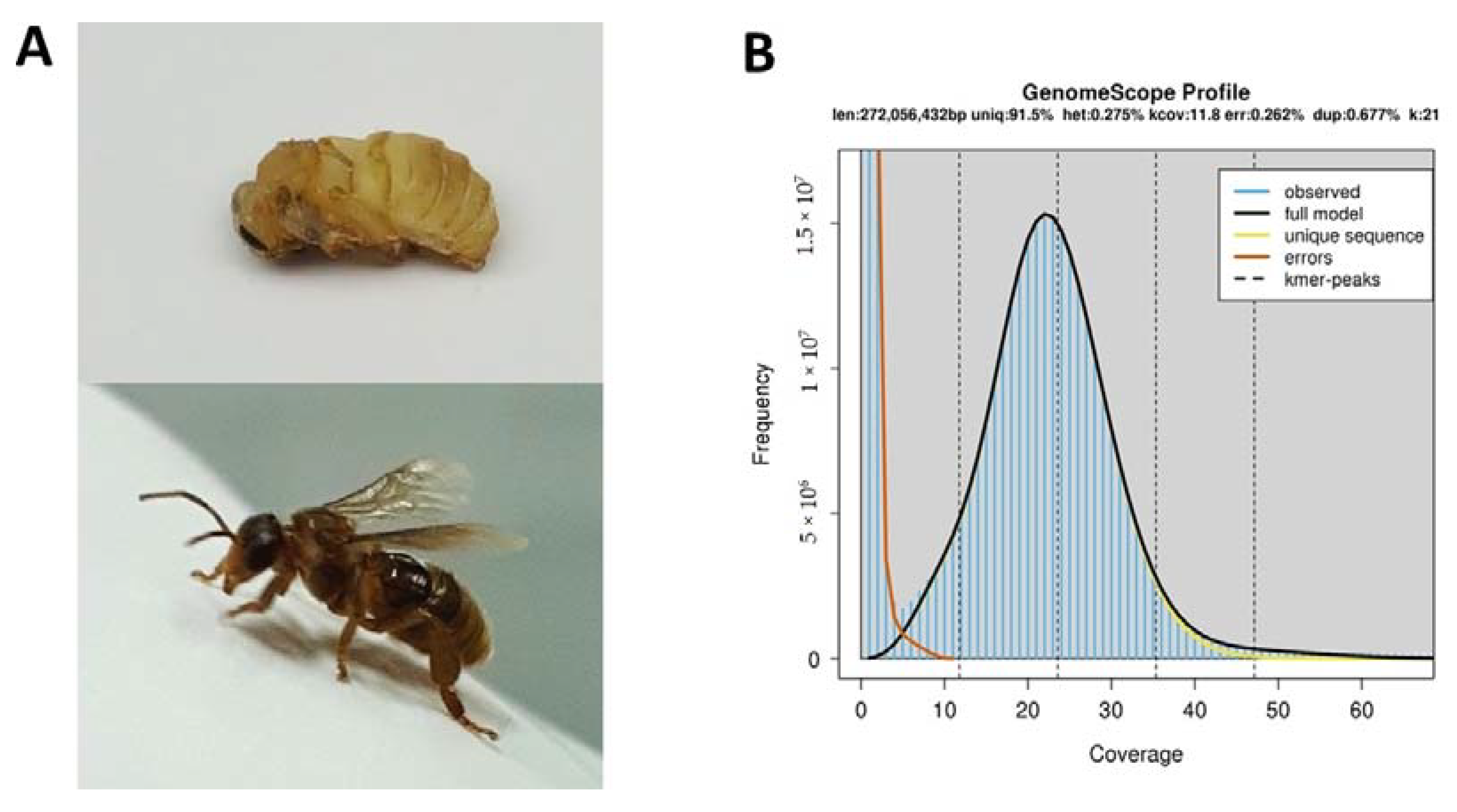
2.1. Genome Size Estimation
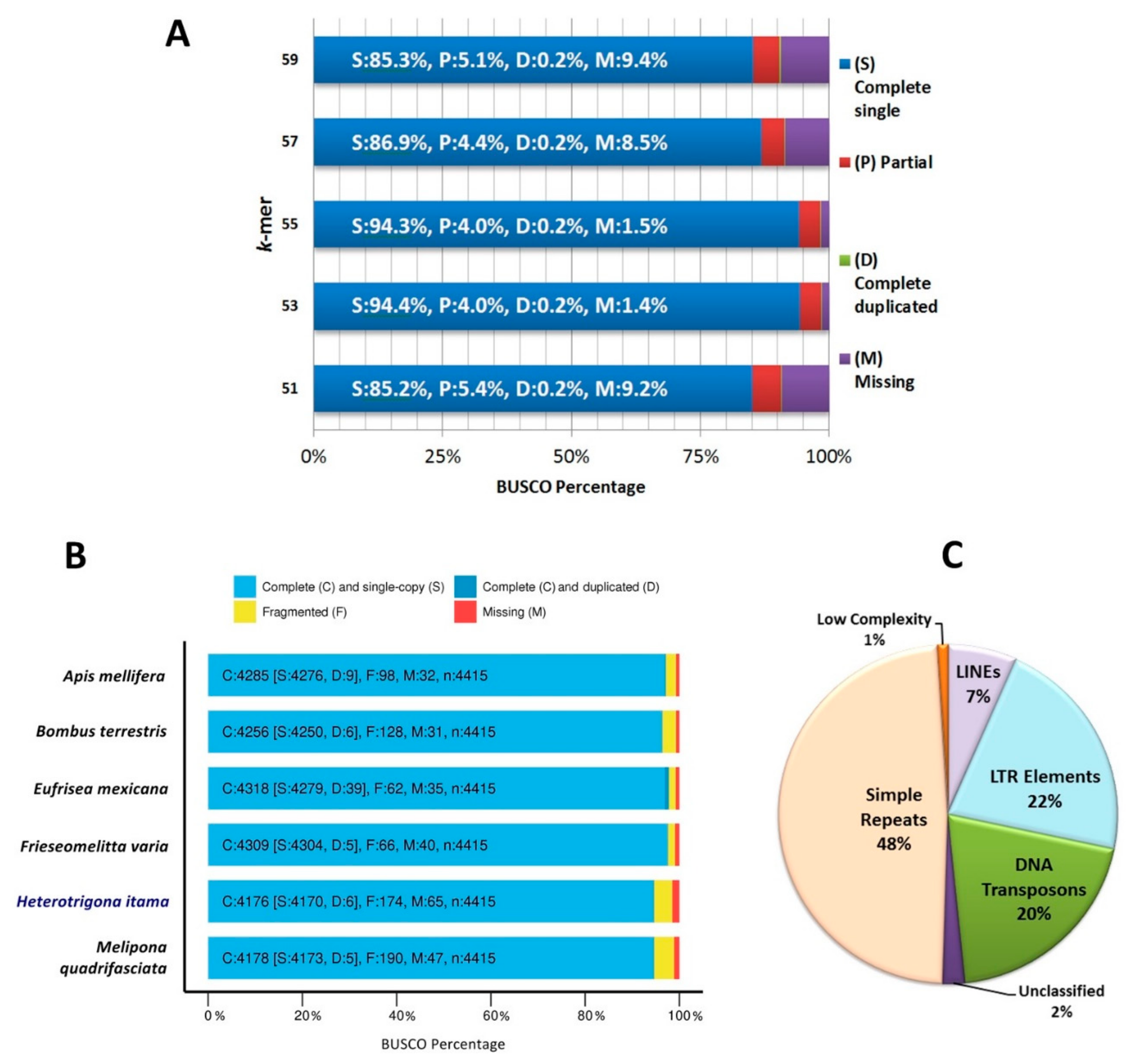
2.2. Genome Assembly and Structural Annotation
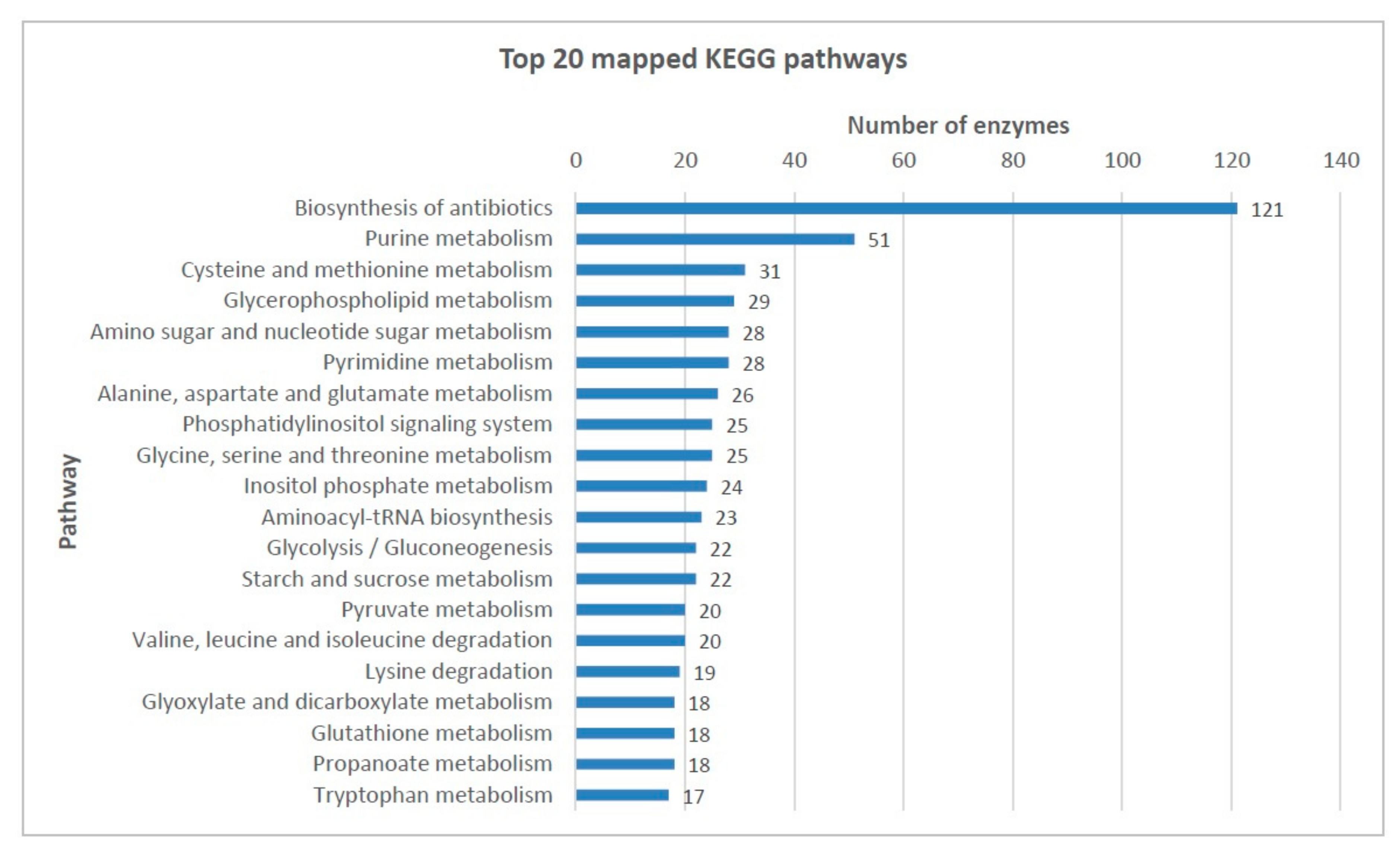
2.3. Functional Annotation
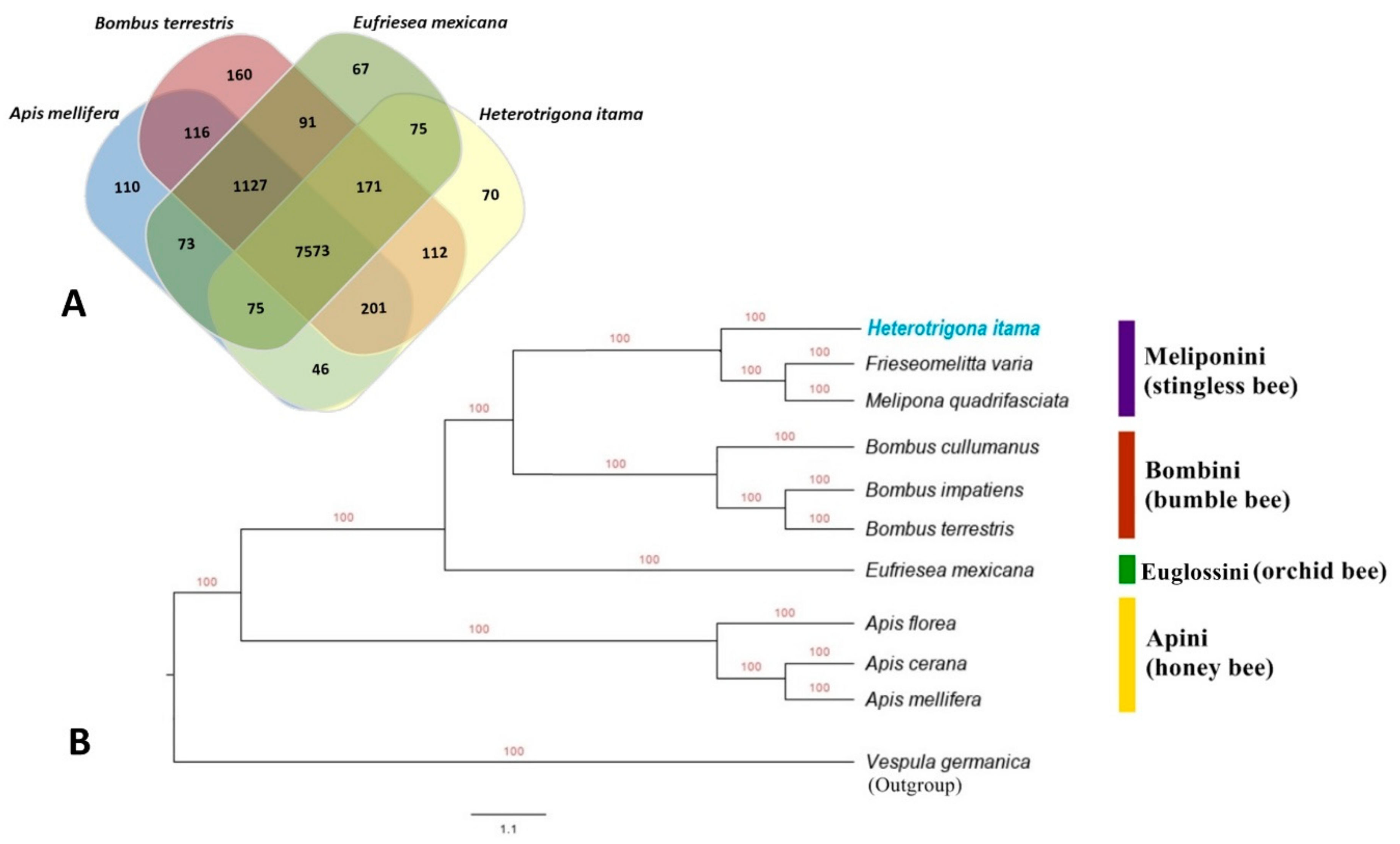
2.4. Orthologous and Phylogenetic Analysis
3. Methods
3.1. Sample Preparation and Sequencing
3.2. Genome Size Estimation and Genome Assembly
3.3. Gene Structural Annotation and Functional Annotation
3.4. Orthologous and Phylogenetic Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rasmussen, C. Catalog of the Indo-Malayan/Australasian stingless bees (Hymenoptera: Apidae: Meliponini). Zootaxa 2008, 1935, 1–80. [Google Scholar] [CrossRef]
- Engel, M.S.; Rasmussen, C. A new subgenus of Heterotrigona from New Guinea (Hymenoptera: Apidae). J. Melittology 2017, 73, 1–16. [Google Scholar] [CrossRef]
- Rasmussen, C.; Thomas, J.C.; Engel, M.S. A new genus of Eastern Hemisphere stingless bees (Hymenoptera, Apidae), with a key to the supraspecific groups of Indomalayan and Australasian Meliponini. Am. Mus. Novit. 2017, 3888, 1–33. [Google Scholar] [CrossRef]
- Brummitt, R.K. World Geographical Scheme for Recording Plant Distributions, 2nd ed.; International Working Group on Taxonomic Databases for Plant Sciences (TDWG); Hunt Institute for Botanical Documentation, Carnegie Mellon University: Pittsburgh, UK, 2001; pp. 43–115. [Google Scholar]
- Ghazalli, M.N.; Tamizi, A.A.; Talip, N. Impak Ekonomi Famili Sapindaceae. In Debunga Rambutan Hutan (Sapindaceae) dan Kepentingan Taksonomi; Jaafar, S., Ed.; UKM Press: Bangi, Malaysia; Universiti Kebangsaan Malaysia: Bangi, Malaysia, 2020; pp. 112–114. [Google Scholar]
- Ahmad-Jailani, N.M.; Mustafa, S.; Mustafa, M.Z.; Mariatulqabtiah, A.R. Nest characteristics of stingless bee Heterotrigona itama (Hymenoptera: Apidae) upon colony transfer and splitting. Pertanika J. Trop. Agric. Sci. 2019, 42, 861–869. [Google Scholar]
- Wong, P.; Hii, S.L.; Koh, C.C.; Moh, T.S.Y.; Anak Gindi, S.R. Chemical analysis on the honey of Heterotrigona itama and Tetrigona binghami from Sarawak, Malaysia. Sains Malays. 2019, 48, 1635–1642. [Google Scholar] [CrossRef]
- Fahimee, J.; Nursyazwani, N.; Fairuz, K.; Rosliza, J.; Mispan, M.R.; Idris, A.B. Variation the oviposition behavior by the stingless bee, Heterotrigona itama (Hymenoptera, Apidae, Meliponini). J. Asia-Pac. Entomol. 2018, 21, 322–328. [Google Scholar] [CrossRef]
- Md-Zaki, N.N.; Abd-Razak, S.B. Pollen profile by stingless bee (Heterotrigona itama) reared in rubber smallholding environment at Tepoh, Terengganu. Malays. J. Microsc. 2018, 14, 38–54. [Google Scholar]
- Chen, X.; Hu, Y.; Zheng, H.; Cao, L.; Niu, D.; Yu, D.; Sun, Y.; Hu, S.; Hu, F. Transcriptome comparison between honey bee queen- and worker-destined larvae. Insect Biochem. Mol. Biol. 2012, 42, 665–673. [Google Scholar] [CrossRef]
- Barchuk, A.R.; Cristino, A.S.; Kucharski, R.; Costa, L.F.; Simoes, Z.L.P.; Maleszka, R. Molecular determinants of caste differentiation in the highly eusocial honeybee Apis mellifera. BMC Dev. Biol. 2007, 7, 70. [Google Scholar] [CrossRef]
- Evans, J.D.; Wheeler, D.E. Differential gene expression between developing queens and workers in the honey bee, Apis mellifera. Proc. Natl. Acad. Sci. USA 1999, 96, 5575–5580. [Google Scholar] [CrossRef]
- Severson, D.W.; Williamson, J.L.; Aiken, J.M. Caste-specific transcription in the female honey bee. Insect Biochem. 1989, 19, 215–220. [Google Scholar] [CrossRef]
- Tamizi, A.A.; Nazaruddin, N.H.; Yeong, W.C.; Mohd-Radzi, M.F.; Jaafar, M.A.; Sekeli, R. The first dataset of de novo transcriptome assembly of Heterotrigona itama (Apidae, Meliponinae) queen larva. Data Brief 2020, 29, 105235. [Google Scholar] [CrossRef] [PubMed]
- de Paula-Freitas, F.C.; Lourenco, A.P.; Nunes, F.M.F.; Paschoal, A.R.; Abreu, F.C.P.; Barbin, F.O.; Bataglia, L.; Cardoso-Junior, C.A.M.; Cervoni, M.S.; Silva, S.R.; et al. The nuclear and mitochondrial genomes of Frieseomelitta varia-a highly eusocial stingless bee (Meliponini) with a permanently sterile worker caste. BMC Genom. 2020, 21, 386. [Google Scholar] [CrossRef] [PubMed]
- Kapheim, K.M.; Pan, H.; Li, C.; Salzberg, S.L.; Puiu, D.; Magoc, T.; Robertson, H.M.; Hudson, M.E.; Venkat, A.; Fischman, B.J.; et al. Genomic signatures of evolutionary transitions from solitary to group living. Science 2015, 348, 1139–1143. [Google Scholar] [CrossRef] [PubMed]
- Nowak, R.M.; Jastrzebski, J.P.; Kusmirek, W.; Salamatin, R.; Rydzanicz, M.; Sobczyk-Kopciol, A.; Sulima-Celinska, A.; Paukszto, L.; Makowczenko, K.G.; Płoski, R.; et al. Hybrid de novo whole-genome assembly and annotation of the model tapeworm Hymenolepis diminuta. Sci. Data 2019, 6, 302. [Google Scholar] [CrossRef] [PubMed]
- Weinstock, G.M.; Robinson, G.E.; Gibbs, R.A.; Worley, K.C.; Evans, J.D.; Maleszka, R.; Robertson, H.M.; Weaver, D.B.; Beye, M.; Bork, P.; et al. Insights into social insects from the genome of the honeybee Apis mellifera. Nature 2006, 443, 931–949. [Google Scholar] [CrossRef]
- Sadd, B.M.; Barribeau, S.M.; Bloch, G.; de Graaf, D.C.; Dearden, P.; Elsik, C.G.; Gadau, J.; Grimmelikhuijzen, C.J.P.; Hasselmann, M.; Lozier, J.D.; et al. The genomes of two key bumblebee species with primitive eusocial organization. Genome Biol. 2015, 16, 76. [Google Scholar] [CrossRef]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef]
- Bairoch, A.; Apweiler, R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 2000, 28, 45–48. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Bomtorin, A.D.; Mackert, A.; Rosa, G.C.C.; Moda, L.M.; Martins, J.R.; Bitondi, M.M.G.; Hartfelder, K.; Simoes, Z.L.P. Juvenile hormone biosynthesis gene expression in the corpora allata of Honey Bee (Apis mellifera) Female Castes. PLoS ONE 2014, 9, e86923. [Google Scholar] [CrossRef] [PubMed]
- Murray, M.G.; Thompson, W.F. Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res. 1980, 8, 4321–4325. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 13 September 2019).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Marcais, G.; Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 2011, 27, 764–770. [Google Scholar] [CrossRef]
- Vurture, G.W.; Sedlazeck, F.J.; Nattestad, M.; Underwood, C.J.; Fang, H.; Gurtowski, J.; Schatz, M.C. GenomeScope: Fast reference-free genome profiling from short reads. Bioinformatics 2017, 33, 2202–2204. [Google Scholar] [CrossRef]
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18, 821–829. [Google Scholar] [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Simao, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef]
- Cantarel, B.L.; Korf, I.; Robb, S.M.; Parra, G.; Ross, E.; Moore, B.; Holt, C.; Sánchez-Alvarado, A.; Yandell, M. MAKER: An easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 2008, 18, 188–196. [Google Scholar] [CrossRef]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef]
- Lagesen, K.; Hallin, P.; Rodland, E.A.; Staefeldt, H.-H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and rapid annotation of Ribosomal RNA genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef] [PubMed]
- Smit, A.F.A.; Hubley, R.; Green, P. RepeatMasker Open-4.0. 2013–2015. Available online: http://www.repeatmasker.org/ (accessed on 8 November 2019).
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualisation and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Stoeckert, C.J.; Roos, D.S. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 2003, 12, 2178–2189. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Huelsenbeck, J.P.; Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef]
- Rambaut, A. FigTree v1.4.4: A Graphical Viewer of Phylogenetic Trees. 2014. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 10 November 2020).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (A) Sequencing Reads | ||
| Total number | Total bases (bp) | |
| Raw data | 89,154,444 | 13,373,166,600 |
| Pre-processed data | 82,979,630 (93.07%) | 12,146,499,923 (90.83%) |
| (B) Assembly Data | ||
| No. contigs (≥1000 bp) | 13,733 | |
| Total length (≥1000 bp) | 262,450,989 | |
| N50 | 43,263 | |
| N75 | 14,434 | |
| L50 | 1649 | |
| L75 | 4534 | |
| GC content | 37.31% | |
| (C) Structural Annotation | ||
| Number of predicted protein-coding genes | 12,496 | |
| Total length of CDS (bp) | 5,951,300 | |
| Number of predicted protein-coding genes (≥99 bp) | 12,482 | |
| Total length of CDS (bp) (≥99 bp) | 5,951,047 | |
| Number of tRNA | 398 | |
| Number of rRNA | 14 | |
| (D) Functional Annotation | ||
| Predicted protein-coding genes (≥99bp) | 12,496 (100%) | |
| Protein with RefSeq blast hits | 10,388 (83.22%) | |
| Protein with Swiss-Prot blast hits | 8,214 (65.81%) | |
| Protein with GO assignments | 7,999 (64.08%) | |
| Total GO annotation | 175,576 | |
| Total EC annotation | 613 | |
| Total KEGG pathway annotation | 142 | |
| (E) Annotated Gene Ontology Related to Caste Differentiation and Insect Hormone (Note: Number in the parentheses indicates the number of the genes of interest) | ||
| Caste differentiation | GO:0048650 | Caste determination, influenced by environmental factors (1) |
| Insect hormone (Top 5) | GO:0009725 | Response to hormone (77) |
| GO:0045433 | Male courtship behavior (30) | |
| GO:0048749 | Compound eye development (25) | |
| GO:0045169 | Fusome (21) | |
| GO:0060562 | Epithelial tube morphogenesis (35) | |
| BUSCO ID | Description |
|---|---|
| EOG091G00GQ | thyroid hormone receptor interactor 12 |
| EOG091G00L0 | integrator complex subunit 1 |
| EOG091G01TH | CSE1 chromosome segregation 1-like (yeast) |
| EOG091G01VH | Aminoacyl-tRNA synthetase, class Ia |
| EOG091G01WY | SCY1-like, kinase-like 2 |
| EOG091G01XI | vacuolar protein sorting 18 homolog (S. cerevisiae) |
| EOG091G02C8 | component of oligomeric golgi complex 1 |
| EOG091G02LY | cleavage stimulation factor, 3’ pre-RNA, subunit 3, 77kDa |
| EOG091G02O6 | Vacuole morphology and inheritance protein 14 |
| EOG091G02P2 | component of oligomeric golgi complex 4 |
| EOG091G02V5 | eukaryotic translation initiation factor 3, subunit B |
| EOG091G03AN | UFM1-specific ligase 1 |
| EOG091G03I1 | SAC1 suppressor of actin mutations 1-like (yeast) |
| EOG091G03JX | mediator complex subunit 17 |
| EOG091G03M4 | nucleolar protein 10 |
| EOG091G03P0 | component of oligomeric golgi complex 6 |
| EOG091G03PD | Protein arginine N-methyltransferase |
| EOG091G03QC | mitochondrial translational initiation factor 2 |
| EOG091G03RW | exocyst complex component 5 |
| EOG091G03XO | tyrosyl-tRNA synthetase |
| EOG091G03Z1 | asunder spermatogenesis regulator |
| EOG091G03ZS | Radical SAM |
| EOG091G04FV | SMG8 nonsense mediated mRNA decay factor |
| EOG091G04GK | PDZ domain containing 8 |
| EOG091G04JK | neurochondrin |
| EOG091G04WH | negative elongation factor complex member B |
| EOG091G017G | Ribosomal protein S5 domain 2-type fold |
| EOG091G017T | N-acetyltransferase 10 (GCN5-related) |
| EOG091G024J | vacuolar protein sorting 53 homolog (S. cerevisiae) |
| EOG091G025T | Integrator complex subunit 2 |
| EOG091G040D | methyltransferase like 13 |
| EOG091G046H | USO1 vesicle transport factor |
| EOG091G049W | lin-9 DREAM MuvB core complex component |
| EOG091G0262 | excision repair cross-complementation group 2 |
| EOG091G0321 | vacuolar protein sorting 51 homolog (S. cerevisiae) |
| EOG091G0349 | exocyst complex component 8 |
| EOG091G0495 | zinc finger, RAN-binding domain containing 1 |
| EOG091G0525 | NOP9 nucleolar protein |
| EOG091G03Z1 | asunder spermatogenesis regulator |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wee, C.-Y.; Tamizi, A.-A.; Nazaruddin, N.-H.; Ng, S.-M.; Khoo, J.-S.; Jajuli, R. First Draft Genome Assembly of the Malaysian Stingless Bee, Heterotrigona itama (Apidae, Meliponinae). Data 2020, 5, 112. https://doi.org/10.3390/data5040112
Wee C-Y, Tamizi A-A, Nazaruddin N-H, Ng S-M, Khoo J-S, Jajuli R. First Draft Genome Assembly of the Malaysian Stingless Bee, Heterotrigona itama (Apidae, Meliponinae). Data. 2020; 5(4):112. https://doi.org/10.3390/data5040112
Chicago/Turabian StyleWee, Chien-Yeong, Amin-Asyraf Tamizi, Nazrul-Hisham Nazaruddin, Siuk-Mun Ng, Jia-Shiun Khoo, and Rosliza Jajuli. 2020. "First Draft Genome Assembly of the Malaysian Stingless Bee, Heterotrigona itama (Apidae, Meliponinae)" Data 5, no. 4: 112. https://doi.org/10.3390/data5040112
APA StyleWee, C.-Y., Tamizi, A.-A., Nazaruddin, N.-H., Ng, S.-M., Khoo, J.-S., & Jajuli, R. (2020). First Draft Genome Assembly of the Malaysian Stingless Bee, Heterotrigona itama (Apidae, Meliponinae). Data, 5(4), 112. https://doi.org/10.3390/data5040112