1. Introduction
With the characteristics of an insidious onset, rapid progress, and poor prognosis, hepatocellular carcinoma is one of the leading causes of cancer-associated deaths worldwide [
1]. Although more and more treatments for hepatocellular carcinoma are being developed, the 5-year overall survival rate of hepatocellular carcinoma patients has not improved significantly in the past 10 years, and one of the most important reasons for this is that hepatocellular carcinoma is very prone to metastasis [
2,
3,
4]. The metastatic sites of hepatocellular carcinoma include the lungs, bones, adrenal glands, brain, lymph nodes, and peritoneum, among which lungs are the most frequent site [
5,
6]. However, the mechanism underlying lung metastasis from hepatocellular carcinoma is still not very clear.
Weighted gene coexpression network analysis (WGCNA) is a bioinformatics analysis method in which a coexpression network is constructed across the entire microarray or RNA sequencing data. It aims to find gene modules with coexpressed genes and to explore the relationship between gene modules and phenotypes of interest as well as the core genes in the network [
7]. The Cancer Genome Atlas (TCGA) is one of the most widely used databases in cancer research today, and from this, many researchers have used the WGCNA method to discover a variety of molecular mechanisms involved in tumor development [
8,
9]. Although many studies have used hepatocellular carcinoma data from the TCGA to perform WGCNA analysis, no relevant research has investigated lung metastasis from hepatocellular carcinoma [
10,
11].
In our study, WGCNA was firstly used to find the hub genes associated with hepatocellular carcinoma and lung metastasis by comparing hepatocellular carcinoma cases with and without lung metastasis in the TCGA. Then, we used the Gene Expression Omnibus (GEO) database and our own cohort to verify the impact of the FBL gene on the prognosis. In addition, in vitro or in vivo experiments were used to verify the effects of the FBL gene on the proliferation, stemness, migration, and invasion of hepatocellular carcinoma cells (
Figure 1). Our findings may provide new ideas relevant to the exploration of the molecular mechanism responsible for lung metastasis in hepatocellular carcinoma.
2. Materials and Methods
2.1. Data Acquisition and Screening
We downloaded gene expression profile data and clinical data collected from patients with hepatocellular carcinoma from the TCGA database (
https://portal.gdc.cancer.gov/projects/TCGA-LIHC, accessed on 27 February 2020). Transcriptome sequencing data of 370 hepatocellular carcinoma tissues and 47 normal liver tissues were obtained. We focused on the differences between samples of lung metastasis after hepatectomy and samples without recurrence or metastasis within 5 years after hepatectomy. According to the follow-up data, a total of 32 cases screened met the study’s requirements (including 18 cases of lung metastasis after hepatectomy and 14 cases of no recurrence or metastasis within 5 years after hepatectomy). We also downloaded the GSE14520 dataset and corresponding clinical information from the GEO database (
https://www.ncbi.nlm.nih.gov/geo/, accessed on 4 April 2020), which contained 247 hepatocellular carcinoma samples and 241 normal liver tissue samples adjacent to cancer. Besides, hepatocellular carcinoma proteomes containing 165 samples were obtained from Proteomic Data Commons PDC000198 (
https://pdc.cancer.gov/pdc/, accessed on 19 August 2020).
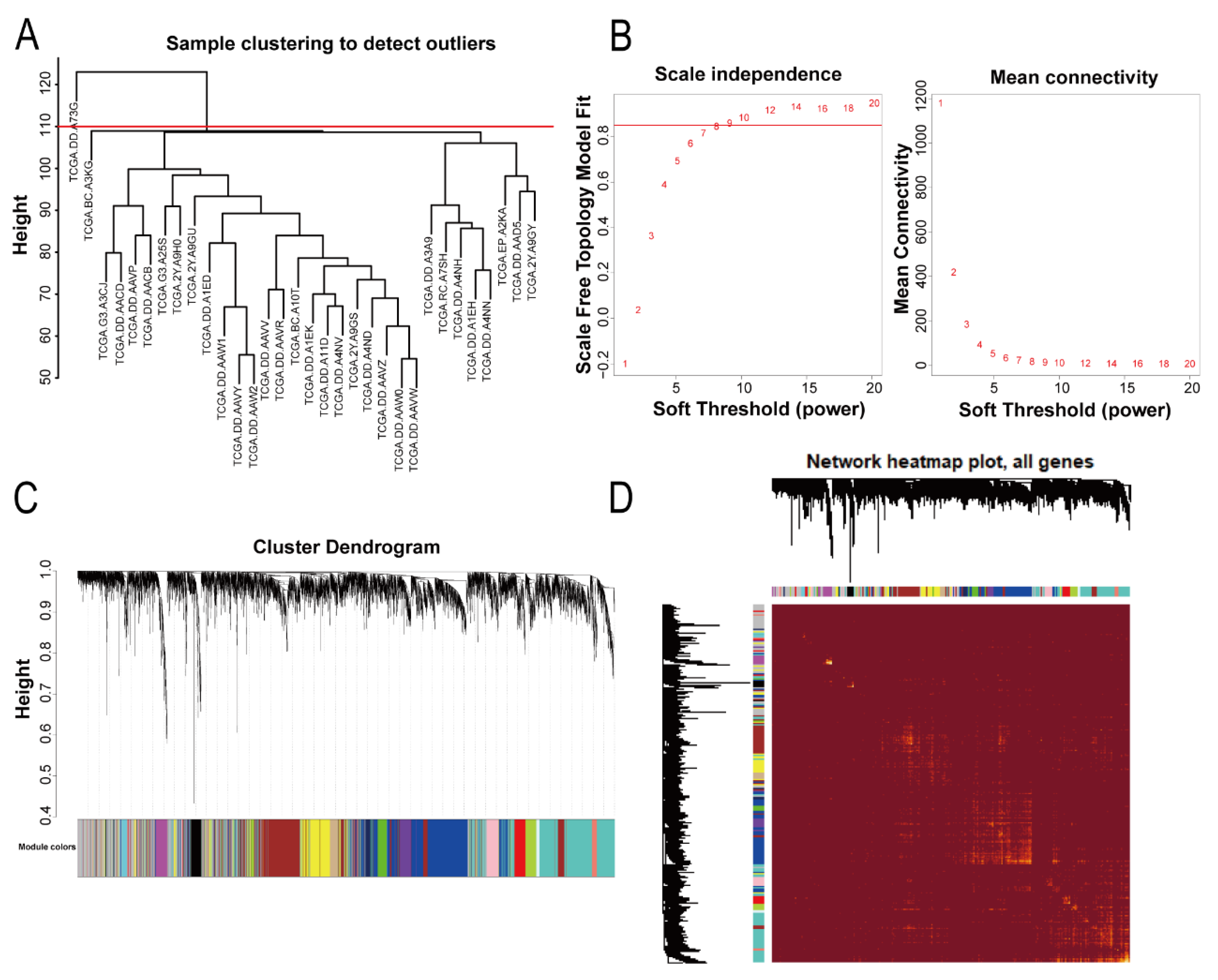
2.2. Construction of the Gene Coexpression Network
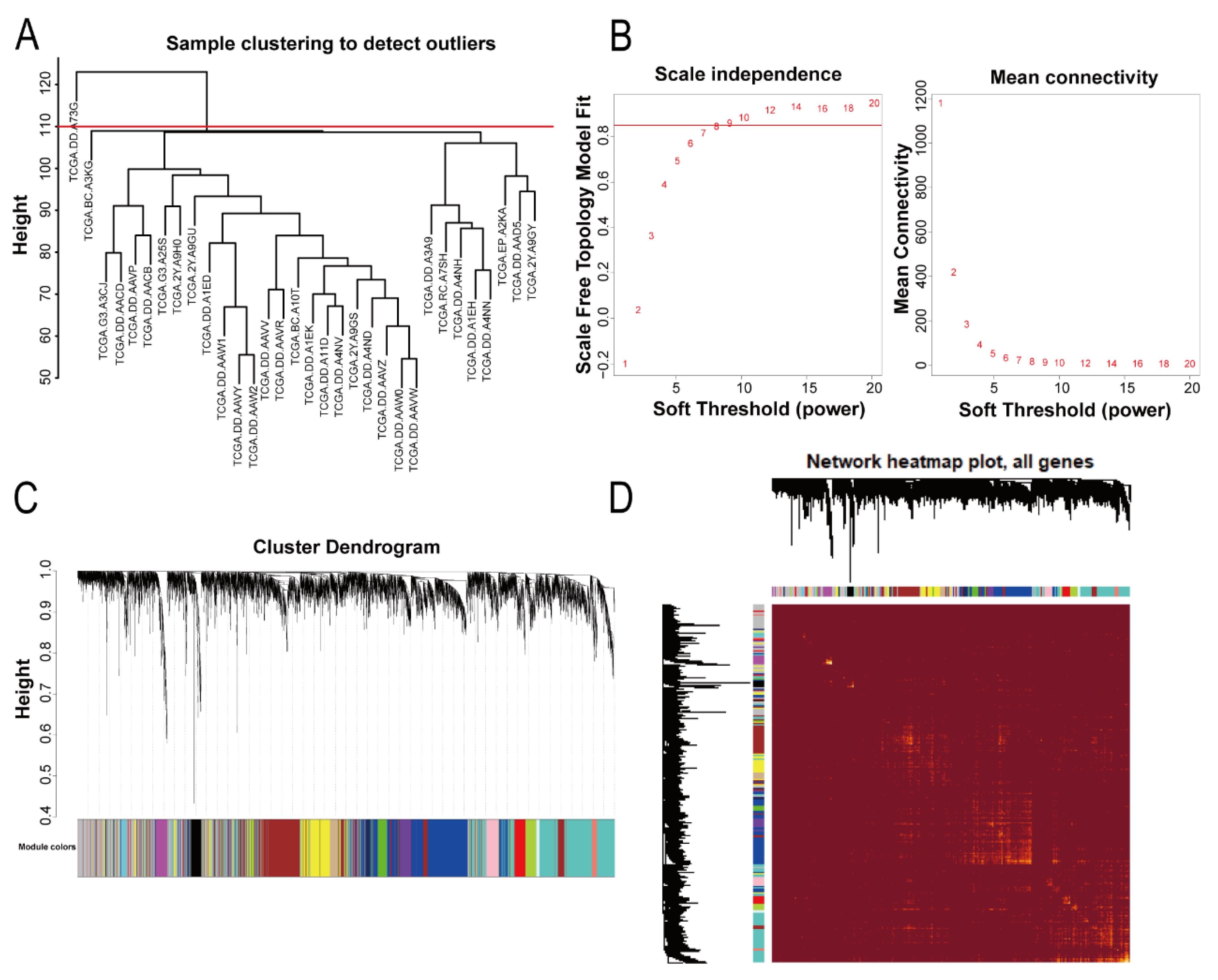
The WGCNA R package (version 1.69) was used to analyze the data [
7]. Firstly, we obtained the top 5000 genes according to the median absolute deviation (MAD) size of each gene in the samples. Then, we clustered the samples, deleted outliers (TCGA.DD.A73G), and performed a gene coexpression network analysis on samples that met the criteria. A topological overlap matrix (TOM) was constructed with a suitable soft threshold β value (β = 9, scale-free R
2 = 0.85). Genes were classified according to the hybrid dynamic shearing tree method, the minimum number of genes in each module was set at 30, and finally the differences of each module were calculated. A heatmap plot was used to visualize the topological overlap matrix (TOM) for all genes.
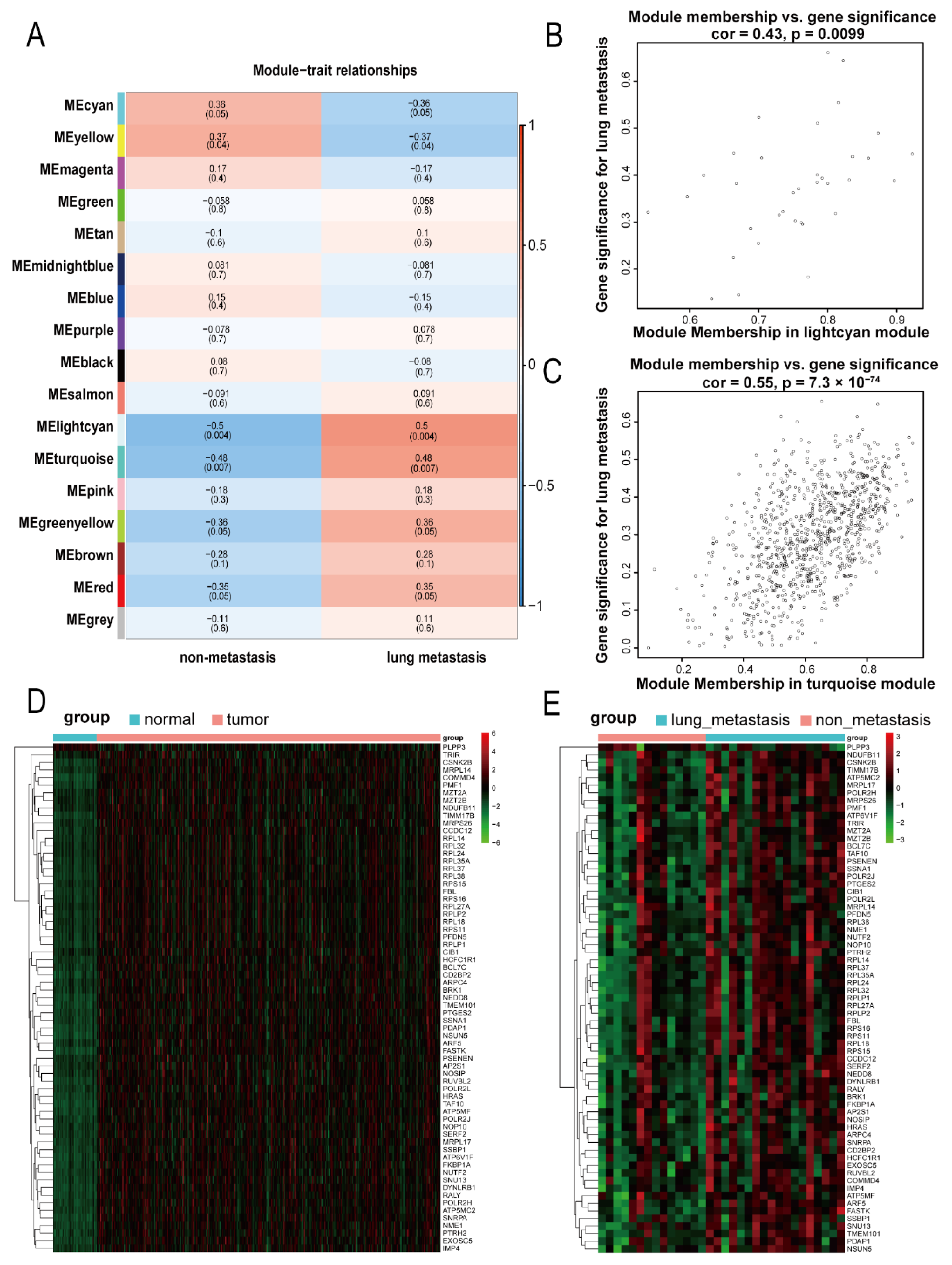
2.3. Identification of Significant Relevant Modules and Hub Genes
The gene modules related to lung metastasis were determined by the correlations between the modules and lung metastasis. In our study, modules whose p value was <0.01 were considered statistically significant. For statistically significant modules, we used the “signedKME” function in the WGCNA package (version 1.69) to calculate the module membership (MM) and gene significance (GS) in the module. The genes with MM > 0.8 and GS > 0.4 were screened as genes related to lung metastasis. Further, we used the pheatmap package (version 1.0.12) to display the differences in the expression of lung-metastasis-related genes in hepatocellular carcinoma tissues and normal liver tissues as well as the differences between hepatocellular carcinoma tissues with lung metastasis and those without metastasis.
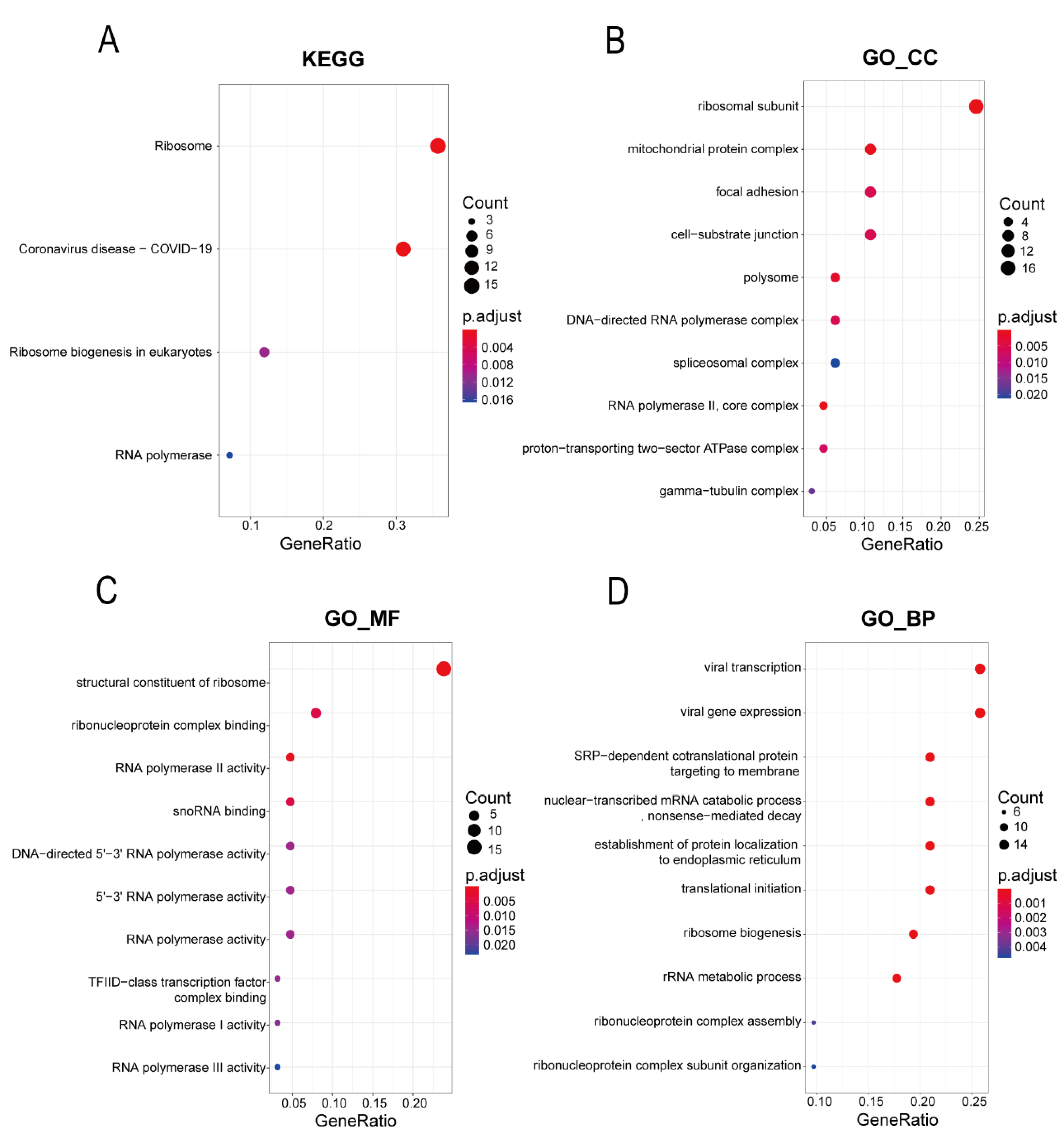
2.4. Gene Ontology Annotations and Kyoto Encyclopedia of Genes and Genomes Pathway Analysis of the Hub Genes
The Clusterprofiler package (version 3.16.0) was used to annotate and enrich the signal pathway of lung-metastasis-related genes using the Kyoto Encyclopedia of Genes and Genomes (KEGG) database and the Gene Ontology (GO) database. This included biological processes (BPs), cellular components (CCs), and molecular functions (MFs). A
p adjusted value of <0.05 was considered statistically significant [
12].
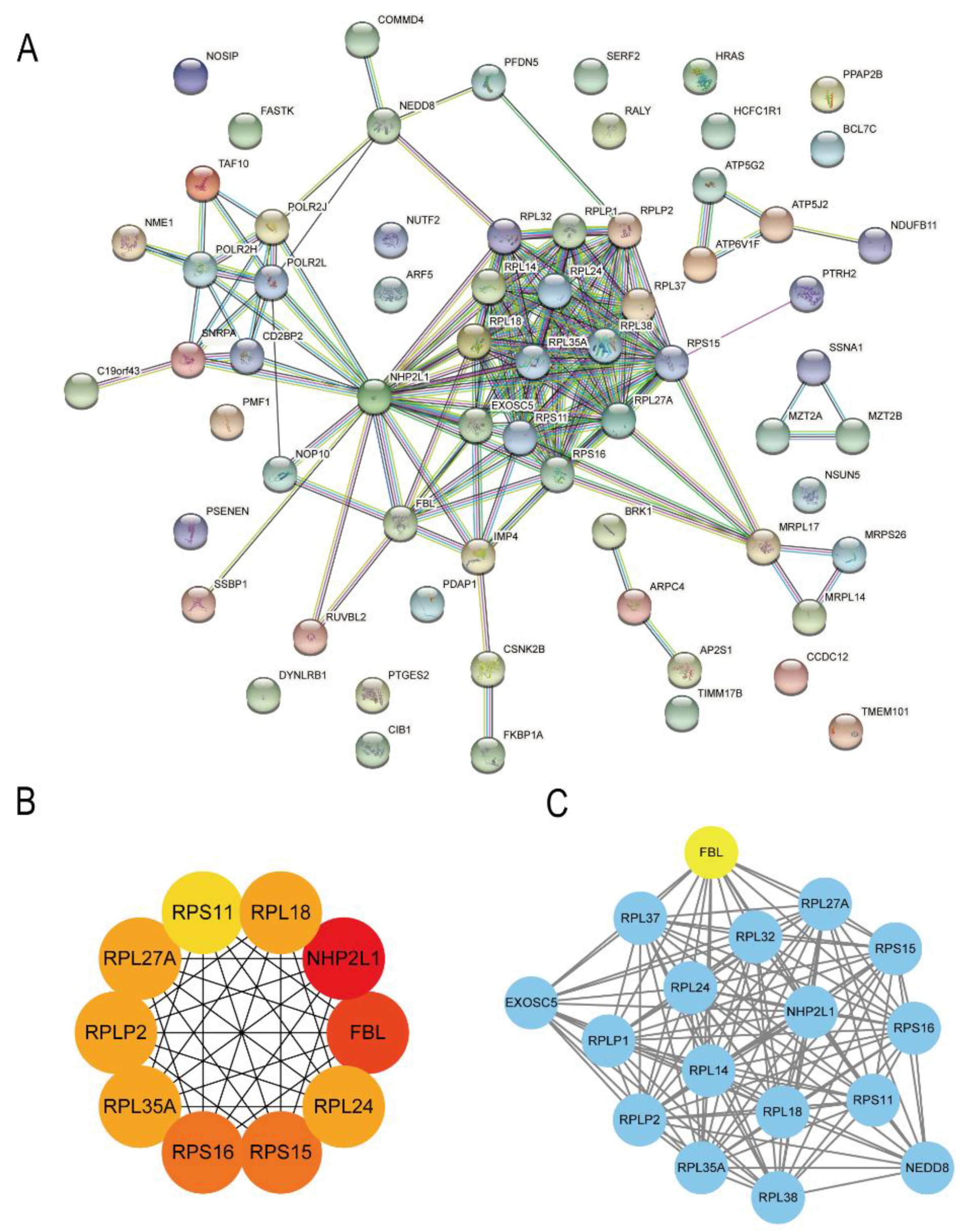
2.5. Protein–Protein Interaction Network Analysis of the Hub Genes
The Search Tool for the Retrieval of Interacting Genes (STRING) database (version 11.0) (
https://www.string-db.org, accessed on 16 April 2021) was used to analyze the interactions between the proteins of target genes to map the Protein–Protein Interaction network with a high level of confidence (minimum required interaction score = 0.700) [
13]. The cytoHubba plug-in was used to screen the top 10 genes according to the Degree score, and the MCODE plug-in was used to screen the core subnetworks to determine the genes in the core area of the interaction network with the Cytoscape software.
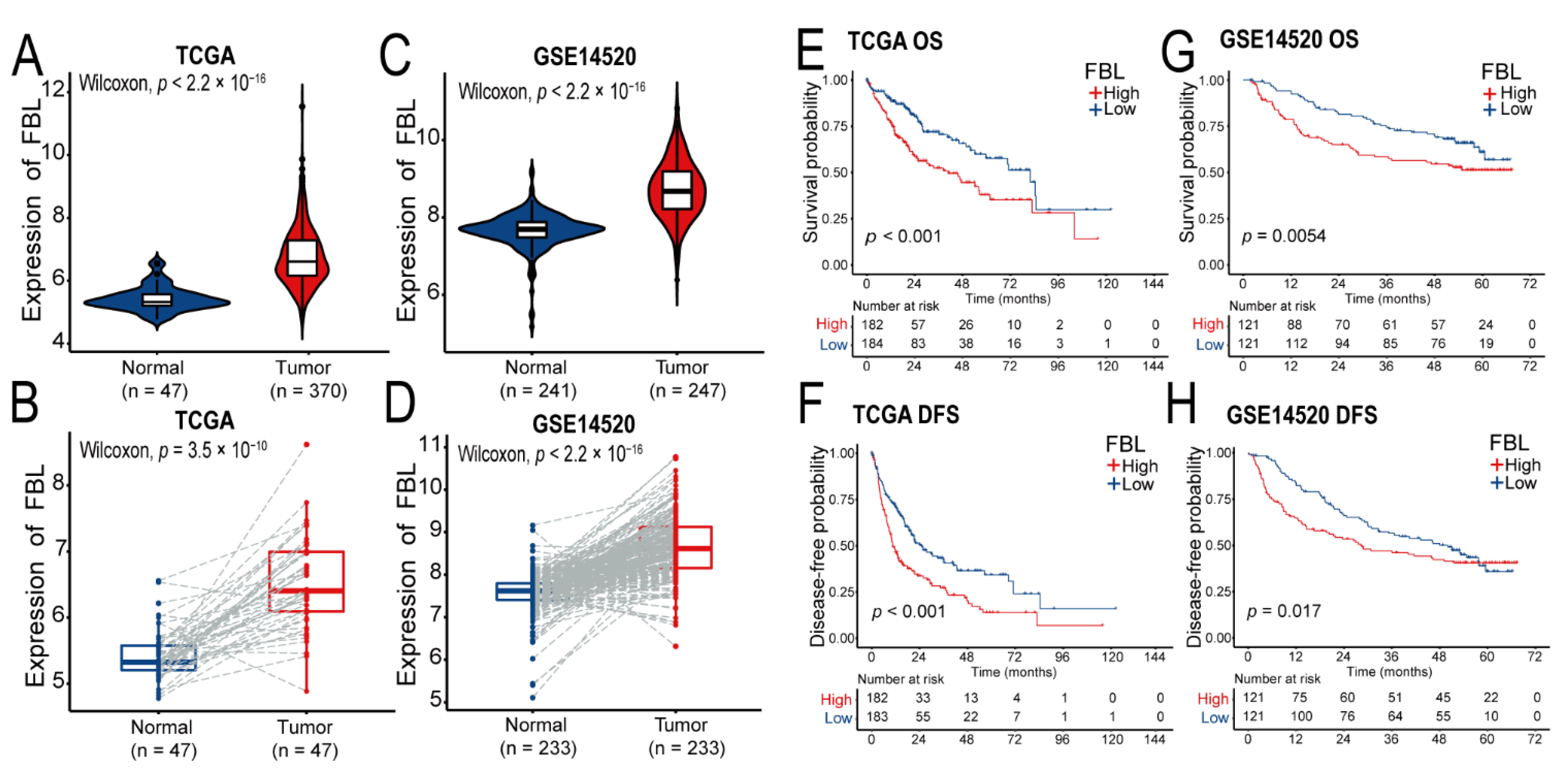
2.6. Expression and Prognosis Analysis of the FBL in TCGA, GSE14520, and Our Cohort
We used the ggpubr package (version 0.4.0) in R to construct violin plots or box plots to show the difference between the expression of FBL in hepatocellular carcinoma tissues versus that in adjacent normal liver tissues. The Wilcoxon rank sum test was used for statistical testing, and p < 0.05 was judged to be statistically significant.
We used the survival package (version 3.1-12) and the survminer package (version 0.4.8) in R to calculate the cumulative survival time of patients through the Kaplan–Meier (KM) method, and the built-in Peto-Peto’s modified survival estimate method in the survminer package was used for statistical testing. The overall survival time was defined as the time from the start of surgery to death due to any cause or until the last follow-up. Disease-free survival was defined as the time from the start of the operation until recurrence of the disease or death (for any reason).
2.7. Clinical Specimens
All HCC tissues and normal liver tissues were collected from HCC patients who underwent liver resection at the Sun Yat-Sen Memorial Hospital, Sun Yat-Sen University. All procedures were conducted with the approval of the Ethical Committee of the Sun Yat-Sen Memorial Hospital, Sun Yat-Sen University. Patient consent was obtained before the collection of tissue samples.
2.8. Cell Lines and Culturing Conditions
The human hepatocellular carcinoma cell lines Huh7 and PLC/PRF/5, and the immortalized liver cell line LO2 were purchased from the America Type Culture Collection. The cell lines were cultured in Dulbecco’s modified Eagle’s medium (BI, Biological Industries Israel Beit Haemek Ltd., Beit Haemek, Israel) supplemented with 10% fetal bovine serum (GIBCO, Life Technologies Corporation, New York, NY, USA), 100 μg/mL penicillin, and 100 μg/mL streptomycin at 37 °C in a humidified incubator containing 5% CO2.
2.9. Western Blotting
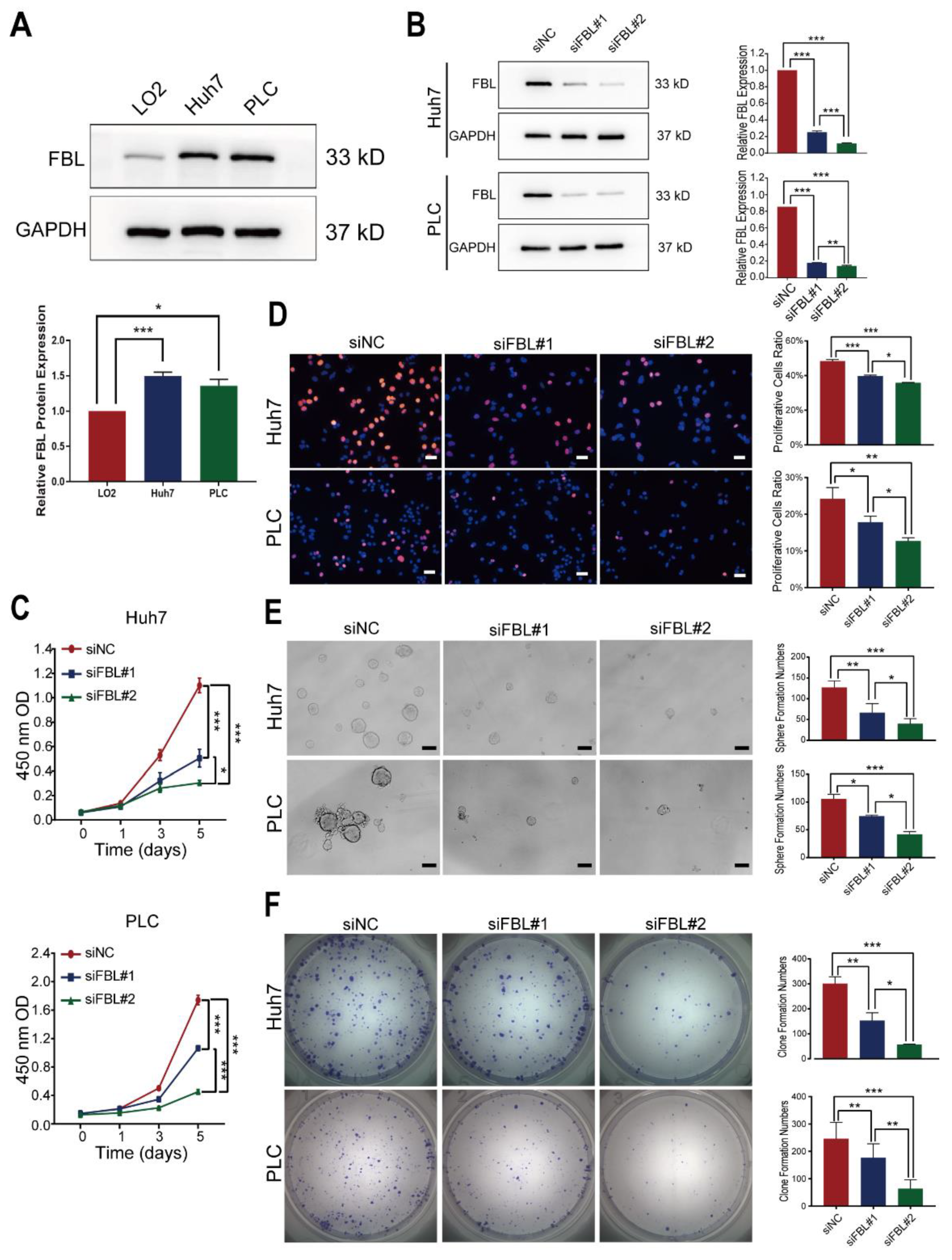
Cells and tissue samples were harvested in RIPA lysis buffer containing protease and phosphatase inhibitors to obtain protein extracts. A bicinchoninic acid (BCA) assay was performed to measure the protein concentrations. Then, equal amounts of the extracts were separated in an SDS-polyacrylamide gel, electrophoretically transferred to the polyvinylidene fluoride membrane (Millipore, Merck KGaA, Darmstadt, Germany), and incubated sequentially with primary and secondary antibodies after being blocked with 5% BSA. Finally, signals of proteins on the membrane were detected by chemiluminescence reagents (Millipore, Merck KGaA, Darmstadt, Germany) as per the manufacturer’s instructions and imaged with an Imaging System. The relative densitometry was calculated with Image J (1.50d version). The antibodies used for the Western blot analysis were FBL antibody (ab166630, Abcam plc, Cambridge Biomedical Campus, United Kingdom; 1:1000) and GAPDH antibody (2118s, Cell Signaling Technology, Inc., Danvers, MA, USA; 1:2000). Except for human specimens, Western blotting was performed in three independent replicates to confirm the reproducibility of the experiments.
2.10. RNA Extraction and RT-qPCR
Total RNA was extracted from cells or tissues using RNAiso Plus (Takara Bio Inc., Kusatsu, Shiga, Japan) in accordance with the manufacturer’s instructions. Reverse transcription was performed using PrimeScript RT Master Mix (Takara Bio Inc., Kusatsu, Shiga, Japan). qPCR was conducted using SYBR Premix Ex Taq II (Takara Bio Inc., Kusatsu, Shiga, Japan) and performed on the LightCycler® 480 Real-Time PCR System (Roche Applied Science, Mannheim, Germany). GAPDH was used as a reference control, and the relative expression level of mRNA was calculated with 2−ΔΔCt methods. Each experiment was performed in triplicate. The primer sequences used were as follows:
FBL Forward: 5′- GTCTTCATTTGTCGAGGAAAGGA-3′;
FBL Reverse: 5′- CTGGGTGAACTCCAAGGTG-3′;
GAPDH Forward: 5′- GGAGCGAGATCCCTCCAAAAT-3′;
GAPDH Reverse: 5′- GGCTGTTGTCATACTTCTCATGG-3′.
2.11. Immunohistochemistry
The paraffin sections were first dewaxed according to the standard procedures, and their endogenous peroxidase was inactivated in 3% H2O2. Then, they were heated up to 100 °C in Sodium Citrate-EDTA Antigen Retrieval Solution (P0085, Beyotime Institute of Biotechnology, Songjiang, Shanghai, China) under high pressure for 20 min for FBL antigen retrieval. Sections were incubated with FBL antibody (ab154806, Abcam plc, Cambridge Biomedical Campus, United Kingdom; 1:200) overnight at 4 °C and then incubated with HRP conjugated secondary antibody (DAKO, Agilent Technologies, Inc., Santa Clara, CA, USA) for 45 min at 37 °C. Finally, the antibody binding was visualized with diaminobenzidine (DAB) solutions, and sections were stained with hematoxylin for 3 min. The results of the staining were scored according to the semiquantitative scoring method. Immunoreactive score (0–12) was equal to the intensity of the staining (0 = not stained, 1 = low intensity, 2 = moderate intensity, 3 = high intensity) multiplied by the percentage of stained cells (0 = 0–5%, 1 = 6–25%, 2 = 26–50%, 3 = 51–75%, 4 = > 75%). The degree of staining was classified according to the final immunoreactive score: negative, 0 (−); weak, 1–4 (+); moderate, 5–8 (++); and strong, 9–12 (+++).
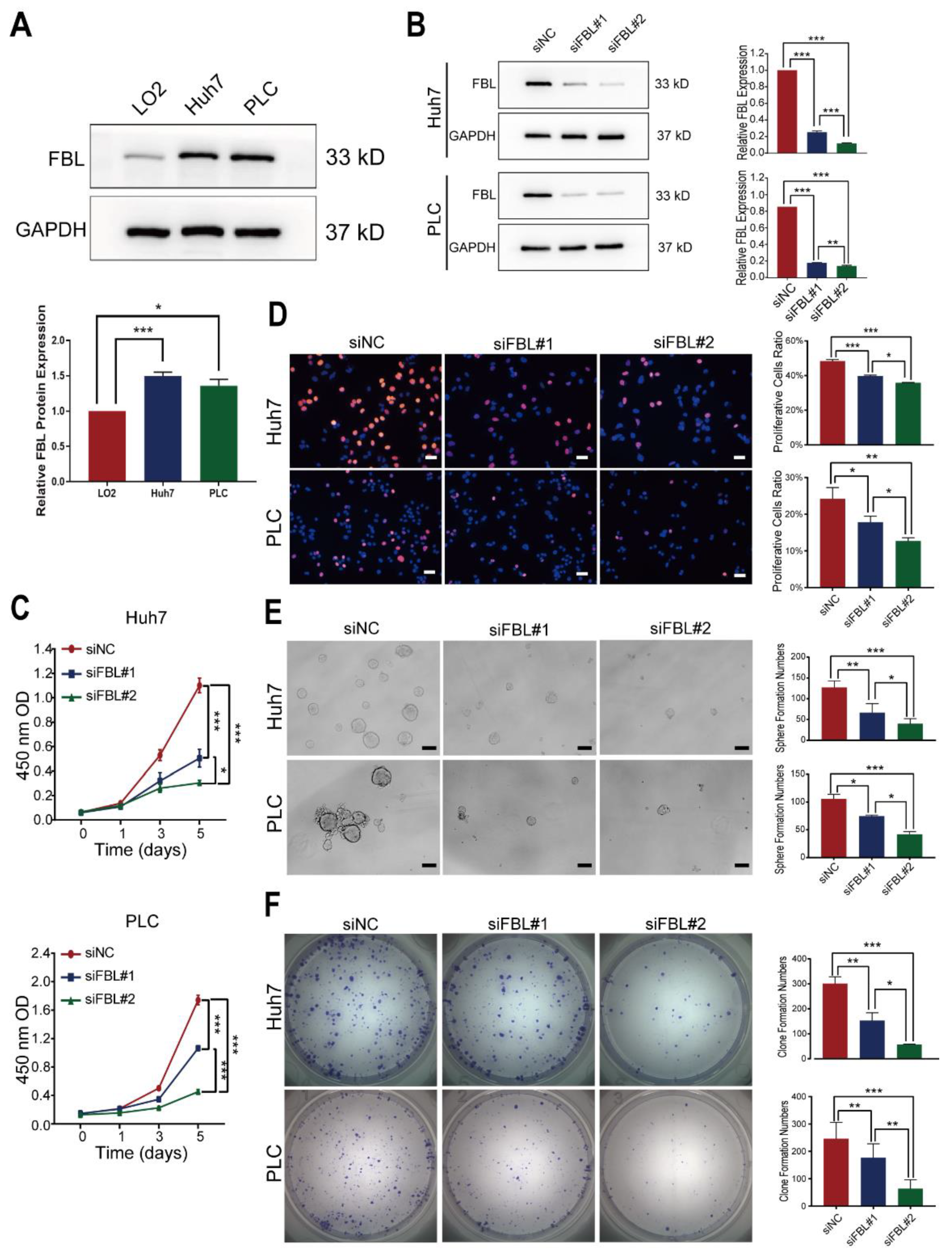
2.12. RNA Interference
FBL siRNA or negative control RNA at a final concentration of 75 nmol/L was transfected into cells with the lipofectamine 3000 transfection kit (Invitrogen, Thermo Fisher Scientific, Carlsbad, CA, USA) in accordance with the manufacturer’s instructions. At 48–72 h after transfection, cells were harvested for further experiments. The sequences of siRNA used were as follows:
Negative control (Sense: 5′-GTCTTCATTTGTCGAGGAAAGGA-3′, Antisense: 5′-CTGGGTGAACTCCAAGGTG-3′);
siFBL#1 (Sense: 5′-GACACUUUGUGAUUUCCAUTT-3′, Antisense: 5′-AUGGAAAUCACAAAGUGUCTT-3′);
siFBL#2 (Sense: 5′-CCTTGAGCCATATGAAAGATT-3′, Antisense: 5′-UCUUUCAUAUGGCUCAAGGTT-3′).
2.13. CCK8 Proliferation Assay
Cells transfected with siRNA for 48 h were seeded into a 96-well plate at a density of 1000 (Huh7) or 1500 (PLC/PRF/5) cells/well. After the planked cells grew adherent to the wall, CCK8 proliferation assay kits (Yeasen Biotechnology Co, Ltd., Pudong New Area, Shanghai, China) were used to test the cell proliferation at different time points (day 0,1,3,5) based on the manufacturer’s instructions. The absorbance at a wavelength of 450 nm was measured on a TS Microplate Reader (Tecan Group Ltd., Seestrasse, Männedorf, Switzerland). These experiments were performed in triplicate.
2.14. EdU Proliferation Assay
A total of 8 × 103 huh7 cells or 1.5 × 104 PLC/PRF/5 cells transfected with siRNA for 72 h were seeded into a 96-well plate. After 24 h, as the normal cell growth cycle was restored, an appropriate EdU solution was added to the 96-well plate with the cells mentioned above. After incubation for 2 h, cells were fixed with 4% paraformaldehyde for 15 min, and the EdU incorporated into the DNA of cells was detected by fluorescent antibodies in accordance with the manufacturer’s instructions. Finally, the proportion of proliferating cells was tested through a High Content Analysis on the ImageXpress Micro Confocal (Molecular Devices, San Jose, CA, USA). These experiments were performed in triplicate.
2.15. Colony Formation Assay
Huh7 or PLC/PRF/5 cells transfected with siRNA for 48 h were seeded into 6-well plates in complete growth medium. Initially, for Huh7, there were 1500 cells per well, and for PLC/PRF/5, there were 5000 cells per well. The culture medium was changed every 3 days. After 12 days, the cells were stained with 0.1% crystal violet after being fixed with 4% paraformaldehyde. The colony number was counted using ELISPOT Reader (AID GmbH, Penzberg, Germany). These experiments were performed in triplicate.
2.16. Sphere Formation Assay
A total of 500 cells transfected with siRNA for 72 h were seeded into low-adhesion 96-well plates in serum-free epithelial basal medium supplemented with B27 (50×) (GIBCO, Life Technologies Corporation, New York, NY, USA), 20 ng/mL EGF (PeproTech Inc., Cranbury, NJ, USA), and 20 ng/mL FGF (PeproTech Inc., Cranbury, NJ, USA) in each well. Fresh microsphere medium was regularly added for supplementary nutrients. Finally, we counted the spheres under a microscope after incubation for 7 days. These experiments were performed in triplicate.
2.17. Scratch Wound Healing Assay
When the cells transfected with siRNA for 72 h in a 6-well plate reached 95% confluence as a monolayer, we gently and slowly scratched the monolayer with a new 200 μL pipette tip across the center of the well to create a cross in each well. After scratching, serum-free medium was replenished into each well. Finally, we took photos of the scratches around the cross in each well under a microscope at 0 h (the time at which the scratching of the monolayer was completed) as well as 48 h later. The wound healing area of each scratch was quantitatively evaluated at different times using ImageJ. These experiments were performed in triplicate.
2.18. Transwell Migration and Invasion Assay
For the Transwell migration assay, 1 × 105 huh7 cells or 2 × 105 PLC/PRF/5 cells transfected with siRNA for 72 h were seeded with serum-free medium in the upper compartments of Transwell inserts (8 μm pore size, Corning, Glendale, AZ, USA), while medium containing 10% FBS was added to the lower bottom compartment. After 24 (huh7) or 36 h (PLC/PRF/5) of incubation, the inserts were taken out. Cells that remained in the upper chamber were gently removed with a cotton swab, and then cells on the lower chamber were fixed with 4% paraformaldehyde for 15 min and stained with 0.1% crystal violet for 20 min. Finally, we counted the number of cells on the lower chamber under a microscope. We randomly chose 5 different views and took the average number. These experiments were performed in triplicate.
For the Transwell invasion assay, 1 × 105 huh7 cells or 2 × 105 PLC/PRF/5 cells transfected with siRNA for 72 h were seeded with serum-free medium in the upper compartment of Transwell inserts (8 μm pore size, Corning, Glendale, AZ, USA) where the diluted Matrigel (diluted ratio = 1:9) was laid in advance, while medium containing 10% FBS was added to the lower bottom compartment. After 36 h of incubation, the inserts were taken out. Cells that remained on the upper chamber were gently removed with a cotton swab, and then cells on the lower chamber were fixed with 4% paraformaldehyde for 15 min and stained with 0.1% crystal violet for 20 min. Finally, the number of cells on the lower chamber was counted under a microscope. We randomly chose 3 different views and took the average number. These experiments were performed in triplicate.
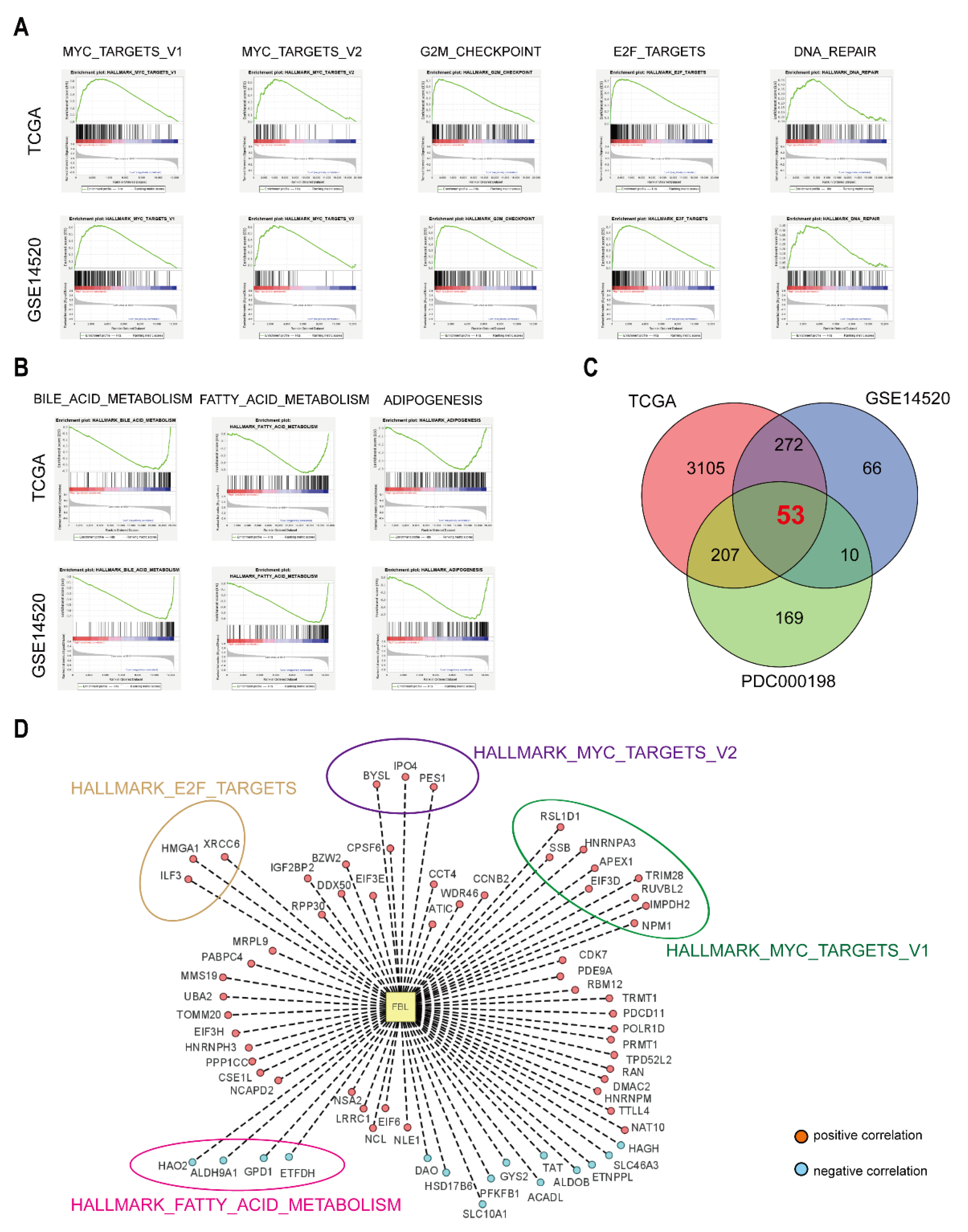
2.19. Gene Set Enrichment Analysis
According to the median expression of FBL in hepatocellular carcinoma tissues in the TCGA and GSE14520 datasets, the samples were divided into high-expressing FBL samples and low-expressing FBL samples. Then, we imported the grouped sample data into the GSEA software (version 4.0.3) and selected “c7.all.v7.1.entrez.gmt” in the Molecular Signatures Database (MSig DB) on the GSEA website as the reference gene set to evaluate the impact of FBL on each reference set [
14]. The default weighted enrichment statistical method was used to perform the Gene Set Enrichment Analysis (GSEA), and the number of random combinations was designed to be 1000. Gene sets that met the criteria of NES ≥ 1, NOM
p-val < 0.05, and FDR q-val < 0.25 were defined as statistically significant enriched gene sets.
2.20. Establishment of the FBL Knockdown Stable Transfectant in Huh7
Huh7 cells were transfected with the FBL knockdown lentivirus and the corresponding negative control lentivirus (IGE Biotechnology Ltd., Guangzhou International Bio Island, Guangzhou, China) at a confluence of 50–70%, respectively. Multiplicity of infection (MOI) was set at 10. The fresh medium was replaced after transfection for 8 h. After 3 days, culture medium with 4 μg/mL puromycin was used to select successfully transfected cells. Finally, Western blotting was used to confirmed the efficacy of the infection 7 days later.
2.21. Xenograft Mouse Model
The 10 male nude mice (6–8 weeks old) were randomly divided into two groups. Then, 5 × 106 Huh7 cells stably transfected with the negative control lentivirus or the FBL knockdown lentivirus were injected subcutaneously into mice with 100 μL PBS-Matrigel mixture (PBS: Matrigel = 1:1) respectively. Tumor sizes and volumes were measured regularly, and the tumor volumes were calculated as follows: tumor volume = width/2 × width × length. After 4–5 weeks, the mice were sacrificed, and the subcutaneous tumors were excised, imaged, and weighted. We strictly followed the guidelines for laboratory animal care of Sun Yat-Sen University, and the following conditions were regarded as the humane end point of this experiment: the maximum diameter of the subcutaneous tumor in nude mice exceeded 1.5 cm, or the weight loss of nude mice was greater than 10%, or there were obvious endangered manifestations in mice (such as significantly reduced food intake, significantly reduced activity, obvious respiratory depression, etc.).
2.22. Statistical Analysis
In this study, SPSS 24.0 was used for the statistical analysis, and the GraphPad Prism 7.0 software, Adobe Illustrator software, and R language were used for mapping. All in vitro experiments were performed in triplicate to confirm the reproducibility of the results obtained. All the experimental data are presented as the mean ± standard error (S.E.M.). Comparisons between two or more groups were statistically analyzed by the Student’s t test, Wilcoxon rank sum test, paired t-test, or random block design analysis of variance depending on the corresponding data type (Student’s t test: Western blotting analysis of FBL protein expression in hepatocellular carcinoma and adjacent liver tissues, tumor growth curve chart comparison, weight of gross tumors comparison; Wilcoxon rank sum test: FBL expression in hepatocellular carcinoma and adjacent liver tissues in TCGA and GEO; paired t-test: RT-qPCR analysis of FBL mRNA expression in hepatocellular carcinoma and adjacent liver tissues; random block design analysis: CCK8 proliferation assay, EdU Proliferation Assay, colony formation assay, sphere formation assay, scratch wound healing assay, Transwell migration and invasion assay between the control and two experimental groups). Four-grid table data were statistically analyzed by the χ2 test. p < 0.05 was considered statistically significant. Log-rank test was used for overall survival and disease-free survival without curved intersection. Peto-Peto’s modified survival estimate test was used for overall survival and disease-free survival with curved intersection. Pearson correlation test was performed to analyze the correlation amongst FBL and other genes.
4. Discussion
Lung metastasis is an important factor leading to poor prognosis in hepatocellular carcinoma patients. In-depth research on the molecular mechanism associated with lung metastasis in hepatocellular carcinoma will help us to predict the prognosis of hepatocellular carcinoma more accurately and provide a scientific basis for its prevention and treatment.
Many previous studies found that the phenomenon of the over-synthesis of ribosomes exists in a variety of tumors, and it plays an important role in the occurrence and development of tumors [
18,
19]. As the “factories” of protein synthesis, ribosomes are mainly composed of ribosomal proteins and ribosomal RNA. Ribosomal proteins are divided into large ribosomal proteins (RPL) and small ribosomal proteins (RPS). Previous studies showed that ribosomal proteins mainly participate in the process of protein synthesis. However, with the deepening of research, researchers have found that ribosomal proteins are also involved in the activation of proto-oncogenes or tumor suppressor genes that regulate the cell cycle through multiple signal pathways to promote the growth and proliferation of cells [
20]. In addition, a variety of ribosomal proteins have been reported to be associated with tumor metastasis. For instance, the high expression of
RPS6 in lung cancer is significantly related to the increased risk of early metastasis [
21]. Knocking down
RPL39 could significantly inhibit the occurrence of lung metastasis of breast cancer in immunodeficiency mouse models, and most breast cancer patients with lung metastasis have acquired mutations of
RPL39 [
22]. Knocking out
RPL15 using CRISPR technology significantly weakened lung metastasis in breast cancer circulating tumor cells in mouse models, and with
RPL15 overexpression, breast cancer circulating tumor cells had stronger lung and ovarian metastasis ability [
23]. However, the tumor metastasis mechanism affected by the ribosomal protein has not been clearly reported in the past research. According to the clues provided in the existing research, there are two types of mechanisms: the first is that a variety of ribosomal proteins could promote the composition and activity of ribosomes, which might affect the translation efficiency of specific mRNA types [
24]; the other is that it might affect the activation or inhibition of signal pathways through the binding of specific proteins, thereby affecting tumor metastasis [
20]. Our study found that ribosomal protein genes account for a large proportion of the modules related to lung metastasis analyzed by WGCNA, such as
RPS15,
RPS16,
RPL18,
RPL35A,
RPLP2,
RPL27A,
RPL24, and
RPS11. These ribosomal proteins are likely to play important roles in lung metastasis associated with hepatocellular carcinoma, but the related mechanisms need further verification through in vivo and in vitro experiments.
Fibrillarin (FBL) is a 2′-O methyltransferase that is involved in ribosomal biogenesis [
25]. It is the main constituent protein of box C/D snoRNP (small nucleolar ribonucleoprotein particles). It is upregulated in prostate cancer, breast cancer, and other tumors, and it can promote tumor cell growth, proliferation, and drug resistance [
26,
27,
28]. Additionally, recent research suggested that increased Fibrillarin expression is related to poor tumor prognosis in hepatocellular carcinoma [
29]. However, few studies have investigated whether
FBL can promote the progression of hepatocellular carcinoma. Our research preliminarily proves that
FBL could affect the proliferation, stemness, migration, and invasion of hepatocellular carcinoma cells, which might be involved in the activation of MYC and E2F signaling. In previous research, it was shown that the tumor suppressor gene
TP53 and the proto-oncogene
MYC are upstream regulators of
FBL [
16,
27]. In breast cancer cells,
TP53 can inhibit the expression of
FBL. In prostate cancer, however, the expression of
FBL can be upregulated by
MYC. As for hepatocellular carcinoma, further study is required to determine which gene plays the main regulatory role in
FBL expression. Regarding the downstream regulation of
FBL, in addition to participating in the regulation of ribosome synthesis, the latest research shows that
FBL could directly modify specific types of mRNA by 2′-O methylation, thereby inhibiting its translation efficiency and achieving the regulation of specific gene expression, which suggests that the effect of
FBL on the biological function of hepatocellular carcinoma may also play a role in this regard [
30]. Besides, our study demonstrated that genes related to MYC and E2F signaling pathway, such as
BYSL,
IPO4,
PES1,
SSB,
HNRNPA3,
RSL1D1,
APEX1,
NPM1,
EIF3D,
RUVBL2,
TRIM28,
IMPDH2,
ILF3,
HMGA1, and
XRCC6, were strongly positively correlated to FBL, which means that they may be the downstream molecules responsible for
FBL-induced lung metastasis.
In general, our research provides ideas for the prediction of the prognosis of hepatocellular carcinoma patients as well as a scientific basis for research on the mechanisms associated with lung metastasis occurrence in hepatocellular carcinoma.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}