1. Introduction
Skeletal muscle makes up 40% of the body by mass and is a highly regenerative tissue due to its reservoir of muscle satellite stem cells (MuSCs). Extremity trauma, such as the type incurred during warfare, car accidents, tumor resection, and other blunt trauma can result in volumetric muscle loss (VML) injury. In VML injuries, the MuSC pool is depleted through the loss of muscle volume, infringing on muscle’s inherent regenerative capacity [
1,
2]. In addition, extracellular matrix (ECM) accounts for ~10% of the skeletal muscle mass and coats muscle fibers and MuSCs [
3]. Loss of the ECM eliminates a key element in muscle regeneration. Indeed, when the ECM is present following injury, muscle’s regenerative capacity remains intact [
4,
5]. A strategy to overcome a loss of both the MuSC pool and the ECM is to implant a muscle-derived ECM seeded with muscle-derived stem cells [
6]. Our lab has previously established a method to decellularize skeletal muscle, removing the cellular components that would induce an aberrant immune response, and using this as a platform for new cell ingrowth in the injury area [
7,
8]. Other research groups have also explored the use of decellularized tissues in VML injuries and most have determined that decellularized matrices result in functional fibrosis and not in regeneration [
9,
10,
11], owing a lack luster regeneration to an inability to support MuSC pool expansion. However, our research showed that our DMM preparation is capable of supporting MuSCs and nascent muscle fiber formation within the graft area [
8].
Regenerative medicine strategies often use cellular therapies to enhance tissue regeneration, where the stem and progenitor cells used are typically derived from a single source. These cell therapies have focused on myoblasts, satellite cells, mesoangioblasts, pericytes, and mesenchymal stem cells [
12,
13]. Stem-cell-based therapies represent a promising VML treatment option because they have the ability to self-renew and differentiate into various types of functional progeny, including skeletal myoblasts [
14]. Muscle satellite cells are tissue resident stem cells in skeletal muscle and are the primary cell type used to regenerate new muscle fibers. In uninjured muscle, these cells reside underneath the basal lamina in a quiescent state. Following injury, mitogenic signals activate them into a proliferating state, whereby parent satellite cells give rise to daughter cells often referred to as myogenic precursor cells (or myoblasts), and their activation initiates the myogenic program [
15]. While these cells are the gold standard for muscle regeneration studies, few have been able to effectively deliver satellite cells to VML injuries without eliminating the inherent ability of satellite cells to proliferate and self-renew, a key advantage in their use.
While satellite cells are crucial for effective muscle regeneration and functional recovery, the number of cells harvested from muscle is far less than other mesenchymal tissues. This may require the addition of other mesenchymal stem cells to regenerate enough tissue for VML. ASCs (Adipose-Derived Stromal Cells) are a potent source of mesenchymal stem cells, but their ability to enhance muscle regeneration has been limited [
16,
17,
18]. ASCs have been shown to support muscle repair [
19], and our preliminary data demonstrate expression of myogenic genes, indicating their potential to enter the myogenic program. Despite the work done to characterize an optimal biomaterial delivery system, the current solutions available for VML have had mixed results with low rates of improvement and the risk of adverse immune responses [
12]. Thus, the field continues to investigate a variety of potential scaffolds, cells, and molecular signaling solutions.
The challenge with current scaffold options is that some natural polymers are easily degraded, and synthetic materials risk stimulation of a foreign body response [
20,
21,
22]. This study will use DMM which has been shown to provide a suitable cellular environment and remains within the wound site to allow the native tissue to reform in a VML injury [
7,
8].
Use of ASCs provides an alternative solution with potential to acquire an appropriate number of cells capable of contributing to muscle regeneration. We have data that indicate an ability for ASCs to enter the myogenic program, and these findings were supported by others [
12,
13,
14]. In other recent studies, ASCs cultured with myoblast-conditioned media increased levels of myogenic differentiation [
15] or co-cultured with muscle satellite cells stimulated ASC fusion into multinucleated myotubes [
16]. In addition, ASCs were implanted into a muscle defect with an angiotensin inhibitor that stimulated ASC fusion into new myofibers [
17], suggesting that ASCs can communicate and fuse with muscle progenitors and aid in regeneration of new muscle fibers in VML injuries.
For the current study, we hypothesized that delivering either ASCs or early-stage myoblasts to the injury site using DMM would improve muscle regeneration. In addition to satellite cells, it has been shown that ASCs participate in the myogenic process. Moreover, these cells are easy to harvest from liposuction procedures, making their use in the clinic relevant for skeletal muscle tissue engineering. We used human ASCs or human myoblasts to test this hypothesis in an athymic rat gastrocnemius defect model as a step toward clinical translation in humans.
2. Materials and Methods
Decellularized muscle preparation. All animal studies were performed in accordance with approved VCU protocols (IACUC #AD10000675). Male Sprague Dawley rats weighing 250–300 g (Envigo, Huntingdon, UK) were euthanized using CO2. Gastrocnemius muscles were isolated bilaterally frozen at −80 °C, and shipped to MTF Biologics (Edison, NJ, USA) to be decellularized. Decellularization was performed via a proprietary method composed of multiple saline, detergent, and disinfection soaks using American Association of Tissue Banking and Food and Drug Administration approved protocols developed by MTF. The rat tissue was processed aseptically and without any terminal sterilization. Frozen decellularized muscle matrices (DMM) were shipped back to Virginia Commonwealth University (VCU) and were kept frozen at −80 °C until surgery.
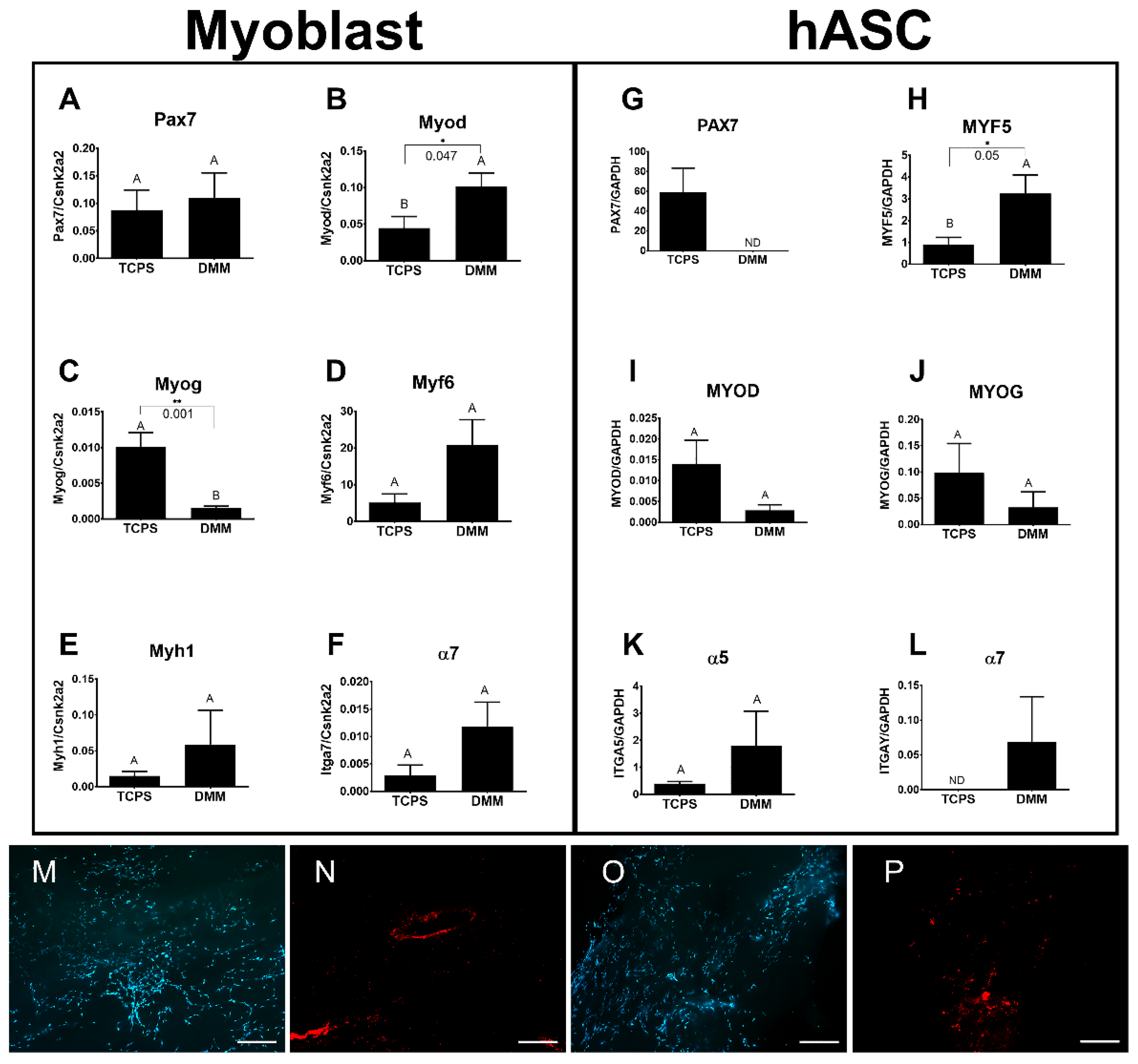
Cell culture. Human myoblasts (Cook Myosite, NJ, USA) were subcultured in myoblast growth media (Cook Myosite, NJ, USA) and passaged at 50% confluence. Cells were maintained in subculture until passage 3 and were used for DMM seeding. Human ASCs were subcultured in αMEM supplemented with 10% fetal bovine serum (FBS), 1% penicillin/streptomycin (P/S), and 1% L-glutamine. Cells were maintained in subculture and passaged at 70% confluence to be seeded onto DMM scaffolds. Cell seeding on DMM was first optimized for cell response data using both murine myoblasts and human ASCs. Murine myoblasts (C2C12, ATCC) were cultured on DMM using DMEM supplemented with 10% FBS and 1% P/S, and hASCs were cultured as described above. An optimal seeding density of 50,000 cells/cm2 was determined. Seeding density was checked using Live/Dead Viability staining (Thermo Scientific, L3224). Live/Dead staining was prepared by diluting 20 µL of 2 mM EthD-1 into 10 mL of 1x PBS followed by a dilution of 5 µL of 4 mM calcein AM stock solution into the 4 µM EthD-1 solution. Then, 100 µL of Live/Dead stain was added to each cell seeded sample and incubated at room temperature for 30 min. Labeled cells were imaged using a Zeiss AXIO Observer, Z1 fluorescence microscope. Prior to surgery, DMM was seeded with 2,000,000 cells/scaffold and allowed to subculture for 24 h. At surgery, seeded DMM scaffolds were prepared by washing in 1x PBS just prior to implantation.
Volumetric muscle loss surgery. In total, 40 male Foxn1
RNU (RNU) rats (250–300 g, ~4 months old) were obtained from Envigo (Huntingdon, UK) and were divided into five groups (N = 8 rats/group unless specified): sham surgery (n = 6); empty defect surgery, DMM surgery, DMM + hASC surgery, and DMM + h − myoblast surgery. All rats were given ad libitum access to standard pellet and water and provided environmental enrichment. Animals were housed individually. All surgical procedures were performed under an approved protocol at VCU (IACUC #AD10000675) as previously described [
8]. Briefly, rats were anesthetized using 4% isoflurane/400 mL/minute O
2 and prepared for surgery. Rats were transferred to the operating table and anesthesia was continued at 1–3% isoflurane in O
2. An oblique anterolateral incision extending from the patella to the calcaneus was made. After an incision in the biceps femoris muscle to expose the gastrocnemius was made, the left lateral gastrocnemius muscle was isolated, and a 1.5 × 1 cm defect was cut in the lateral gastrocnemius, taking care to preserve the sural and tibial nerve. At the end of surgery, biceps femoris was sutured closed using 5-0 nylon and skin was stapled closed thereafter.
Sham surgeries were performed as described except when the left lateral gastrocnemius was exposed, no incision was made to create a defect. Instead, biceps femoris and skin were closed without a defect. Empty defect surgeries were performed as described and left untreated, closing the biceps femoris and skin following tissue harvest. Rats that received DMM, DMM + ASC, or DMM + myoblast were sutured into muscle defects, using a modified Kessler technique and taking care to orient the anisotropic features in the direction of the muscle fibers. One rat from the empty defect group and one from the DMM + ASC group died or was euthanized (due to significant weight loss) prior to the end of the study. All remaining rats were euthanized at 8 weeks following muscle physiology tests.
Ultrasound Imaging. Rats were anesthetized with isoflurane (4% with 400 mL/min O
2) and hindlimbs shaved. Ultrasound gel was applied to an L15-7io Broadband compact linear array transducer from Philips (Bothell, WA, USA), and the transducer was applied to the gastrocnemius muscle to visualize the injury area (cross-sectional orientation). Suture landmarks identified the edges of the injury. A cross-section from the injury site’s center was used for analysis. We applied Heckmatt’s Scale [
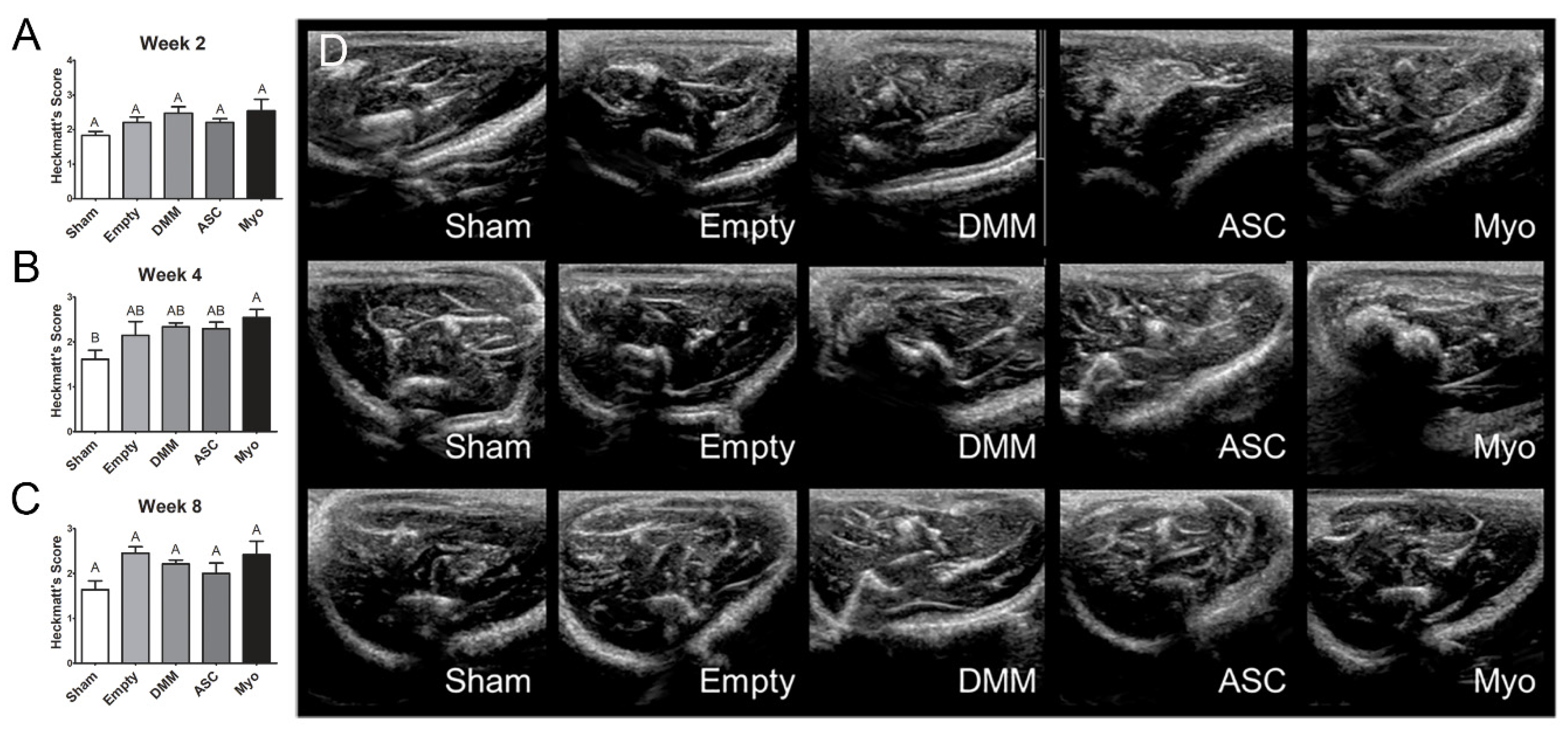
23] to grade the ultrasound images qualitatively. The grading criteria are as follows: Grade I—normal, darker muscle, normal fascia thickness/brightness, and the tibia/fibula will be clearly visible; Grade II—brighter muscle, thicker/brighter fascia, and the tibia/fibula will be clearly visible; Grade III—markedly brighter muscle, much thicker/brighter fascia, and less visible tibia/fibula; Grade IV—very bright muscle, considerably thicker/brighter fascia, and no visible tibia/fibula. Three independent, blinded observers scored each image. The three independent ordinal scores for each image were averaged together, and the resulting data was treated parametrically by applying a one-way ANOVA with Tukey’s post test (α = 0.05).
Muscle physiology. Peak tetanic force, peak twitch force, maximum rate of contraction, force–time curve integration, and maximum relaxation were measured. Rats were chosen at random and anesthetized using a vaporizer at 4% isoflurane/400 mL/minute O2. Following induction of general anesthesia, the sciatic nerve was isolated, and sural and peroneal branches ligated. Sciatic nerve was then stimulated using platinum electrodes connected to a Grass stimulator model SD9 (Astro-Med, Inc., Westwarwick, RI, USA) at 2 msec duration and 2 msec delay at varying voltages and frequencies. The knee and ankle joints were immobilized, and the Achilles tendon was cut from its insertion and connected to a MLT500/A force transducer (ADInstruments, Inc., Colorado Springs, CO, USA) with 2–0 silk sutures. Output was collected digitally using LabChart 8 software (ADInstruments, Colorado Springs, CO, USA). Optimal muscle length, stimulating voltage, and tetanic frequency were determined. During muscle lengthening, muscle was stimulated to tetanus for 3 s at each interval. Once optimal length was determined, tetanic contraction was stimulated at 3 s intervals until peak tetanic force dropped, indicating fatigue. Immediately following this force drop, 3 separate submaximal stimulations were measured. Those stimulations were used for muscle physiology assessments. Peak tetanic force was measured at maximum force for optimal frequency. Peak twitch force was measured at the peak of the submaximal force–time curve. Measurements in injured and treated limbs were the result of total force output from the posterior crural muscles (medial gastrocnemius, lateral gastrocnemius, soleus, and plantaris).
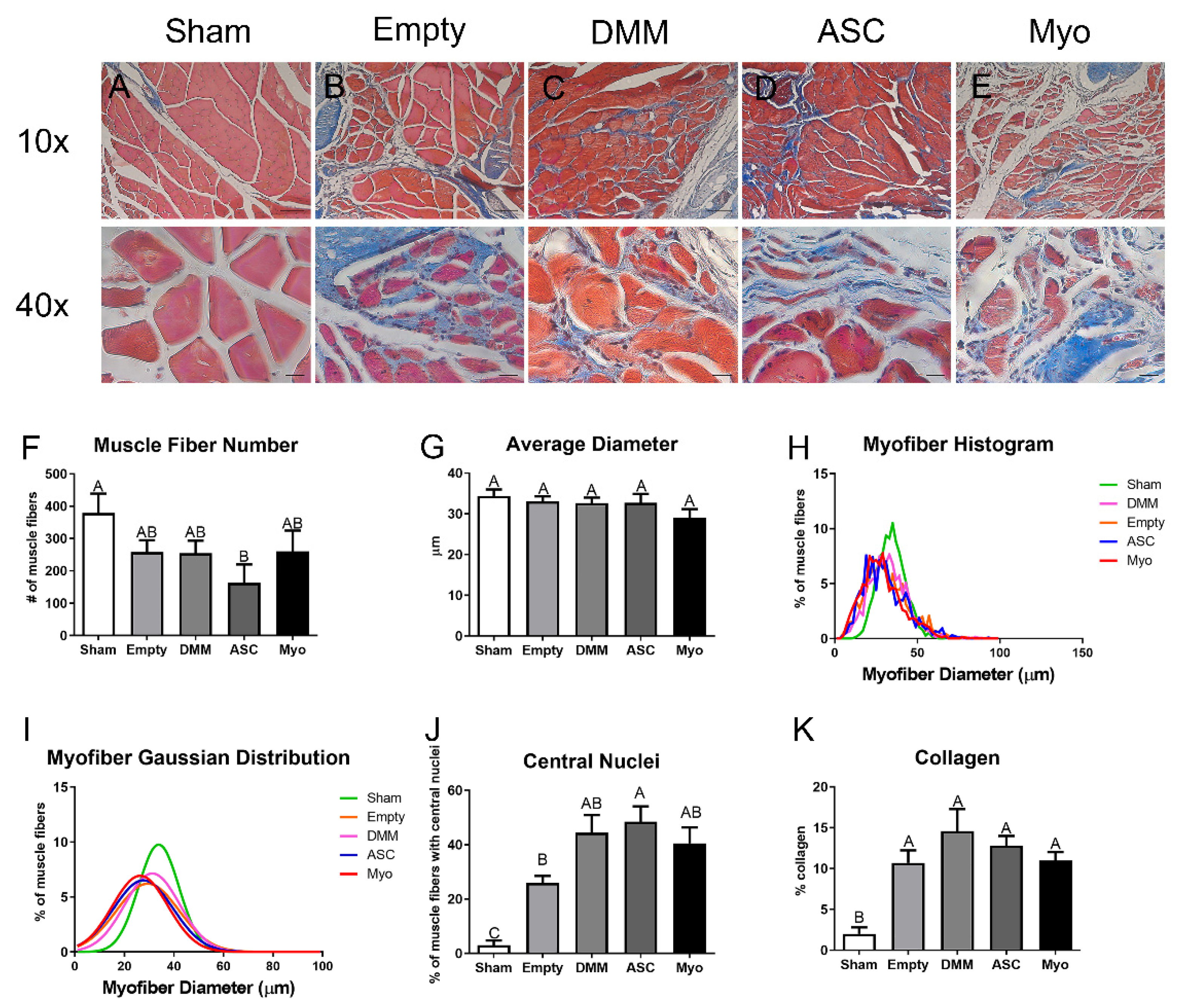
Histology. Whole gastrocnemius muscles were removed and fixed in 10% neutral buffer formalin, dehydrated, and embedded in paraffin. Muscles were cross-sectioned approximately 0.5 cm from the margins. Sections (5 μm) were placed on Histobond slides (VWR, Radnor, PA, USA), deparaffinized and rehydrated, and stained with Masson’s trichrome using Weighert’s hematoxylin (Sigma-Aldrich, St. Louis, MO, USA), Biebrich’s scarlet-acid fuschin (Sigma-Aldrich, St. Louis, MO, USA), and aniline blue (Sigma-Aldrich, St. Louis, MO, USA). Coverslips were mounted with xylene-based mounting media and allowed to dry flat before imaging.
Histomorphometry. Histomorphometry was used to quantify the percent of centrally located nuclei, myofiber diameter, and ratio of muscle to collagen. Histological sections stained with Masson’s trichrome were imaged using 10× and 40× objectives, and were assessed as previously reported [
8]. Healthy muscle from sham operated animals was used as a positive control. Histological analysis was performed on each muscle (n = 6 for sham, n = 7 for Empty, n = 7 for DMM-ASC, n = 8 for DMM, n = 8 for DMM-Myo) in 3 different locations within the graft area. Field sizes of 650 × 870 μm and 165 × 220 μm were used for histomorphometry. DMM locations were identified by first locating the zone of injury using injury margins. Once the zone was established, images were taken from three locations within the injury site, one at the margin, a second within the middle of the graft, and a third within an additional area of the graft away from the margins.
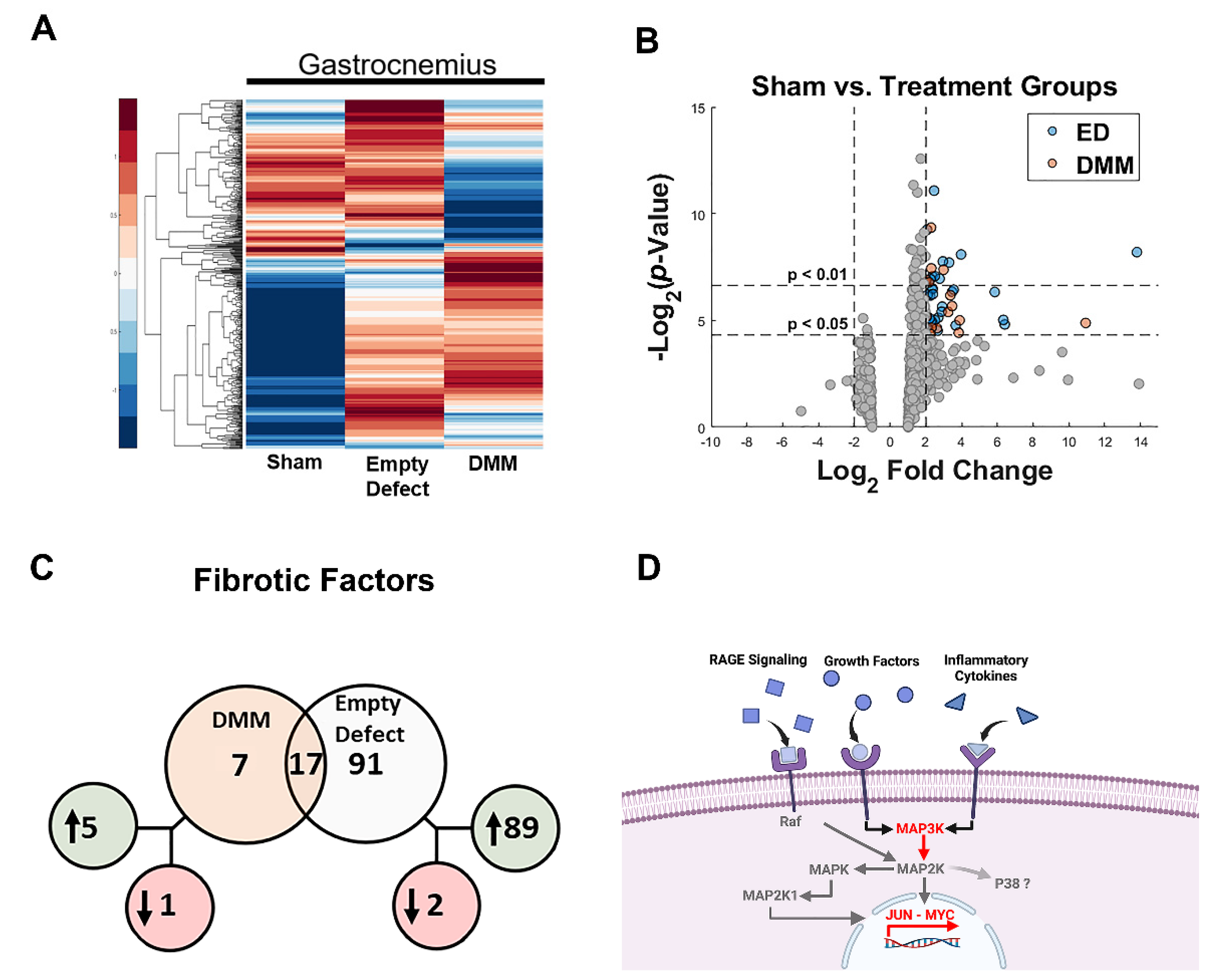
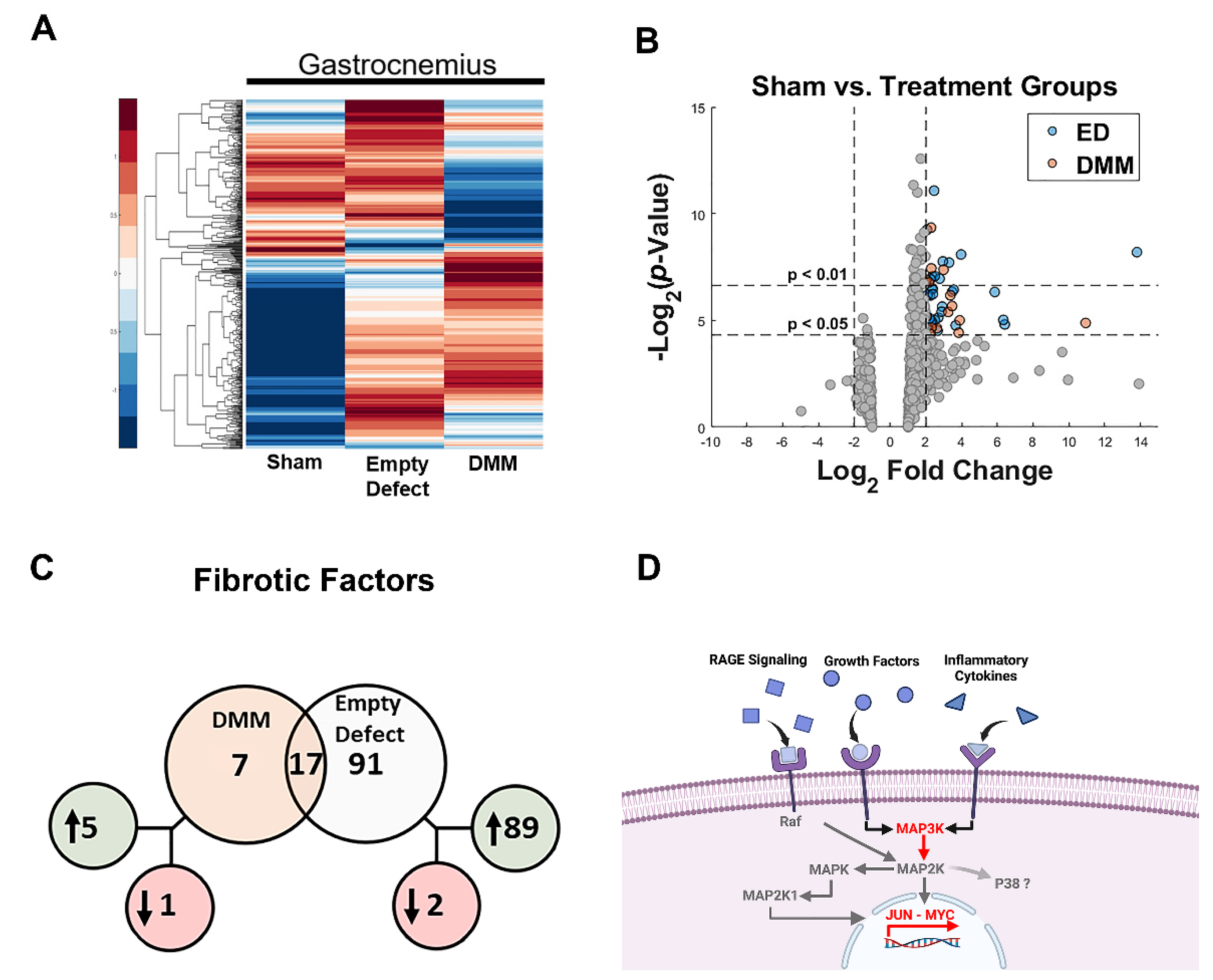
Nanostring. Gene expression of 817 total RNAs were measured using the NanoString fluorescent RNA hybridization and counting method. The custom panel measured genes related to muscle and nerve expression from the mouse genome matching at least 90% homology to the rat genome of interest. Samples were diluted to 55 ng/uL and checked for 260/280 purity using spectrophotometry. In total, 14 genes were used as reference genes while 10 others were used as housekeeping normalization genes. Counting was performed using the nCounter® Digital Analyzer while raw data analysis was accomplished using nSolverTM Analysis Software 4.0. Background thresholding was used to eliminate potential noise caused by low count (<20) genes followed by housekeeping normalization using geometric means. Genes found outside of the normalization factor range of 0.1–10 were flagged for QC. Fold change from each group was calculated against sham. Genes were considered differentially expressed if their log2 fold change was greater than |±2| and p-value ≤ 0.05.
Western blot. In total, 30 mg of gastrocnemius muscles were homogenized in NP-40 lysis buffer (BP-119, Boston BioProducts, Ashland, MA, USA) with a PI cocktail and 25 mM NaF with a 6.0 mm zirconium bead in a beadbug homogenizer (BeadBug™ Cat #: 31-212, Genesee Scientific, San Diego, CA, USA) at 4000 rpm for 60 s 5 times while keeping the tubes on ice for at least 5 min between runs. The homogenate was centrifuged at 9703 rpm (10,000×
g) (Centrifuge 5427 R, Eppendorf, Hamburg, Germany) for 10 min, and the supernatant was used for Western blotting. Briefly, the supernatant was run on the BCA assay. Equal amounts of protein were denatured with Laemmli buffer at 100 °C for 10 min, then electrophoresed on polyacrylamide gels, transferred to a PVDF low fluorescence membrane, blocked for 1 h at room temperature, and stained overnight with primary antibodies listed in
Table 1. The membranes were then incubated with secondary antibodies (926-68073, 926-32210, and 926-32211, LI-COR Biosciences, Lincoln, NE, USA) for 40 min at room temperature, then they were imaged on the LI-COR Odyssey and quantified. Each blot was analyzed separately and results were normalized to GAPDH levels. The normalization factor for samples on one blot was determined by identifying the highest GAPDH signal and dividing each GAPDH signal intensity by the highest GAPDH signal, producing a value of 1 for the strongest signal and less than 1 relative to that stronger signal. Target protein signals were divided by the normalization factor to obtain the normalized signal.
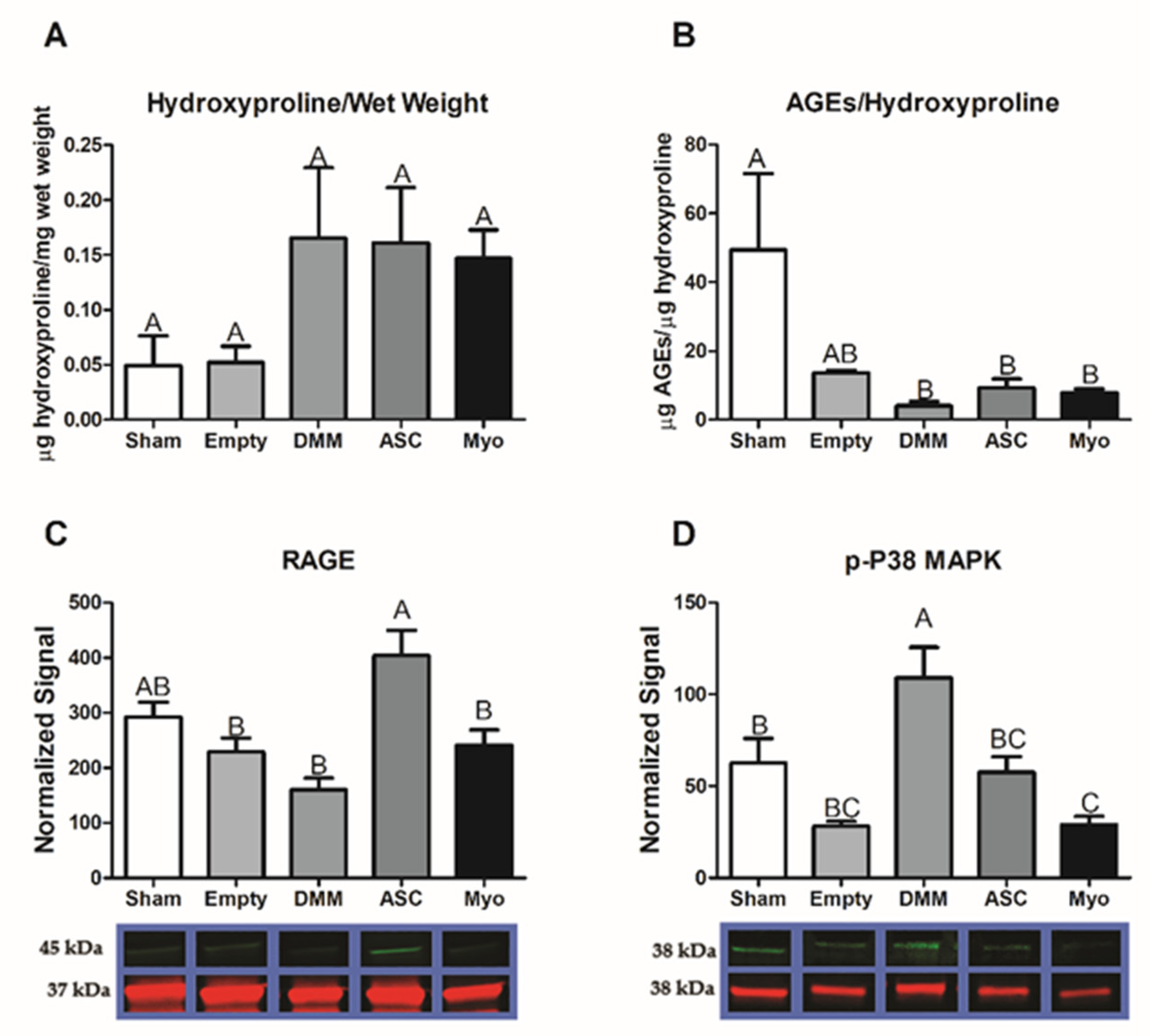
AGEs. Muscle samples were minced and homogenized with the MinuteTM Total Protein Extraction Kit for Muscles (using the Denaturing Buffer) and ran on an AGE ELISA (Cell Biolabs, STA-817) to determine AGE levels. For DMM, homogenization was completed with a 6.0 mm zirconium bead in a beadbug homogenizer (Genesee Scientific, BeadBug™ Cat #: 31-212) at 4000 rpm for 60 s 20 times while keeping the tubes on ice for at least 5 min between runs, to pulverize the tough ECM, followed by centrifugation at 13,000 rpm (17,949× g) (Eppendorf, Centrifuge 5427 R) for 3 min. The Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific, 23225 and 23227) and hydroxyproline assays were used for normalization purposes.
Statistical analysis. Each variable was tested using N = 8 independent animals. The animal number was chosen based on a power analysis using an alpha of 0.05 and a power of 80% (delta = 5, sigma = 3, m = 1) to reveal a minimum of n = 7 per group to yield statistical significance. Data are presented as mean ± SEM with analysis completed using GraphPad Prism 6.0 (GraphPad, La Jolla, CA, USA). Analyses comparing more than 2 groups used one-way analysis of variance with Tukey post hoc test to determine differences between rat treatments, while comparisons of 2 groups used Student’s unpaired t-test. For Heckmatt’s Scoring, the three independent ordinal scores for each image were averaged together, and the resulting data was treated parametrically by applying one-way ANOVA with Tukey’s post hoc test (α = 0.05). All p values < 0.05 were considered significant.
4. Discussion
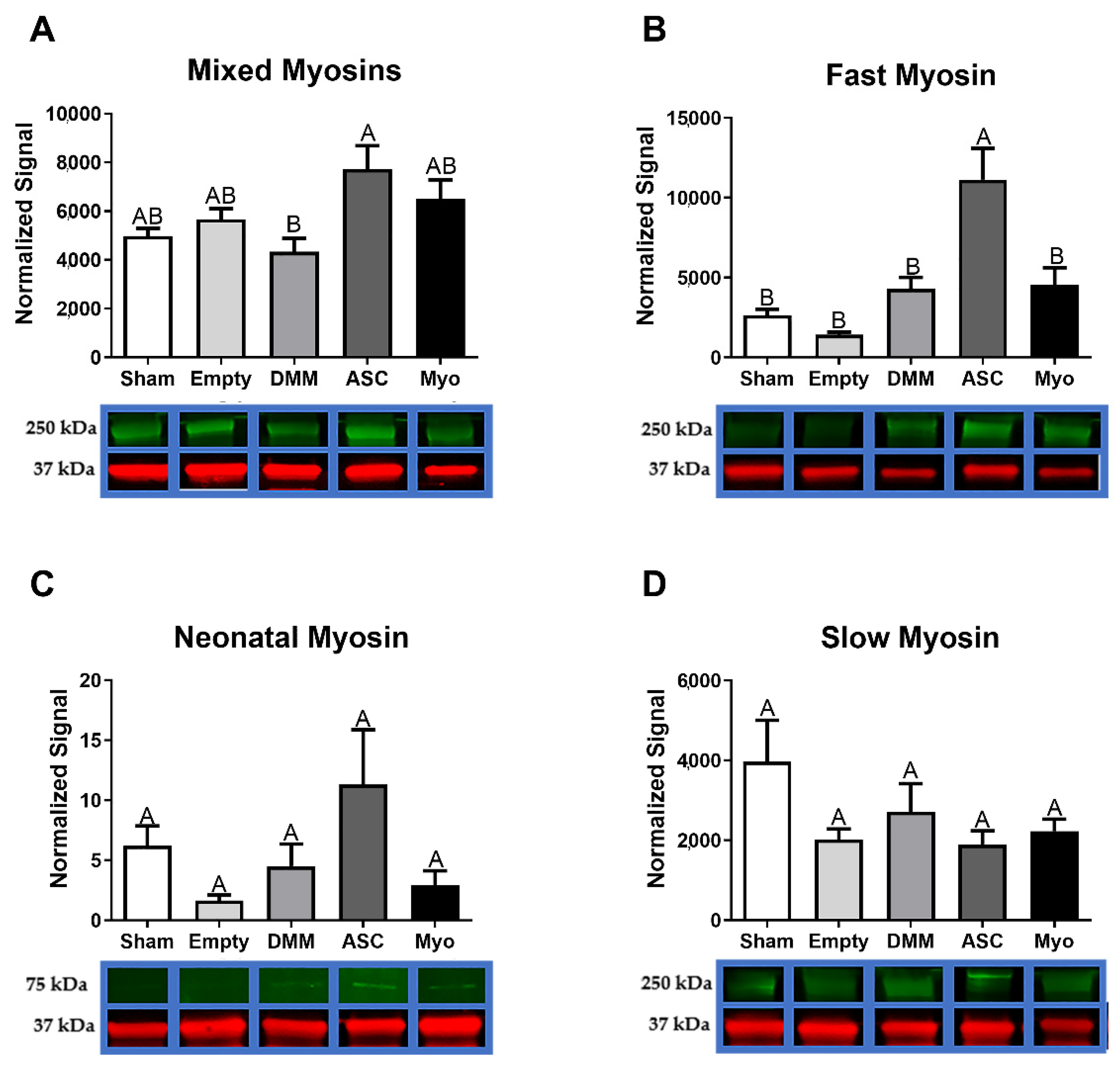
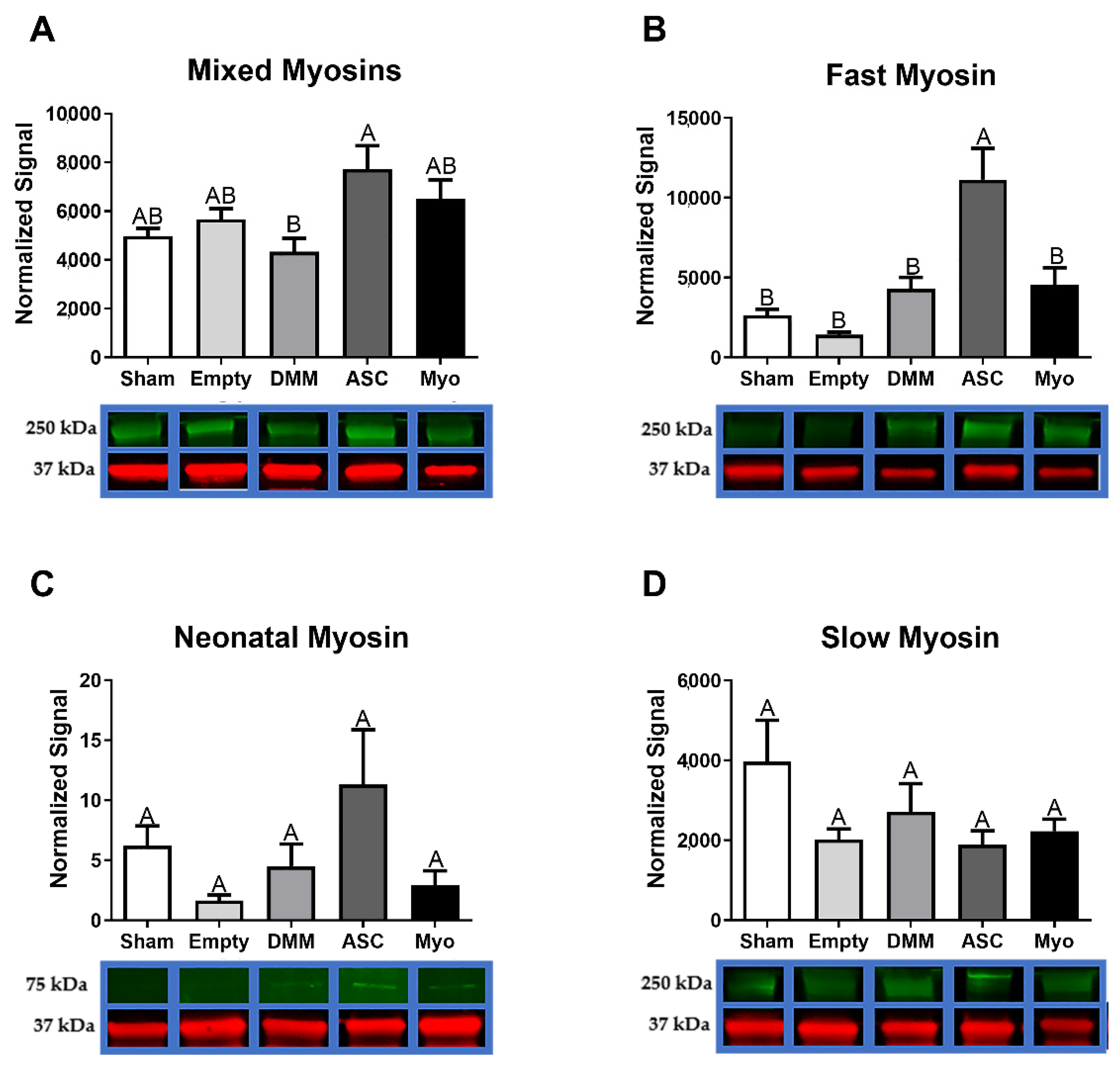
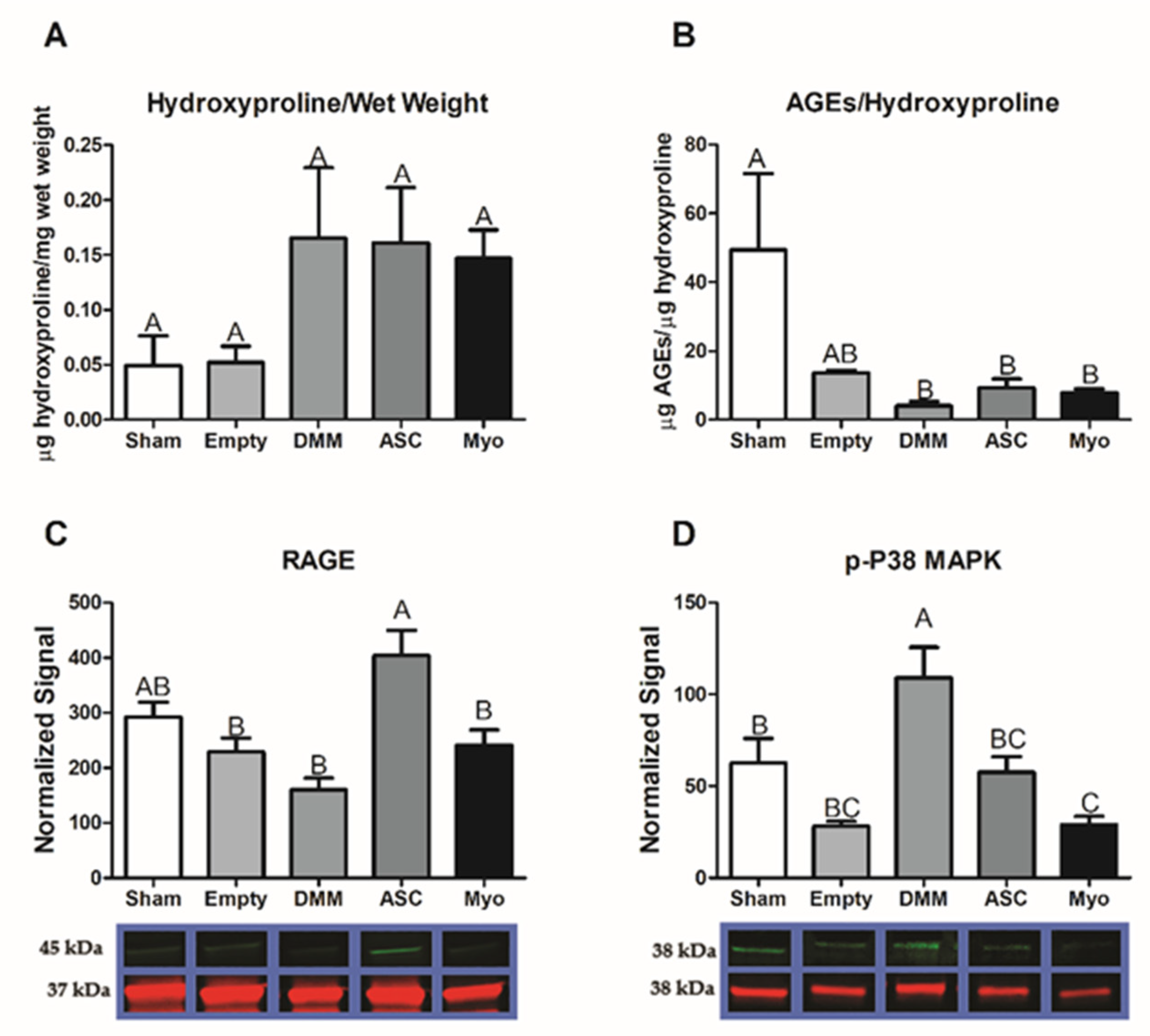
In this study, we first assessed differences in gene expression amongst DMM-treated VML sites, ED VML sites, and sham operated animals and determined that MAPK signaling was strongly affected including inflammatory associated pathways. These initial studies helped to identify specific pathways related to our injury model. DMM scaffolds were then seeded with human ASCs or myoblasts to determine if delivery of stem cells or progenitor cells would improve muscle regeneration in a VML model. While functional differences were unremarkable, differences in centrally located nuclei were elevated in ASC-delivered cells versus DMM and empty defect animals. In contrast, myoblast-delivered injury sites were unchanged compared to DMM and lower than empty defect animals. Western blotting confirmed the observed increase in central nuclei in ASC-treated injuries, showing increased fast twitch myosin heavy chain. In addition, collagen levels were quantified using Masson’s Trichrome staining and showed increased collagen across all injury sites. We hypothesized that increased fibrosis was related to aberrant AGE/RAGE signaling based on our initial gene analysis of VML wounds treated with DMM. Interestingly, AGE cross-links were downregulated in injured animals while RAGE signaling increased in ASC-treated sites. These data correlated with increased regenerative markers in ASC-treated injury sites, suggesting that RAGE signaling occurred in a normal physiologic manner. Downstream p38MAPK signaling was assessed and found to be lower in DMM- and myoblast-treated animals but similar to empty and sham in ASC, further confirming normal RAGE signaling. This is significant because other studies indicated that muscle trauma aberrantly increases RAGE signaling [
25,
26], contributing to a prolonged immune response; however, our data show a different effect in a rodent VML model. Overall, our data suggest that ASCs improved levels of muscle regenerative markers compared to myoblasts and that RAGE signaling also appears to be involved in ASC-mediated regeneration.
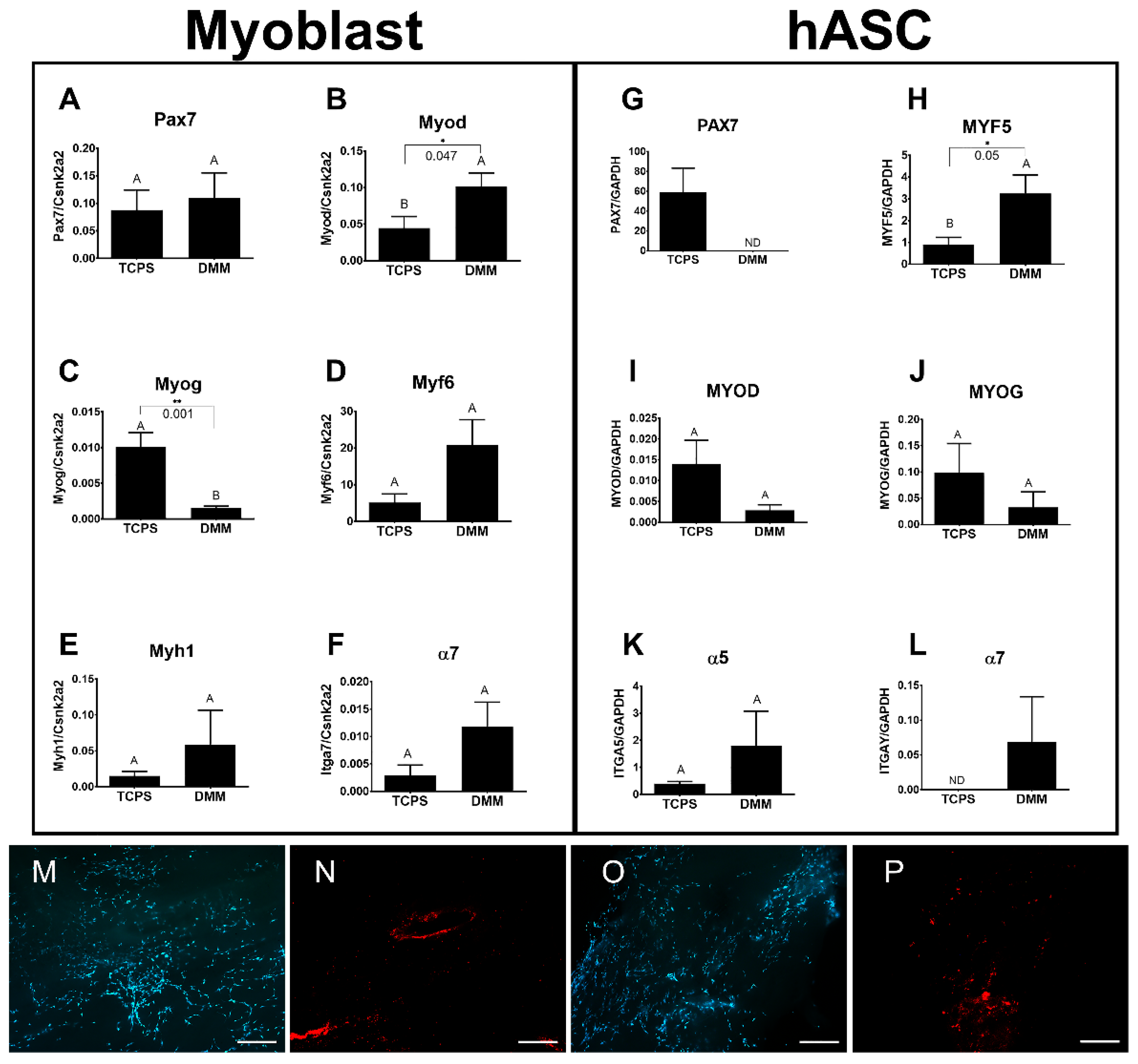
ASCs are an abundant source of multipotent mesenchymal stromal cells with myogenic potential. These cells are capable of expressing several myogenic factors including Pax7, Myf5, MyoD, and myogenin. Indeed, Di Rocco et al. showed that a small population of ASCs was capable of sporadically converting into a myogenic lineage, suggesting an inherent myogenic potential [
14]. Moreover, this same group demonstrated that ASCs injected into an ischemic injury model fused with existing muscle fibers, supporting the idea that ASCs are a suitable stem cell source for muscle regeneration studies. Prior studies also compared ASCs and myoblasts to each other to determine any differences in their regenerative potential when seeded onto a decellularized bladder matrix [
27]. While the study did not remark about ASCs versus myoblasts, they demonstrated histological evidence that ASCs and myoblasts were identified in the decellularized matrix.
We first demonstrated that ASCs and myoblasts could be cultured on DMM, and that ASCs expressed myogenic markers when seeded on DMM in vitro. We then determined whether ASCs or myoblasts would change the muscle’s response to VML injury and showed that regenerative markers were unaffected by myoblast seeding whereas ASCs showed improvements in regeneration when assessing MyHC II, centrally located nuclei, and RAGE signaling.
Myosin heavy chains follow a re-expression pattern during muscle regeneration that is important when analyzing de novo muscle fiber growth in a scaffold area [
28]. In cardiotoxin-induced injury models, neonatal MyHC was detected within 2–3 days after injury and persisted for up to 3 weeks [
29]. The switch from neonatal to adult fast myosins is independent of innervation [
30] and is a likely cause for the increase observed in ASC-treated sites. Since cardiotoxin injuries are a fully regenerative model, it is not surprising for us to detect neonatal MyHC in injured critical-sized muscle injury 8 weeks after implantation. Moreover, only in the presence of nerve is slow myosin upregulated and fast downregulated, suggesting that while we showed elevated levels of neonatal and fast myosins in ASC-treated sites they likely remained denervated.
We explored the role of advanced glycation end-products in our VML injury model and its involvement in fibrosis. Previously, we showed that AGEs were associated with fibrosis in older muscle [
24]. Those data coupled with our pathway analysis (
Figure 1) led us to hypothesize that fibrosis development in VML was also associated with AGEs. Other research showed that AGEs were elevated following chronic muscle injury with long-term RAGE signaling triggered by high concentrations of RAGE ligands producing a deleterious effect [
26]. We first tested AGE cross-link levels and determined that AGEs per collagen were suppressed in all VML groups. Moreover, AGEs levels in whole muscle lysates were lower in DMM-treated sites but were at sham levels for all other groups. This suggested that RAGE ligands were not chronically elevated as shown in prior literature studies, where RAGE ligands were explored in myopathies [
31,
32].
Interestingly, RAGE was elevated in ASC-treated injury sites compared to all other groups. These data coupled with low AGEs and elevated levels of regenerative markers suggested normal physiologic RAGE signaling in the newly regenerating areas. This is supported by studies that demonstrated delayed regeneration in an acute injury model using Ager knockout mice compared to other studies that explored chronic pathologic conditions [
26]. These opposing effects were ascribed to the levels of RAGE ligands available to activate the receptor. In addition, RAGE is highly dependent on its co-activators that help direct a particular signaling pathway. We explored whether p38 MAPK was involved given its known role in AGE/RAGE/p38 MAPK signaling [
33], and determined that p38 MAPK was elevated in DMM- and ASC-treated injury sites compared to empty defect while myoblast-treated sites were similar to empty.
P38 MAPK plays a critical role in muscle regeneration [
34]. It was first described as a transducer of the response to environmental stress conditions, and in muscle was found to regulate muscle fiber formation via satellite cell differentiation, slow myosin heavy chain gene repression [
35], and is involved in muscle pathologies [
36]. When taken into context with our observed increase in fast MyHC, centrally located nuclei, and RAGE signaling, use of p38 MAPK as a marker of regeneration in a VML model becomes more intriguing. As a disease mechanism, cell delivery reduced p38 levels and this could aid in regeneration [
37]. As a repressor of slow myosin heavy chain gene expression, p38 MAPK could explain the increases observed in fast myosin heavy chain protein levels in DMM-, ASC-, and Myo-treated injuries compared to empty sites, but more study would be needed to fully elucidate the role of p38 MAPK in a VML model.





 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}