1. Summary
The primary concern of ecotoxicology is the impact of chemicals on the environment [
1]. To assess this impact at a large-scale, i.e., continental or global context, data of environmental concentrations, effects, use types or application rates have to be incorporated into a consistent structure. Today, science can rely on numerous databases providing these data (
Table 1) for potential environmental impact chemicals (PEICs, e.g., pesticides, industrial chemicals, flame retardants, and solvents). However, the process of linking them takes significant harmonization efforts, even after a common semantic framework has been established, i.e., even after their integration into a coherent base. Among the most fundamental reasons hindering instant data linkage and affecting dimensions of the ecotoxicological data are differing nomenclatures and differing levels of specificity (see
Table 2 for examples). For instance, when linking data spatially, some problems typically arise around issues of specificity, i.e., data present at different spatial scales or resolutions, while linking data within chemical dimension is often impeded by the usage of different nomenclatures. For ecotoxicology, however, the specific interest lies in the chemical dimension, as it applies to all core data (
Table 1).
The usage of different nomenclatures is a well-known issue in chemistry [
7,
8]. Although a chemical compound is defined by its molecular structure, there is no exclusive convention for naming or identifying it. Instead, there are many concurring schemes based on two different approaches: naming chemicals based on their molecular structure (International Chemical Identifier (InChI) and Simplified Molecular Input Line Entry Specification (SMILES) [
7,
9]) or assigning arbitrary, yet unique, identifiers to them (e.g., Chemical Abstract Services registry number (CAS RN) and Distributed Structure-Searchable Toxicity substance identifier (DTXSID) [
10,
11]). In addition, many chemicals, such as pesticides, also have various other names (e.g., trivial, brand, and formulation names) that may also differ among languages (
Table 2). The co-existence of these naming schemes results in a high number of synonyms making nomenclature an important issue, particularly when linking larger datasets. This issue further aggravates in analyses that operate in a trans-national or global context, consider many PEICs and require harmonizing many different data sources.
Differing data specificity is also an issue when linking chemical data. Measured environmental concentrations or effect endpoints may be provided in different databases specifically for any kind of isomers, including data specific to enantiomers or diastereomers, or at the level of unique compound structures, ignoring stereo-compositions and, thus, including isomeric and racemic mixtures. Differing specificity, if not addressed, substantially hinders the integration of data from different sources for some of the ecotoxicologically most important groups of compounds, e.g., insecticides [
12], that may act substantially differently based on their stereo-chemical composition [
13].
For analyses that cover only relatively small sets of PEICs, problems of nomenclature and specificity can be handled manually by expert judgment. Knowledge of PEICs thereby allows constructing data analysis workflows that cover all deviations in chemical names and that reasonably span different levels of specificity. Larger analyses that cover several dozens, or more, of PEICs are often based on relational data representations [
12]. In the case only two different naming schemes are involved, e.g., if only two data sources are linked, differing identifiers of the same chemical can still be resolved by establishing a synonym table. However, relational database joins are costly, and linking more than two different data sources by joining their chemical identifiers (e.g., chemical name) with synonym tables increases the processing complexity significantly, quickly reaching points where complex data analyses become cumbersome [
14]. Moreover, resolving different levels of specificity within and among relational datasets requires sophisticated techniques that entail even more effort to develop and that can hardly be established without significant lack of performance [
15]. At least when combining more than two data sources
—a requirement of many ecotoxicological meta-analyses
—relational data representations are suboptimal due to their constraints in performance and usability [
15,
16,
17].
Labeled property graph databases represent an effective tool to address the aforementioned issues of extendibility, scalability, and flexibility [
14,
18,
19]. Briefly, a labeled property graph consists of nodes (vertices) that are connected through relationships (directed edges) [
19]. Both nodes and relationships can be labeled to distinguish functional roles and can be enriched with properties (see
Appendix A for further details). In contrast to relational database systems, the number and type of relationships between entities is, thus, not strictly defined and allows linking information very flexibly [
18,
20]. This flexibility and the graph’s emphasis on relationships appear well suited for the establishment of a synonym database that can also resolve hierarchical relationships [
14,
15]. Consequently, over the last years, graph databases have evolved as a technical alternative to the established relational database systems, featuring large-scale business (e.g., logistics, social media, and health management) and scientific applications (e.g., web science and sociology) where relational solutions become unfeasible [
21,
22]. However, even after an extensive literature review, we could not find any published approach that uses a graph for managing and analyzing data in ecotoxicology.
The aim of this study was to assess the usability of graph databases for large-scale ecotoxicological meta-analyses that integrate and link a wide range of relevant data (
Table 1) and was conducted by the research group “Meta-Analysis of the Global Impact of Chemicals” (MAGIC). In addition, multiple ecotoxicologically-relevant databases were used to perform a data harmonization, using U.S. EPA Chemical Dashboard (CDDB) [
11] as a synonym provider, demonstrating the method’s applicability in a large-scale ecotoxicological scope and quantifying the method’s advantages compared to relational joins. The data were subsequently reprojected into tabular form, granting easy accessibility to researchers and professionals. The Microsoft
® Excel worksheet published with this data description summarizes the information that is currently contained in the MAGIC graph in a tabular format, while an up-to-date version of the MAGIC graph can be explored using our website (
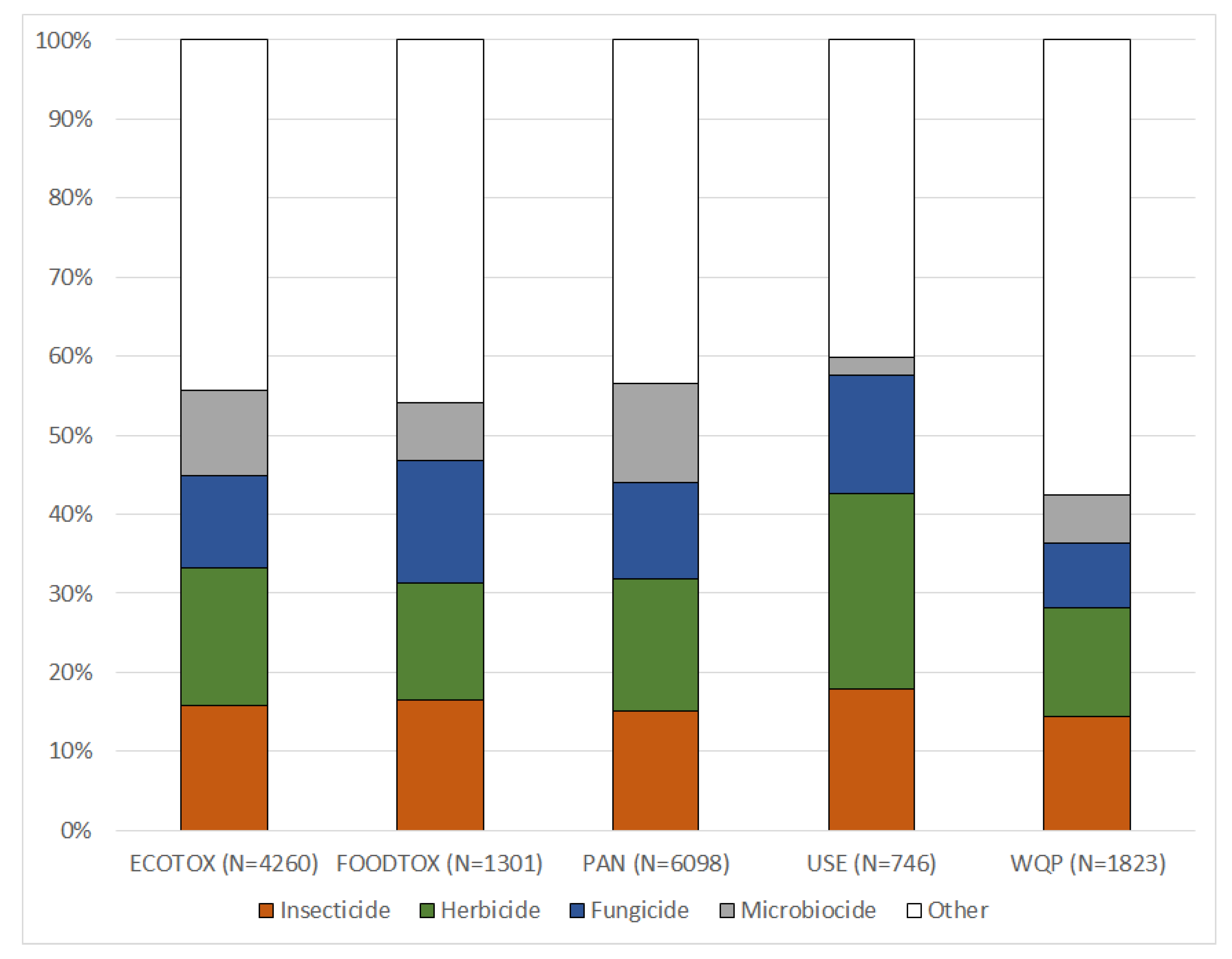
https://magic.eco; see User Notes). Harmonized data for 16,739 PEICs in the MAGIC graph contain information about unique identifiers (CAS RN and DTXSID), valid synonyms, respective chemical classes, use type classification and their inclusion in various databases.
We are positive that the MAGIC graph can serve as a reliable proof that, with graph databases, one already has a suited data integration tool at hand. With it already being actively integrated in large-scale risk analysis at the national level [
23], the MAGIC graph will find further applications and become a central tool in trans-national or global risk analyses in the future (DFG SCHU 2271/6-2). The MAGIC graph is publicly available and will provide a continuously expanding feature set, allowing researchers to take advantage of graph database solutions.
3. Methods
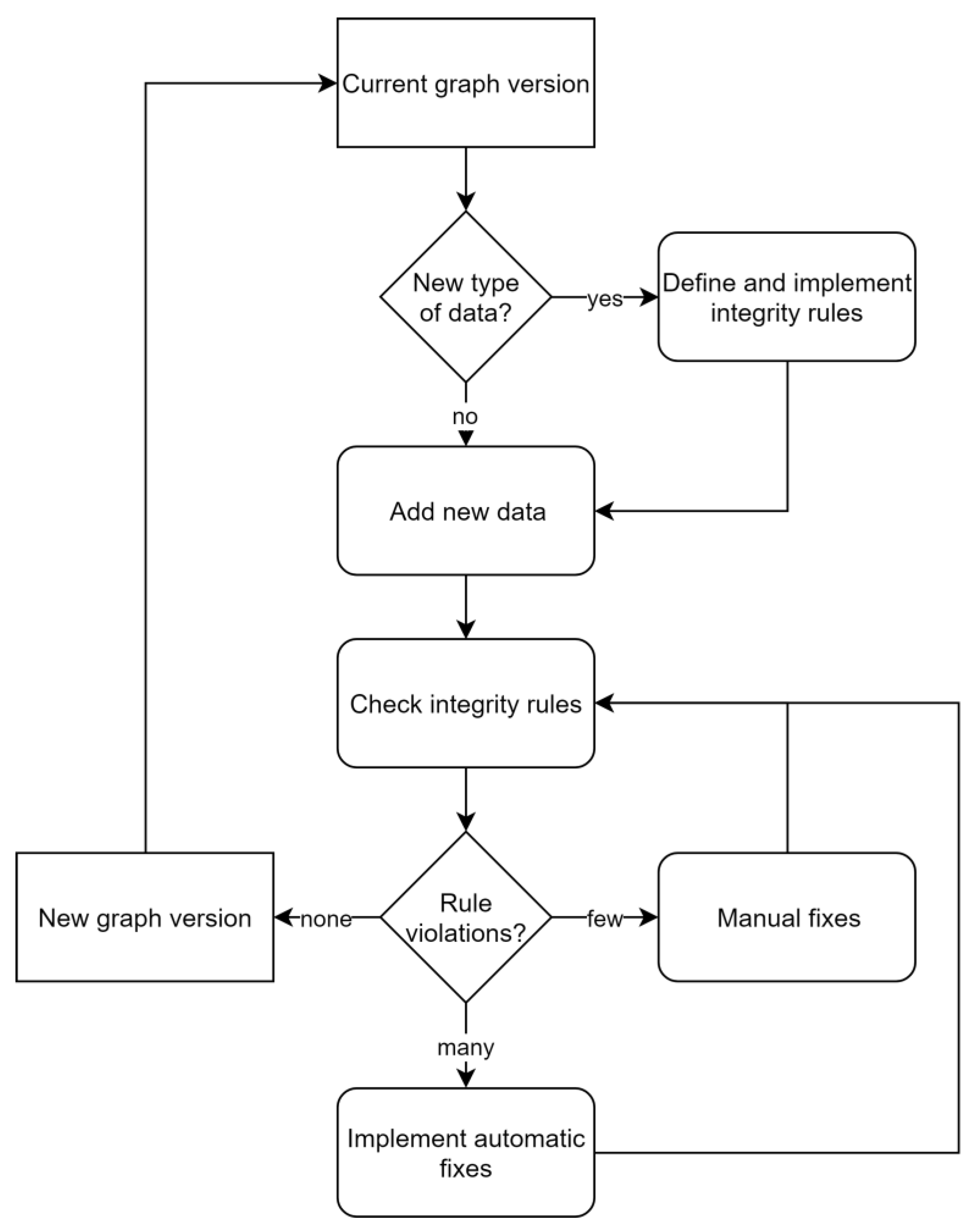
In contrast to relational database management systems, graph databases do not depend on predefined schemata. Briefly, nodes, relationships, labels, types and properties can be added, modified and removed ad hoc and as needed. While this tremendously facilitates the management of changing and growing heterogeneous datasets, it also complicates the usage of these data. Without a static and technically binding schema, the current semantics have to be discovered dynamically: it has to be found what kinds of nodes there are, what properties they have, how nodes are related to other nodes, etc. These concerns were addressed by specifying features of the data model informally outside the database and included semantics of node labels, relationships between nodes and restrictions of properties. To maintain consistency between this specification and the content of the database, as well as safeguard data integrity, 32 rules, checking specific aspects of the data model, were implemented (
Appendix B,
Table A1). Rules were iteratively formulated by expert judgment whenever new conceptual or technical requirements arose, while it was generally aimed at maintaining a small set of rules. Violations of the rules result in notifications that have to be resolved manually or semi-automatically (
Figure 5). This rule-based approach provides a balanced tradeoff between benefits of an agreed schema and flexibility of a graph database. Turning the a priori schema known from relational databases into a posteriori applied consistency rules also resulted in work-flows that resemble those of test-driven developments [
24]. For instance, extension of the domain of the graph database application, e.g., by additionally linking taxonomic data to effect data, is achieved in the two following steps. First, one specifies and implements a set of additional rules, e.g., “species and genus are allowed labels”, “an effect must be linked to a species”, “a species belongs to a genus”, etc., and afterwards modifies the database by adding nodes and relationships until all rules are fulfilled. Fulfillment of all rules then marks a new version of the database application that provides additional information.
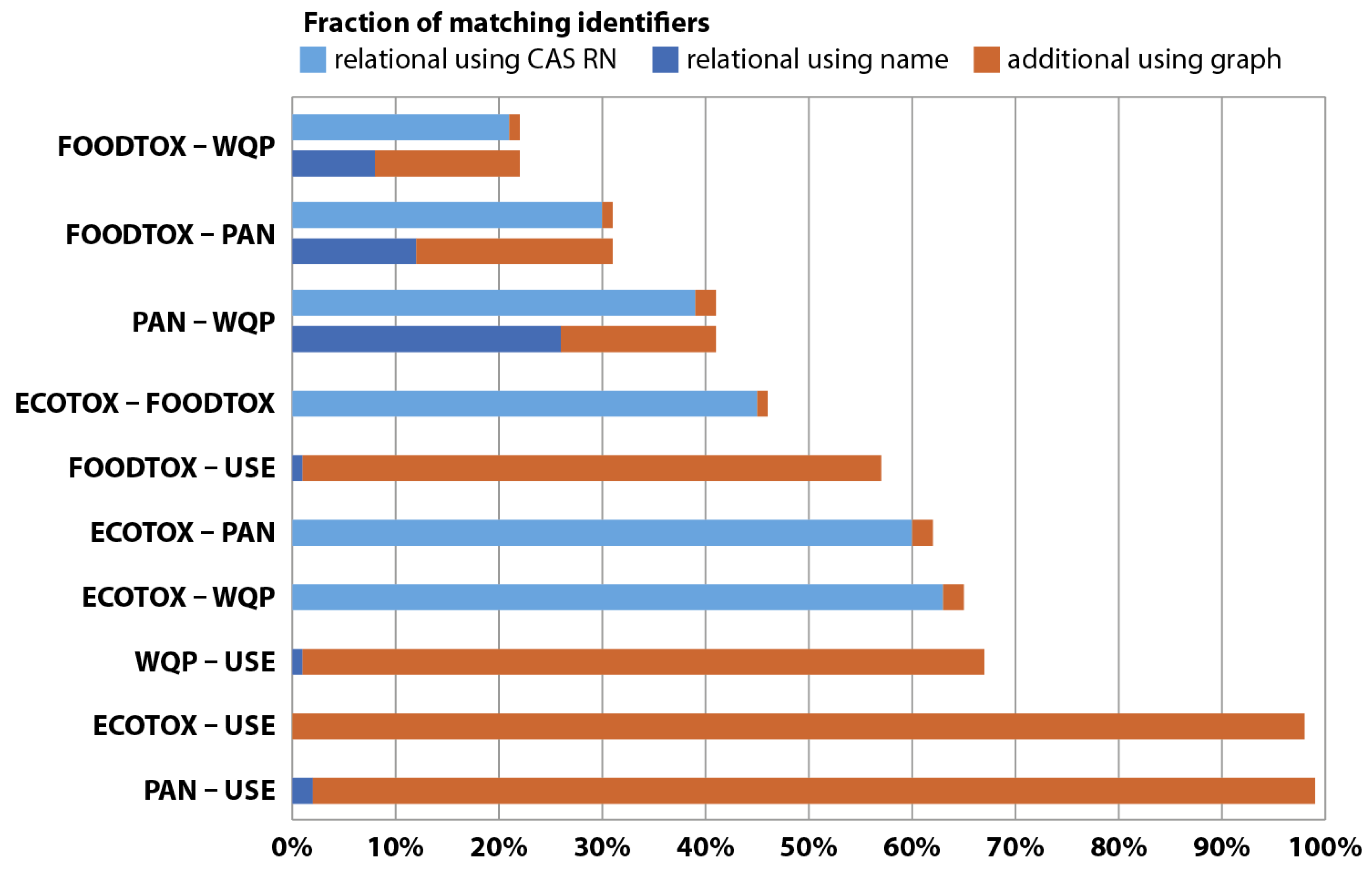
Initially, six publicly available chemical databases were identified and compared regarding quality of synonyms they provide for an array of organic pesticides (
n = 655). After extensive quantity and quality assessments of the generated synonym links, the U.S. EPA Chemical Dashboard [
11], containing approximately 765,000 chemical entries, was chosen as a synonym provider (see
Appendix C).
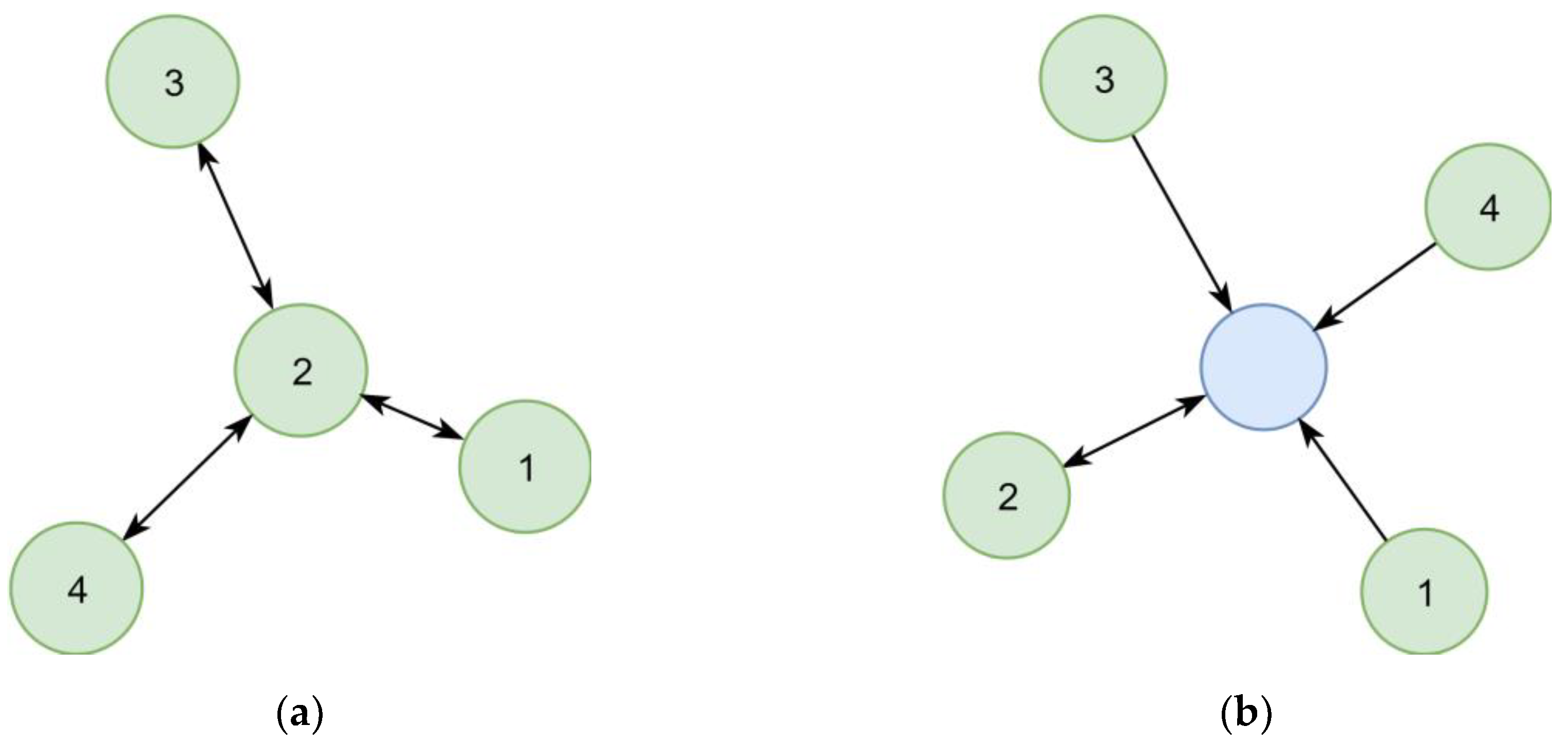
In the first implementation, synonymous chemical identifiers (e.g., substance names) were interlinked directly (
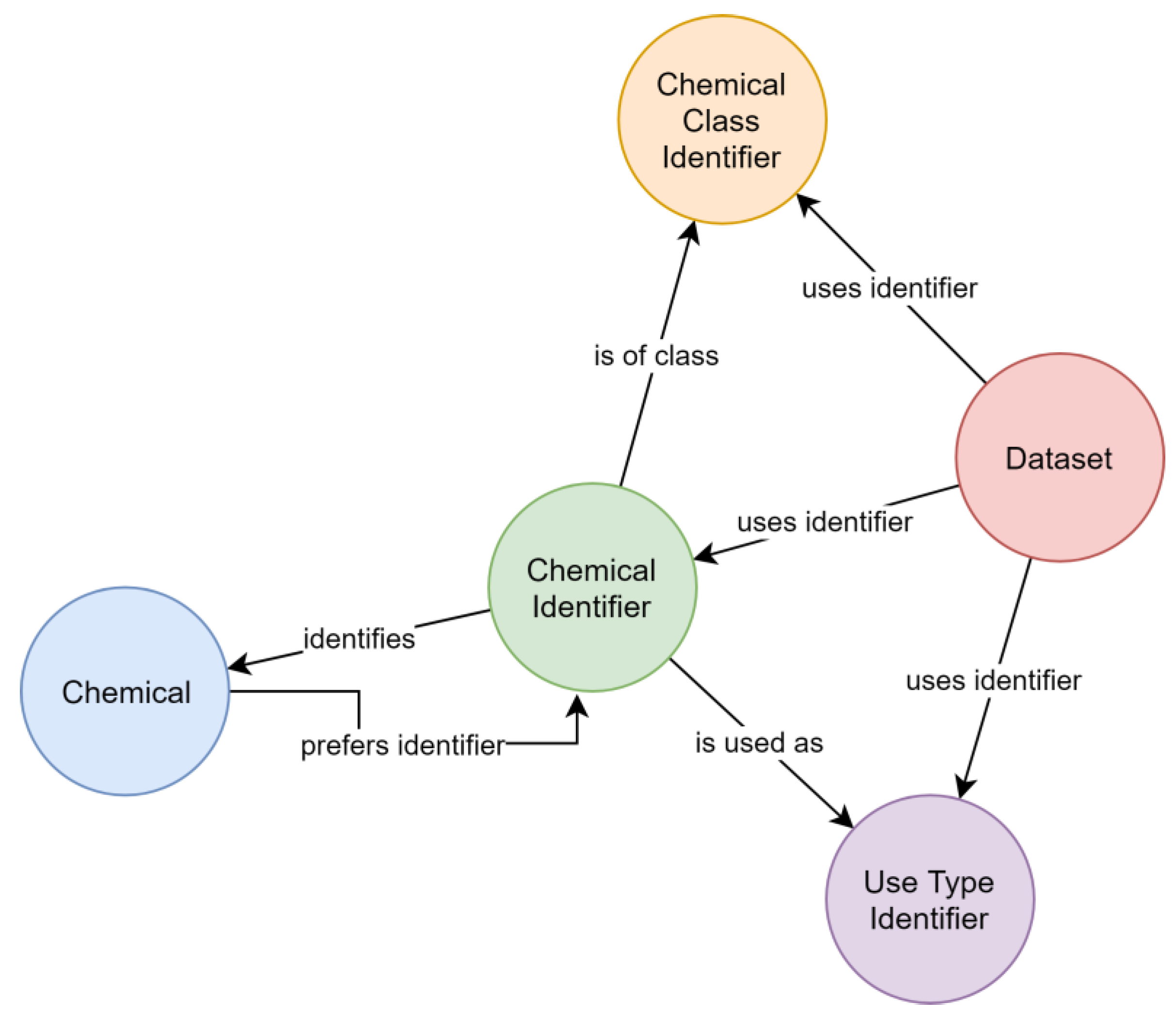
Figure 6a). However, following this concept, the number of steps necessary to collect all synonyms of a given identifier varied between queries, which resulted in complex queries. This concept also complicated the estimation of the quality of synonym relationships, as two distant identifiers could be linked over relationships of different certainty. A later refined representation distinguished between the chemical itself and its identifiers (
Figure 6b), leading to a representation where the step sequence for collecting all synonyms of a chemical is well defined and only requires two steps. This adjustment improved the computational efficacy, at the same time allowing to add further chemical identifiers without increasing the maximum number of steps required.
Consistency between chemical query results was achieved by attributing each chemical a preferred identifier (used by the CDDB), so that chemicals can be identified in a default way. A descriptive property was attributed to relationships between identifiers and chemicals to reflect the identification type, e.g., CAS RN. Data output for chemicals can, thus, be restricted to specific types of relationships for identifying chemicals.
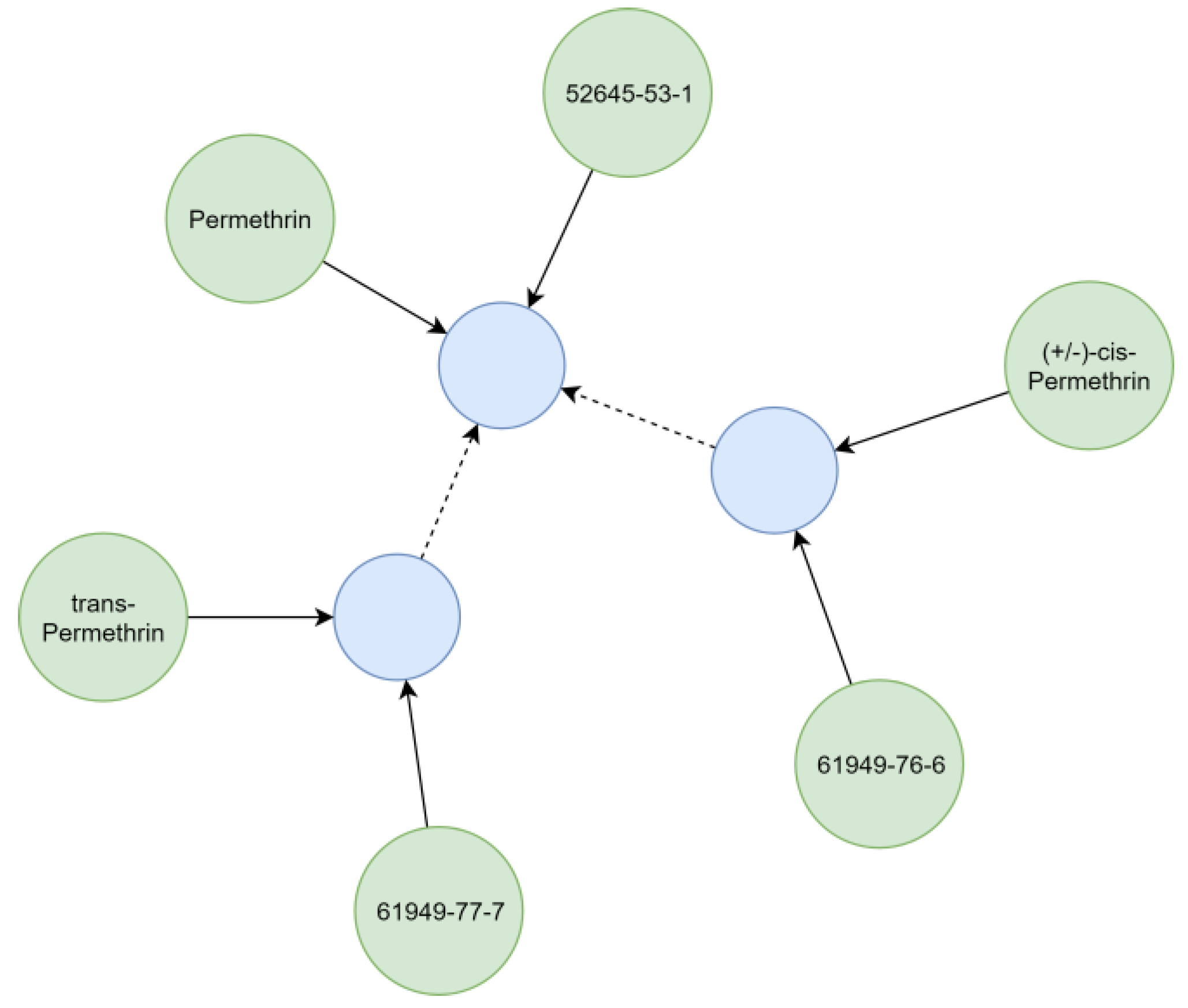
Substances relevant in ecotoxicological contexts can be described by varying detail of specificity (e.g., isomerism), which was addressed by creating hierarchical chemical sub-graphs. For example, permethrin (
Figure 7), an insecticidal compound, represents a stereoisomeric mixture of cis- and trans-permethrin isomers. The respective relationships between chemicals were resolved considering the presence of stereo-layers in their standard InChI strings [
25], creating a hierarchical sub-graph (
Figure 7). Further distinction of hierarchical levels (e.g., enantiomers and diastereomers) currently is not technically possible, as standard InChI strings do not support this operation [
25]. However, it is also rarely needed for ecotoxicological assessments using field concentrations.
After evaluation of different solutions, the MAGIC graph was hosted using the Neo4j native graph database, a mature, actively developed and widespread graph database product that is available as an Open Source Community Edition (GPLv3 license) and as an extended Enterprise Edition. The MAGIC graph was implemented using versions 3.4.5 to 3.5.0 of the Community Edition (updates were applied as soon as available). Validation rules (
Appendix B), as well as tools for automatic rule violation fixes, were implemented using PHP and integrated into an Apache 2 web server. For conducting manual fixes, a set of graphical tools was evaluated (
Appendix D). The web server hosts a publicly available website (
https://magic.eco) that allows, among other functions, to access the data of the MAGIC graph (see user notes).
Currentness of data is accomplished by synchronization routines, which update the MAGIC graph when some external data sources, especially the synonym provider, change. To detect differences between the graph and external sources, we reapplied the rule-based approach by implementing a set of synchronization rules (
Table A2). Violations of these rules indicated differences between databases, prompting a synchronization routine.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






