
Novel Gene-Correction-Based Therapeutic Modalities for Monogenic Liver Disorders

 ,
,  , ,
, ,  ,
, 
Abstract
1. Introduction
2. Familial Hypercholesterolemia
3. Gaucher Disease
4. Mucopolysaccharidosis
5. Urea Cycle Defects
5.1. Ornithine Transcarbamylase Deficiency
5.2. Citrullinemia Type I
6. Alpha-1-Anti Trypsin Deficiency
7. Tyrosinemia Type I
8. Galactosemia
9. Acute Intermittent Porphyria
10. Hemophilia
10.1. Hemophilia A
10.2. Hemophilia B
11. Phenylketonuria
12. Maple Syrup Urine Disease
13. Progressive Familial Intrahepatic Cholestasis
14. Wilson Disease
15. Glycogen Storage Diseases
16. Crigler–Najjar Syndrome
17. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Maestro, S.; Weber, N.D.; Zabaleta, N.; Aldabe, R.; Gonzalez-Aseguinolaza, G. Novel vectors and approaches for gene therapy in liver diseases. JHEP Rep. 2021, 3, 100300. [Google Scholar] [CrossRef]
- Zabaleta, N.; Hommel, M.; Salas, D.; Gonzalez-Aseguinolaza, G. Genetic-based approaches to inherited metabolic liver diseases. Hum. Gene Ther. 2019, 30, 1190–1203. [Google Scholar] [CrossRef]
- Zabulica, M.; Srinivasan, R.C.; Akcakaya, P.; Allegri, G.; Bestas, B.; Firth, M.; Hammarstedt, C.; Jakobsson, T.; Jakobsson, T.; Ellis, E.; et al. Correction of a urea cycle defect after ex vivo gene editing of human hepatocytes. Mol. Ther. 2021, 29, 1903–1917. [Google Scholar] [CrossRef]
- Heydari, Z.; Zarkesh, I.; Ghanian, M.-H.; Aghdaei, M.H.; Kotova, S.; Zahmatkesh, E.; Farzaneh, Z.; Piryaei, A.; Akbarzadeh, I.; Shpichka, A.; et al. Biofabrication of size-controlled liver microtissues incorporated with ECM-derived microparticles to prolong hepatocyte function. Bio-Des. Manuf. 2021, 4, 790–805. [Google Scholar] [CrossRef]
- Ruoß, M.; Vosough, M.; Königsrainer, A.; Nadalin, S.; Wagner, S.; Sajadian, S.; Huber, D.; Heydari, Z.; Ehnert, S.; Hengstler, J.G.; et al. Towards improved hepatocyte cultures: Progress and limitations. Food Chem. Toxicol. 2020, 138, 111188. [Google Scholar] [CrossRef] [PubMed]
- Vosough, M.; Moslem, M.; Pournasr, B.; Baharvand, H. Cell-based therapeutics for liver disorders. Br. Med. Bull. 2011, 100, 157–172. [Google Scholar] [CrossRef]
- Shahriari Felordi, M.; Torabi, S.; Shokoohian, B.; Farzaneh, Z.; Mohamadnejad, M.; Malekzadeh, R.; Baharvand, H.; Vosough, M. Novel Cell-Based Therapies in Hepatic Disorders. J. Maz. Univ. Med. Sci. 2020, 30, 184–208. [Google Scholar]
- Bilheimer, D.W.; Goldstein, J.L.; Grundy, S.M.; Starzl, T.E.; Brown, M.S. Liver transplantation to provide low-density-lipoprotein receptors and lower plasma cholesterol in a child with homozygous familial hypercholesterolemia. N. Engl. J. Med. 1984, 311, 1658–1664. [Google Scholar] [CrossRef] [PubMed]
- Grossman, M.; Raper, S.E.; Wilson, J.M. Transplantation of genetically modified autologous hepatocytes into nonhuman primates: Feasibility and short-term toxicity. Hum. Gene Ther. 1992, 3, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Ayto, R.M.; Hughes, D.A.; Jeevaratnam, P.; Rolles, K.; Burroughs, A.K.; Mistry, P.K.; Mehta, A.B.; Pastores, G.M. Long-term outcomes of liver transplantation in type 1 Gaucher disease. Am. J. Transplant. 2010, 10, 1934–1939. [Google Scholar] [CrossRef] [PubMed]
- Toyama, S.; Migita, O.; Fujino, M.; Kunieda, T.; Kosuga, M.; Fukuhara, Y.; Nagahara, Y.; Li, X.-K.; Okuyama, T. Liver transplantation: New treatment for mucopolysaccharidosis type VI in rats. Pediatr. Int. 2019, 61, 180–189. [Google Scholar]
- Busuttil, A.A.; Goss, J.A.; Seu, P.; Dulkanchainun, T.S.; Yanni, G.S.; McDiarmid, S.V.; Busuttil, R.W. The role of orthotopic liver transplantation in the treatment of ornithine transcarbamylase deficiency. Liver Transplant. Surg. 1998, 4, 350–354. [Google Scholar] [CrossRef] [PubMed]
- Stéphenne, X.; Najimi, M.; Smets, F.; Reding, R.; De Goyet, J.D.V.; Sokal, E. Cryopreserved liver cell transplantation controls ornithine transcarbamylase deficient patient while awaiting liver transplantation. Am. J. Transplant. 2005, 5, 2058–2061. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Luo, Y.; Xia, L.; Qiu, B.; Zhou, T.; Feng, M.; Wang, C.; Xue, F.; Chen, X.; Han, L.; et al. Outcome of liver transplantation for neonatal-onset citrullinemia type I. Transplantation 2021, 105, 569–576. [Google Scholar] [CrossRef]
- Meyburg, J.; Das, A.M.; Hoerster, F.; Lindner, M.; Kriegbaum, H.; Engelmann, G.; Schmidt, J.; Ott, M.; Pettenazzo, A.; Luecke, T.; et al. One liver for four children: First clinical series of liver cell transplantation for severe neonatal urea cycle defects. Transplantation 2009, 87, 636–641. [Google Scholar] [CrossRef] [PubMed]
- Chandler, R.J.; Tarasenko, T.N.; Cusmano-Ozog, K.; Sun, Q.; Sutton, V.R.; Venditti, C.P.; McGuire, P.J. Liver-directed adeno-associated virus serotype 8 gene transfer rescues a lethal murine model of citrullinemia type 1. Gene Ther. 2013, 20, 1188–1191. [Google Scholar] [CrossRef]
- Hood, J.M.; Koep, L.J.; Peters, R.L.; Schröter, G.P.J.; Weil, R.; Redeker, A.G.; Starzl, T.E. Liver transplantation for advanced liver disease with alpha-1-antitrypsin deficiency. N. Engl. J. Med. 1980, 302, 272. [Google Scholar] [CrossRef] [PubMed]
- Kay, M.A.; Baley, P.; Rothenberg, S.; Leland, F.; Fleming, L.; Ponder, K.P.; Liu, T.; Finegold, M.; Darlington, G.; Pokorny, W. Expression of human alpha 1-antitrypsin in dogs after autologous transplantation of retroviral transduced hepatocytes. Proc. Natl. Acad. Sci. USA 1992, 89, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Freese, D.K.; Tuchman, M.; Schwarzenberg, S.J.; Sharp, H.L.; Rank, J.M.; Bloomer, J.R.; Ascher, N.L.; Payne, W.D. Early liver transplantation is indicated for tyrosinemia type I. J. Pediatr. Gastroenterol. Nutr. 1991, 13, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Ribes-Koninckx, C.; Ibars, E.P.; Agrasot, M.C.; Centelles, A.B.; Miquel, B.P.; Carbó, J.J.V.; Aliaga, E.D.; Pallardó, J.M.; Gómez-Lechón, M.J.; Castell, J.V. Clinical outcome of hepatocyte transplantation in four pediatric patients with inherited metabolic diseases. Cell Transplant. 2012, 21, 2267–2282. [Google Scholar] [CrossRef]
- VanLith, C.; Guthman, R.; Nicolas, C.T.; Allen, K.; Du, Z.; Joo, D.J.; Nyberg, S.L.; Lillegard, J.B.; Hickey, R.D. Curative ex vivo hepatocyte-directed gene editing in a mouse model of hereditary tyrosinemia type 1. Hum. Gene Ther. 2018, 29, 1315–1326. [Google Scholar] [CrossRef] [PubMed]
- Otto, G.; Herfarth, C.H.; Senninger, N.; Feist, G.; Post, S.; Gmelin, K. Hepatic transplantation in galactosemia. Transplantation 1989, 47, 902. [Google Scholar] [CrossRef]
- Rasmussen, S.A.; Daenzer, J.M.; Fridovich-Keil, J.L. A pilot study of neonatal GALT gene replacement using AAV9 dramatically lowers galactose metabolites in blood, liver, and brain and minimizes cataracts in GALT-null rat pups. J. Inherit. Metab. Dis. 2021, 44, 272–281. [Google Scholar] [CrossRef]
- Soonawalla, Z.F.; Orug, T.; Badminton, M.N.; Elder, G.H.; Rhodes, J.M.; Bramhall, S.R.; Elias, E. Liver transplantation as a cure for acute intermittent porphyria. Lancet 2004, 363, 705–706. [Google Scholar] [CrossRef]
- Kurian, C.J.; Drelich, D.A.; Rizk, S. Successful liver transplant from a hemophilia A donor with no development of hemophilia A in recipient. J. Thromb. Haemost. 2020, 18, 853–856. [Google Scholar] [CrossRef]
- Tatsumi, K.; Ohashi, K.; Shima, M.; Nakajima, Y.; Okano, T.; Yoshioka, A. Therapeutic effects of hepatocyte transplantation on hemophilia B. Transplantation 2008, 86, 167–170. [Google Scholar] [CrossRef]
- Ozelo, M.C.; Mahlangu, J.; Pasi, K.J.; Giermasz, A.; Leavitt, A.D.; Laffan, M.; Symington, E.; Quon, D.V.; Wang, J.-D.; Peerlinck, K.; et al. Valoctocogene roxaparvovec gene therapy for hemophilia A. N. Engl. J. Med. 2022, 386, 1013–1025. [Google Scholar] [CrossRef]
- Vajro, P.; Strisciuglio, P.; Houssin, D.; Huault, G.; Laurent, J.; Alvarez, F.; Bernard, O. Correction of phenylketonuria after liver transplantation in a child with cirrhosis. N. Engl. J. Med. 1993, 329, 363. [Google Scholar] [CrossRef]
- Stéphenne, X.; Debray, F.G.; Smets, F.; Jazouli, N.; Sana, G.; Tondreau, T.; Menten, R.; Goffette, P.; Boemer, F.; Schoos, R.; et al. Hepatocyte transplantation using the domino concept in a child with tetrabiopterin nonresponsive phenylketonuria. Cell Transplant. 2012, 21, 2765–2770. [Google Scholar] [CrossRef]
- Wendel, U.; Saudubray, J.M.; Bodner, A.; Schadewaldt, P. Liver transplantation in maple syrup urine disease. Eur. J. Pediatr. 1999, 158, S060–S064. [Google Scholar] [CrossRef]
- Skvorak, K.J.; Hager, E.J.; Arning, E.; Bottiglieri, T.; Paul, H.S.; Strom, S.; Homanics, G.; Sun, Q.; Jansen, E.E.; Jakobs, C.; et al. Hepatocyte transplantation (HTx) corrects selected neurometabolic abnormalities in murine intermediate maple syrup urine disease (iMSUD). Biochim. Biophys. Acta-Mol. Basis Dis. 2009, 1792, 1004–1010. [Google Scholar] [CrossRef] [PubMed]
- Aydogdu, S.; Cakir, M.; Arikan, C.; Tumgor, G.; Yuksekkaya, H.A.; Yilmaz, F.; Kilic, M. Liver transplantation for progressive familial intrahepatic cholestasis: Clinical and histopathological findings, outcome and impact on growth. Pediatr. Transplant. 2007, 11, 634–640. [Google Scholar] [CrossRef]
- De Vree, J.M.L.; Ottenhoff, R.; Bosma, P.J.; Smith, A.J.; Aten, J.; Elferink, R.P.J.O. Correction of liver disease by hepatocyte transplantation in a mouse model of progressive familial intrahepatic cholestasis. Gastroenterology 2000, 119, 1720–1730. [Google Scholar] [CrossRef] [PubMed]
- Weber, N.D.; Odriozola, L.; Martínez-García, J.; Ferrer, V.; Douar, A.; Bénichou, B.; González-Aseguinolaza, G.; Smerdou, C. Gene therapy for progressive familial intrahepatic cholestasis type 3 in a clinically relevant mouse model. Nat. Commun. 2019, 10, 5694. [Google Scholar] [CrossRef] [PubMed]
- Bellary, S.; Hassanein, T.; van Thiel, D.H. Liver transplantation for Wilson’s disease. J. Hepatol. 1995, 23, 373–381. [Google Scholar] [CrossRef]
- Allen, K.J.; Cheah, D.M.; Wright, P.F.; Gazeas, S.; Pettigrew-Buck, N.E.; Deal, Y.H.; Mercer, J.F.; Williamson, R. Liver cell transplantation leads to repopulation and functional correction in a mouse model of Wilson’s disease. J. Gastroenterol. Hepatol. 2004, 19, 1283–1290. [Google Scholar] [CrossRef]
- Li, X. Liver transplantation for glycogen storage diseases. In Transplantation 2018; Lippincott Williams Wilkins: Philadelphia, PA, USA, 2001. [Google Scholar]
- Malhi, H.; Irani, A.N.; Volenberg, I.; Schilsky, M.L.; Gupta, S. Early cell transplantation in LEC rats modeling Wilson’s disease eliminates hepatic copper with reversal of liver disease. Gastroenterology 2002, 122, 438–447. [Google Scholar] [CrossRef]
- Rela, M.; Muiesan, P.; Vilca-Melendez, H.; Dhawan, A.; Baker, A.; Mieli-Vergani, G.; Heaton, N.D. Auxiliary partial orthotopic liver transplantation for Crigler-Najjar syndrome type I. Ann. Surg. 1999, 229, 565. [Google Scholar] [CrossRef]
- Ambrosino, G.; Varotto, S.; Strom, S.; Guariso, G.; Franchin, E.; Miotto, D.; Caenazzo, L.; Basso, S.M.M.; Carraro, P.; Valente, M.; et al. Isolated hepatocyte transplantation for Crigler-Najjar syndrome type 1. Cell Transplant. 2005, 14, 151–157. [Google Scholar] [CrossRef]
- Kay, M.A.; Woo, S.L. Gene therapy for metabolic diseases. ILAR J. 1994, 36, 47–53. [Google Scholar] [CrossRef][Green Version]
- Baruteau, J.; Waddington, S.; Alexander, I.E.; Gissen, P. Gene therapy for monogenic liver diseases: Clinical successes, current challenges and future prospects. J. Inherit. Metab. Dis. 2017, 40, 497–517. [Google Scholar] [CrossRef] [PubMed]
- Aravalli, R.N.; Belcher, J.D.; Steer, C.J. Liver-targeted gene therapy: Approaches and challenges. Liver Transplant. 2015, 21, 718–737. [Google Scholar] [CrossRef]
- Ramamoorth, M.; Narvekar, A. Non viral vectors in gene therapy-an overview. J. Clin. Diagn. Res. JCDR 2015, 9, GE01. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhao, P.; Zhang, Y.; Wang, J.; Wang, C.; Liu, Y.; Yang, G.; Yuan, L. Exosome-based Ldlr gene therapy for familial hypercholesterolemia in a mouse model. Theranostics 2021, 11, 2953. [Google Scholar] [CrossRef] [PubMed]
- Senís, E.; Fatouros, C.; Große, S.; Wiedtke, E.; Niopek, D.; Mueller, A.-K.; Börner, K.; Grimm, D. CRISPR/Cas9-mediated genome engineering: An adeno-associated viral (AAV) vector toolbox. Biotechnol. J. 2014, 9, 1402–1412. [Google Scholar] [CrossRef]
- Arslan, A.; Şimşek, Ö.; Turhan, A.; Çarlioğlu, A.; Arikan Durmaz, Ş.; Utlu, M.; Kartal, E. Non-alcoholic fatty liver disease in patients with familial hypercholesterolemia. Ortadoğu Tıp Derg. 2020, 12, 219–224. [Google Scholar] [CrossRef]
- Al-Allaf, F.A.; Coutelle, C.; Waddington, S.N.; David, A.L.; Harbottle, R.; Themis, M. LDLR-Gene therapy for familial hypercholesterolaemia: Problems, progress, and perspectives. Int. Arch. Med. 2010, 3, 36. [Google Scholar] [CrossRef]
- Rodriguez-Calvo, R.; Masana, L. Review of the scientific evolution of gene therapy for the treatment of homozygous familial hypercholesterolaemia: Past, present and future perspectives. J. Med. Genet. 2019, 56, 711–717. [Google Scholar] [CrossRef]
- Mlinaric, M.; Bratanic, N.; Dragos, V.; Skarlovnik, A.; Cevc, M.; Battelino, T.; Groselj, U. Case Report: Liver Transplantation in Homozygous Familial Hypercholesterolemia (HoFH)—Long-Term Follow-up of a Patient and Literature Review. Front. Pediatr. 2020, 8, 567895. [Google Scholar] [CrossRef]
- Kassim, S.H.; Li, H.; Vandenberghe, L.H.; Hinderer, C.; Bell, P.; Marchadier, D.; Wilson, A.; Cromley, D.; Redon, V.; Yu, H.; et al. Gene therapy in a humanized mouse model of familial hypercholesterolemia leads to marked regression of atherosclerosis. PLoS ONE 2010, 5, e13424. [Google Scholar] [CrossRef]
- Chowdhury, J.R.; Grossman, M.; Gupta, S.; Baker, J.R.; Wilson, J.M. Long-term improvement of hypercholesterolemia after ex vivo gene therapy in LDLR-deficient rabbits. Sci. Adv. 1991, 254, 1802–1805. [Google Scholar] [CrossRef] [PubMed]
- Tomita, N.; Morishita, R.; Koike, H.; Hashizume, M.; Notake, M.; Fujitani, B.; Kaneda, Y.; Horiuchi, M.; Ogihara, T. Therapeutic approach to familial hypercholesterolemia by HVJ-liposomes in LDL receptor knockout mouse. Int. J. Mol. Med. 2002, 10, 137–143. [Google Scholar] [CrossRef]
- Bissig-Choisat, B.; Wang, L.; Legras, X.; Saha, P.K.; Chen, L.; Bell, P.; Pankowicz, F.P.; Hill, M.C.; Barzi, M.; Leyton, C.K.; et al. Development and rescue of human familial hypercholesterolaemia in a xenograft mouse model. Nat. Commun. 2015, 6, 7339. [Google Scholar] [CrossRef] [PubMed]
- Rader, D.J.; Kastelein, J.J. Lomitapide and mipomersen: Two first-in-class drugs for reducing low-density lipoprotein cholesterol in patients with homozygous familial hypercholesterolemia. Circulation 2014, 129, 1022–1032. [Google Scholar] [CrossRef]
- Ray, K.K.; Wright, R.S.; Kallend, D.; Koenig, W.; Leiter, L.A.; Raal, F.J.; Bisch, J.A.; Richardson, T.; Jaros, M.; Wijngaard, P.L.; et al. Two phase 3 trials of inclisiran in patients with elevated LDL cholesterol. N. Engl. J. Med. 2020, 382, 1507–1519. [Google Scholar] [CrossRef]
- Caron, J.; Pène, V.; Tolosa, L.; Villaret, M.; Luce, E.; Fourrier, A.; Heslan, J.-M.; Saheb, S.; Bruckert, E.; Gómez-Lechón, M.J.; et al. Low-density lipoprotein receptor-deficient hepatocytes differentiated from induced pluripotent stem cells allow familial hypercholesterolemia modeling, CRISPR/Cas-mediated genetic correction, and productive hepatitis C virus infection. Stem Cell Res. Ther. 2019, 10, 221. [Google Scholar] [CrossRef]
- Hong, Y.B.; Kim, E.Y.; Yoo, H.-W.; Jung, S.-C. Feasibility of gene therapy in Gaucher disease using an adeno-associated virus vector. J. Hum. Genet. 2004, 49, 536–543. [Google Scholar] [CrossRef][Green Version]
- Dahl, M.; Doyle, A.; Olsson, K.; Månsson, J.-E.; A Marques, A.R.; Mirzaian, M.; Aerts, J.M.; Ehinger, M.; Rothe, M.; Modlich, U.; et al. Lentiviral gene therapy using cellular promoters cures type 1 Gaucher disease in mice. Mol. Ther. 2015, 23, 835–844. [Google Scholar] [CrossRef]
- Nalysnyk, L.; Rotella, P.; Simeone, J.C.; Hamed, A.; Weinreb, N. Gaucher disease epidemiology and natural history: A comprehensive review of the literature. Hematology 2017, 22, 65–73. [Google Scholar] [CrossRef]
- Enquist, I.B.; Nilsson, E.; Ooka, A.; Månsson, J.-E.; Olsson, K.; Ehinger, M.; Brady, R.O.; Richter, J.; Karlsson, S. Effective cell and gene therapy in a murine model of Gaucher disease. Proc. Natl. Acad. Sci. USA 2006, 103, 13819–13824. [Google Scholar] [CrossRef] [PubMed]
- Kohn, D.B.; Nolta, J.A.; Weinthal, J.; Bahner, I.; Yu, X.J.; Lilley, J.; Crooks, G.M. Toward gene therapy for Gaucher disease. Hum. Gene Ther. 1991, 2, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Massaro, G.; Hughes, M.P.; Whaler, S.M.; Wallom, K.-L.; Priestman, D.A.; Platt, F.; Waddington, S.N.; Rahim, A.A. Systemic AAV9 gene therapy using the synapsin I promoter rescues a mouse model of neuronopathic Gaucher disease but with limited cross-correction potential to astrocytes. Hum. Mol. Genet. 2020, 29, 1933–1949. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Cui, Y.; Wang, J.; Shi, L.; Qi, Z.; Luan, J.; Zhang, Y.; Zhou, X.; Han, J. Development of a human iPSC line (SMBCi004-A) from a patient with Gaucher disease. Stem Cell Res. Ther. 2020, 48, 101989. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Font, A.; Chabás, A.; Grinberg, D.; Vilageliu, L. RNAi-mediated inhibition of the glucosylceramide synthase (GCS) gene: A preliminary study towards a therapeutic strategy for Gaucher disease and other glycosphingolipid storage diseases. Blood Cells Mol. Dis. 2006, 37, 197–203. [Google Scholar] [CrossRef]
- Sawamoto, K.; Chen, H.-H.; Alméciga-Díaz, C.; Mason, R.W.; Tomatsu, S. Gene therapy for Mucopolysaccharidoses. Mol. Genet. Metab. 2018, 123, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Puckett, Y.; Mallorga-Hernández, A.; Montaño, A.M. Epidemiology of mucopolysaccharidoses (MPS) in United States: Challenges and opportunities. Orphanet J. Rare Dis. 2021, 16, 241. [Google Scholar] [CrossRef]
- Visigalli, I.; Delai, S.; Politi, L.S.; Di Domenico, C.; Cerri, F.; Mrak, E.; D’Isa, R.; Ungaro, D.; Stok, M.; Sanvito, F.; et al. Gene therapy augments the efficacy of hematopoietic cell transplantation and fully corrects mucopolysaccharidosis type I phenotype in the mouse model. Blood J. Am. Soc. Hematol. 2010, 116, 5130–5139. [Google Scholar] [CrossRef] [PubMed]
- Domenico, C.D.; Villani, G.R.D.; Di Napoli, D.; Reyero, E.G.Y.; Lombardo, A.; Naldini, L.; Di Natale, P. Gene therapy for a mucopolysaccharidosis type I murine model with lentiviral-IDUA vector. Hum. Gene Ther. 2005, 16, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Sawamoto, K.; Karumuthil-Melethil, S.; Khan, S.; Stapleton, M.; Bruder, J.T.; Danos, O.; Tomatsu, S. Liver-targeted AAV8 gene therapy ameliorates skeletal and cardiovascular pathology in a Mucopolysaccharidosis IVA murine model. Mol. Ther. -Methods Clin. Dev. 2020, 18, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Ou, L.; DeKelver, R.C.; Rohde, M.; Tom, S.; Radeke, R.; St. Martin, S.J.; Santiago, Y.; Sproul, S.; Przybilla, M.J.; Koniar, B.L.; et al. ZFN-mediated in vivo genome editing corrects murine hurler syndrome. Mol. Ther. 2019, 27, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Muenzer, J.; Prada, C.E.; Burton, B.; Lau, H.A.; Ficicioglu, C.; Foo, C.W.P.; Vaidya, S.A.; Whitley, C.B.; Harmatz, P. CHAMPIONS: A phase 1/2 clinical trial with dose escalation of SB-913 ZFN-mediated in vivo human genome editing for treatment of MPS II (Hunter syndrome). Mol. Genet. Metab. 2019, 126, S104. [Google Scholar] [CrossRef]
- de Carvalho, T.G.; Schuh, R.; Pasqualim, G.; Pellenz, F.M.; Filippi-Chiela, E.C.; Giugliani, R.; Baldo, G.; Matte, U. CRISPR-Cas9-mediated gene editing in human MPS I fibroblasts. Gene Ther. 2018, 678, 33–37. [Google Scholar] [CrossRef]
- Ou, L.; Przybilla, M.J.; Ahlat, O.; Kim, S.; Overn, P.; Jarnes, J.; O’Sullivan, M.G.; Whitley, C.B. A highly efficacious PS gene editing system corrects metabolic and neurological complications of mucopolysaccharidosis type I. Mol. Ther. 2020, 28, 1442–1454. [Google Scholar] [CrossRef]
- Gomez-Ospina, N.; Scharenberg, S.G.; Mostrel, N.; Bak, R.O.; Mantri, S.; Quadros, R.M.; Gurumurthy, C.B.; Lee, C.; Bao, G.; Suarez, C.J.; et al. Human genome-edited hematopoietic stem cells phenotypically correct Mucopolysaccharidosis type I. Nat. Commun. 2019, 10, 4045. [Google Scholar] [CrossRef] [PubMed]
- Leal, A.F.; Alméciga-Díaz, C.J. Efficient CRISPR/Cas9 nickase-mediated genome editing in an in vitro model of mucopolysaccharidosis IVA. Gene Ther. 2022, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Miki, T.; Vazquez, L.; Yanuaria, L.; Lopez, O.; Garcia, I.M.; Ohashi, K.; Rodriguez, N.S. Induced pluripotent stem cell derivation and ex vivo gene correction using a mucopolysaccharidosis type 1 disease mouse model. Stem Cells Int. 2019, 2019, 6978303. [Google Scholar] [CrossRef]
- Sriramoju, M.K.; Yang, T.-J.; Hsu, S.-T.D. Comparative folding analyses of unknotted versus trefoil-knotted ornithine transcarbamylases suggest stabilizing effects of protein knots. Biochem. Biophys. Res. Commun. 2018, 503, 822–829. [Google Scholar] [CrossRef] [PubMed]
- Lichter-Konecki, U.; Caldovic, L.; Morizono, H.; Simpson, K.; Mew, A.; MacLeod, E. Ornithine Transcarbamylase Deficiency; University of Washington: Seattle, WA, USA, 2021. [Google Scholar]
- Brusilow, S.W.; Maestri, N.E. Urea cycle disorders: Diagnosis, pathophysiology, and therapy. Adv. Pediatr. 1996, 43, 127–170. [Google Scholar] [PubMed]
- Guan, J.; Yan, B.; Zhang, H.; Liu, C.; Li, Y.; Yang, X.; Li, Z.; Gai, Z.; Liu, Y. Generation of a human induced pluripotent stem cell line (SDQLCHi036-A) from a patient with ornithine transcarbamylase deficiency carrying a deletion involving 3–9 exons of OTC gene. Stem Cell Res. Ther. 2021, 52, 102220. [Google Scholar] [CrossRef] [PubMed]
- Prieve, M.G.; Harvie, P.; Monahan, S.D.; Roy, D.; Li, A.G.; Blevins, T.L.; Paschal, A.E.; Waldheim, M.; Bell, E.C.; Galperin, A.; et al. Targeted mRNA therapy for ornithine transcarbamylase deficiency. Mol. Ther. 2018, 26, 801–813. [Google Scholar] [CrossRef]
- Wang, L.; Yang, Y.; Breton, C.; Bell, P.; Li, M.; Zhang, J.; Che, Y.; Saveliev, A.; He, Z.; White, J.; et al. A mutation-independent CRISPR-Cas9—Mediated gene targeting approach to treat a murine model of ornithine transcarbamylase deficiency. Sci. Adv. 2020, 6, eaax5701. [Google Scholar] [CrossRef]
- Zabulica, M.; Jakobsson, T.; Ravaioli, F.; Vosough, M.; Gramignoli, R.; Ellis, E.; Rooyackers, O.; Strom, S. Gene editing correction of a urea cycle defect in organoid stem cell derived hepatocyte-like cells. Int. J. Mol. Sci. 2021, 22, 1217. [Google Scholar] [CrossRef] [PubMed]
- Paunovska, K.; Loughrey, D.; Dahlman, J.E. Drug delivery systems for RNA therapeutics. Nat. Rev. Genet. 2022, 23, 265–280. [Google Scholar] [CrossRef]
- Wang, L.; Warzecha, C.C.; Kistner, A.; Chichester, J.A.; Bell, P.; Buza, E.L.; He, Z.; Pampena, M.B.; Couthouis, J.; Sethi, S.; et al. Prednisolone reduces the interferon response to AAV in cynomolgus macaques and may increase liver gene expression [113/120 characters]. Mol. Ther.-Methods Clin. Dev. 2022, 24, 292–305. [Google Scholar] [CrossRef] [PubMed]
- Baruteau, J.; Cunningham, S.C.; Yilmaz, B.S.; Perocheau, D.P.; Eaglestone, S.; Burke, D.; Thrasher, A.J.; Waddington, S.N.; Lisowski, L.; Alexander, I.E.; et al. Safety and efficacy of an engineered hepatotropic AAV gene therapy for ornithine transcarbamylase deficiency in cynomolgus monkeys. Mol. Ther.-Methods Clin. Dev. 2021, 23, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Zielonka, M.; Kölker, S.; Gleich, F.; Stützenberger, N.; Nagamani, S.C.S.; Gropman, A.L.; Hoffmann, G.F.; Garbade, S.F.; Posset, R.; Sarajlija, A.; et al. Early prediction of phenotypic severity in Citrullinemia Type 1. Ann. Clin. Transl. Neurol. 2019, 6, 1858–1871. [Google Scholar] [CrossRef] [PubMed]
- Laróvere, L.E.; Ruiz, S.M.S.; Angaroni, C.J.; De Kremer, R.D. Molecular epidemiology of Citrullinemia type I in a risk region of Argentina: A first step to preconception heterozygote detection. In JIMD Reports-Case and Research Reports, 2012/3; Springer: Berlin/Heidelberg, Germany, 2012; pp. 27–29. [Google Scholar]
- Quinonez, S.C.; Thoene, J.G. Citrullinemia Type I; Book from University of Washington: Seattle, WA, USA, 2016. [Google Scholar]
- Janwadkar, A.; Shirole, N.; Nagral, A.; Bakshi, R.; Vasanth, S.; Bagde, A.; Yewale, V.; Mirza, D. Citrullinemia type 1: Behavioral improvement with late liver transplantation. Indian J. Pediatr. 2019, 86, 639–641. [Google Scholar] [CrossRef] [PubMed]
- Yoshitoshi-Uebayashi, E.Y.; Toyoda, T.; Yasuda, K.; Kotaka, M.; Nomoto, K.; Okita, K.; Yasuchika, K.; Okamoto, S.; Takubo, N.; Nishikubo, T.; et al. Modelling urea-cycle disorder citrullinemia type 1 with disease-specific iPSCs. Biochem. Biophys. Res. Commun. 2017, 486, 613–619. [Google Scholar] [CrossRef]
- Demarquoy, J.J.E. Retroviral-mediated gene therapy for the treatment of citrullinemia. Transfer and expression of argininosuccinate synthetase in human hematopoietic cells. Experientia 1993, 49, 345–348. [Google Scholar] [CrossRef]
- Patejunas, G.; Bradley, A.; Beaudet, A.L.; O’Brien, W.E. Generation of a mouse model for citrullinemia by targeted disruption of the argininosuccinate synthetase gene. Somat. Cell Mol. Genet. 1994, 20, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Long, G.L.; Chandra, T.; Woo, S.L.C.; Davie, E.W.; Kurachi, K. Complete sequence of the cDNA for human. alpha. 1-antitrypsin and the gene for the S variant. Biochemistry 1984, 23, 4828–4837. [Google Scholar] [CrossRef] [PubMed]
- Leon, C.; Bouchecareilh, M. The autophagy pathway: A critical route in the disposal of alpha 1-antitrypsin aggregates that holds many mysteries. Int. J. Mol. Sci. 2021, 22, 1875. [Google Scholar] [CrossRef]
- Greene, C.M.; McElvaney, N.G. Z α-1 antitrypsin deficiency and the endoplasmic reticulum stress response. World J. Gastrointest. Pharmacol. Ther. 2010, 1, 94. [Google Scholar] [CrossRef]
- Jančiauskienė, S.M.; Nitaa, I.M.; Subramaniyam, D.; Li, Q.; Lancaster, J.R.; Matalon, S. Alpha1-antitrypsin inhibits the activity of the matriptase catalytic domain in vitro. Am. J. Respir. Cell Mol. Biol. 2008, 39, 2008. [Google Scholar] [CrossRef] [PubMed]
- He, N.; Liu, X.; Vegter, A.R.; Evans, T.I.A.; Gray, J.S.; Guo, J.; Moll, S.R.; Guo, L.J.; Luo, M.; Ma, N.; et al. Ferret models of alpha-1 antitrypsin deficiency develop lung and liver disease. JCI Insight 2022, 7, e143004. [Google Scholar] [CrossRef] [PubMed]
- Stoller, J.K.; Aboussouan, L.S. α1-antitrypsin deficiency. Lancet 2005, 365, 2225–2236. [Google Scholar] [CrossRef]
- Sark, A.D.; Fromme, M.; Olejnicka, B.; Welte, T.; Strnad, P.; Janciauskiene, S.; Stolk, J. The Relationship between Plasma Alpha-1-Antitrypsin Polymers and Lung or Liver Function in ZZ Alpha-1-Antitrypsin-Deficient Patients. Biomolecules 2022, 12, 380. [Google Scholar] [CrossRef]
- Mueller, C.; Gernoux, G.; Gruntman, A.M.; Borel, F.; Reeves, E.P.; Calcedo, R.; Rouhani, F.N.; Yachnis, A.; Humphries, M.; Campbell-Thompson, M.; et al. 5 Year expression and neutrophil defect repair after gene therapy in alpha-1 antitrypsin deficiency. Mol. Ther. 2017, 25, 1387–1394. [Google Scholar] [CrossRef]
- Wooddell, C.I.; Blomenkamp, K.; Peterson, R.M.; Subbotin, V.M.; Schwabe, C.; Hamilton, J.; Chu, Q.; Christianson, D.R.; Hegge, J.O.; Kolbe, J.; et al. Development of an RNAi therapeutic for alpha-1-antitrypsin liver disease. JCI Insight 2020, 5, e135348. [Google Scholar] [CrossRef]
- Guo, S.; Booten, S.L.; Aghajan, M.; Hung, G.; Zhao, C.; Blomenkamp, K.; Gattis, D.; Watt, A.; Freier, S.M.; Teckman, J.H.; et al. Antisense oligonucleotide treatment ameliorates alpha-1 antitrypsin–related liver disease in mice. J. Clin. Investig. 2014, 124, 251–261. [Google Scholar] [CrossRef]
- Attaran, M.; Schneider, A.; Grote, C.; Zwiens, C.; Flemming, P.; Gratz, K.F.; Jochheim, A.; Bahr, M.J.; Manns, M.P.; Ott, M. Regional and transient ischemia/reperfusion injury in the liver improves therapeutic efficacy of allogeneic intraportal hepatocyte transplantation in low-density lipoprotein receptor deficient Watanabe rabbits. J. Hepatol. 2004, 41, 837–844. [Google Scholar] [CrossRef]
- Bjursell, M.; Porritt, M.J.; Ericson, E.; Taheri-Ghahfarokhi, A.; Clausen, M.; Magnusson, L.; Admyre, T.; Nitsch, R.; Mayr, L.; Aasehaug, L.; et al. Therapeutic genome editing with CRISPR/Cas9 in a humanized mouse model ameliorates α1-antitrypsin deficiency phenotype. EBioMedicine 2018, 29, 104–111. [Google Scholar] [CrossRef]
- Werder, R.B.; Kaserman, J.E.; Packer, M.S.; Lindstrom-Vautrin, J.; Villacorta-Martin, C.; Young, L.E.; Aratyn-Schaus, Y.; Gregoire, F.; Wilson, A.A. Adenine base editing reduces misfolded protein accumulation and toxicity in alpha-1 antitrypsin deficient patient iPSC-hepatocytes. Mol. Ther. 2021, 29, 3219–3229. [Google Scholar] [CrossRef]
- Shen, S.; Sanchez, M.E.; Blomenkamp, K.; Corcoran, E.M.; Marco, E.; Yudkoff, C.; Jiang, H.; Teckman, J.H.; Bumcrot, D.; Albright, C.F. Amelioration of alpha-1 antitrypsin deficiency diseases with genome editing in transgenic mice. Hum. Gene Ther. 2018, 29, 861–873. [Google Scholar] [CrossRef]
- Chiuchiolo, M.J.; Kaminsky, S.M.; Sondhi, D.; Hackett, N.R.; Rosenberg, J.B.; Frenk, E.Z.; Hwang, Y.; Van de Graaf, B.G.; Hutt, J.A.; Wang, G.; et al. Intrapleural administration of an AAVrh. 10 vector coding for human α1-antitrypsin for the treatment of α1-antitrypsin deficiency. Hum. Gene Ther. Clin. Dev. 2013, 24, 161–173. [Google Scholar] [CrossRef]
- Janosz, E.; Hetzel, M.; Spielmann, H.; Tumpara, S.; Rossdam, C.; Schwabbauer, M.; Kloos, D.; von Kaisenberg, C.; Schambach, A.; Buettner, F.F.R.; et al. Pulmonary transplantation of alpha-1 antitrypsin (AAT)-transgenic macrophages provides a source of functional human AAT in vivo. Gene Ther. 2021, 28, 477–493. [Google Scholar] [CrossRef]
- Sosulski, M.L.; Stiles, K.M.; Frenk, E.Z.; Hart, F.M.; Matsumura, Y.; De, B.P.; Kaminsky, S.M.; Crystal, R.G. Gene therapy for alpha 1-antitrypsin deficiency with an oxidant-resistant human alpha 1-antitrypsin. JCI Insight 2020, 5, e135951. [Google Scholar] [CrossRef]
- Packer, M.S.; Chowdhary, V.; Lung, G.; Cheng, L.-I.; Aratyn-Schaus, Y.; Leboeuf, D.; Smith, S.; Shah, A.; Chen, D.; Zieger, M.; et al. Evaluation of cytosine base editing and adenine base editing as a potential treatment for alpha-1 antitrypsin deficiency. Mol. Ther. 2022, 30, 1396–1406. [Google Scholar] [CrossRef]
- Scriver, C.R. The Metabolic Molecular Bases of Inherited Disease; McGraw-Hill: New York, NY, USA; Montreal, QC, Canada, 2001; Volume 4. [Google Scholar]
- Morrow, G.; Tanguay, R.M. Biochemical and clinical aspects of hereditary tyrosinemia type 1. Hered. Tyrosinemia 2017, 9–21. [Google Scholar] [CrossRef]
- Endo, F.; Sun, M.-S. Tyrosinaemia type I and apoptosis of hepatocytes and renal tubular cells. J. Inherit. Metab. Dis. 2002, 25, 227–234. [Google Scholar] [CrossRef]
- Peng, M.-Z.; Li, X.-Z.; Mei, H.-F.; Sheng, H.-Y.; Yin, X.; Jiang, M.-Y.; Cai, Y.-N.; Su, L.; Lin, Y.-T.; Shao, Y.-X.; et al. Clinical and biochemical characteristics of patients with ornithine transcarbamylase deficiency. Clin. Biochem. 2020, 84, 63–72. [Google Scholar] [CrossRef]
- Lindstedt, S.; Holme, E.; Lock, E.A.; Hjalmarson, O.; Strandvik, B. Treatment of hereditary tyrosinaemia type I by inhibition of 4-hydroxyphenylpyruvate dioxygenase. Lancet 1992, 340, 813–817. [Google Scholar] [CrossRef]
- van Spronsen, F.J.; Bijleveld, C.M.A.; van Maldegem, B.T.; Wijburg, F.A. Hepatocellular carcinoma in hereditary tyrosinemia type I despite 2-(2 nitro-4-3 trifluoro-methylbenzoyl)-1, 3-cyclohexanedione treatment. J. Pediatr. Gastroenterol. Nutr. 2005, 40, 90–93. [Google Scholar] [CrossRef]
- Thompson, W.S.; Mondal, G.; Vanlith, C.J.; Kaiser, R.A.; Lillegard, J.B. The future of gene-targeted therapy for hereditary tyrosinemia type 1 as a lead indication among the inborn errors of metabolism. Expert Opin. Orphan Drugs 2020, 8, 245–256. [Google Scholar] [CrossRef]
- Hickey, R.D.; Nicolas, C.T.; Allen, K.; Mao, S.; Elgilani, F.; Glorioso, J.; Amiot, B.; VanLith, C.; Guthman, R.; Du, Z.; et al. Autologous gene and cell therapy provides safe and long-term curative therapy in a large pig model of hereditary tyrosinemia type 1. Cell Transplant. 2019, 28, 79–88. [Google Scholar] [CrossRef]
- Zhang, Q.-S.; Tiyaboonchai, A.; Nygaard, S.; Baradar, K.; Major, A.; Balaji, N.; Grompe, M. Induced liver regeneration enhances CRISPR/Cas9-mediated gene repair in tyrosinemia type 1. Hum. Gene Ther. 2021, 32, 294–301. [Google Scholar] [CrossRef]
- Cheng, Q.; Wei, T.; Jia, Y.; Farbiak, L.; Zhou, K.; Zhang, S.; Wei, Y.; Zhu, H.; Siegwart, D.J. Dendrimer-based lipid nanoparticles deliver therapeutic FAH mRNA to normalize liver function and extend survival in a mouse model of hepatorenal tyrosinemia type I. Adv. Mater. 2018, 30, 1805308. [Google Scholar] [CrossRef]
- Li, N.; Gou, S.; Wang, J.; Zhang, Q.; Huang, X.; Xie, J.; Li, L.; Jin, Q.; Ouyang, Z.; Chen, F.; et al. CRISPR/Cas9-mediated gene correction in newborn rabbits with hereditary tyrosinemia type I. Mol. Ther. 2021, 29, 1001–1015. [Google Scholar] [CrossRef]
- Zafra, M.P.; Schatoff, E.M.; Katti, A.; Foronda, M.; Breinig, M.; Schweitzer, A.Y.; Simon, A.; Han, T.; Goswami, S.; Montgomery, E.; et al. Optimized base editors enable efficient editing in cells, organoids and mice. Nat. Biotechnol. 2018, 36, 888–893. [Google Scholar] [CrossRef]
- Koblan, L.W.; Doman, J.L.; Wilson, C.; Levy, J.M.; Tay, T.; A Newby, G.; Maianti, J.P.; Raguram, A.; Liu, D.R. Improving cytidine and adenine base editors by expression optimization and ancestral reconstruction. Nat. Biotechnol. 2018, 36, 843–846. [Google Scholar] [CrossRef] [PubMed]
- Song, C.-Q.; Jiang, T.; Richter, M.; Rhym, L.H.; Koblan, L.; Zafra, M.P.; Schatoff, E.M.; Doman, J.L.; Cao, Y.; Dow, L.E.; et al. Adenine base editing in an adult mouse model of tyrosinaemia. Nat. Biomed. Eng. 2020, 4, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Holden, H.M.; Rayment, I.; Thoden, J.B. Structure and function of enzymes of the Leloir pathway for galactose metabolism. J. Biol. Chem. 2003, 278, 43885–43888. [Google Scholar] [CrossRef]
- Rubio-Agusti, I.; Carecchio, M.; Bhatia, K.; Kojovic, M.; Parees, I.; Chandrashekar, H.S.; Footitt, E.J.; Burke, D.; Edwards, M.J.; Lachmann, R.; et al. Movement disorders in adult patients with classical galactosemia. Mov. Disord. 2013, 28, 804–810. [Google Scholar] [CrossRef]
- Kuiper, A.; Grünewald, S.; Murphy, E.; Coenen, M.A.; Eggink, H.; Zutt, R.; Rubio-Gozalbo, M.E.; Bosch, A.M.; Williams, M.; Derks, T.; et al. Movement disorders and nonmotor neuropsychological symptoms in children and adults with classical galactosemia. J. Inherit. Metab. Dis. 2019, 42, 451–458. [Google Scholar] [CrossRef]
- Leslie, N.D.; Yager, K.L.; McNamara, P.D.; Segal, S. A mouse model of galactose-1-phosphate uridyl transferase deficiency. Biochem. Mol. Med. 1996, 59, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Siddiqi, A.; Witt, B.; Yuzyuk, T.; Johnson, B.; Fraser, N.; Chen, W.; Rascon, R.; Yin, X.; Goli, H.; et al. Subfertility and growth restriction in a new galactose-1 phosphate uridylyltransferase (GALT)-deficient mouse model. Eur. J. Hum. Genet. 2014, 22, 1172–1179. [Google Scholar] [CrossRef]
- Ohashi, T. Enzyme replacement therapy for lysosomal storage diseases. Pediatr. Endocrinol. Rev. PER 2012, 10, 26–34. [Google Scholar]
- Lachmann, R.H. Enzyme replacement therapy for lysosomal storage diseases. Curr. Opin. Pediatr. 2011, 23, 588–593. [Google Scholar] [CrossRef]
- McAuley, M.; Mesa-Torres, N.; McFall, A.; Morris, S.; Huang, M.; Pey, A.L.; Timson, D.J. Improving the activity and stability of human galactokinase for therapeutic and biotechnological applications. ChemBioChem 2018, 19, 1088–1095. [Google Scholar] [CrossRef]
- Connock, M.; Burls, A.; Frew, E.; Fry-Smith, A.; Juarez-Garcia, A.; McCabe, C.; Wailoo, A.; Abrams, K.; Cooper, N.; Sutton, A.; et al. The clinical effectiveness and cost-effectiveness of enzyme replacement therapy for Gaucher’s disease: A systematic review. Health Technol. Assess. 2006, 10, iii–iv. [Google Scholar] [CrossRef] [PubMed]
- Delnoy, B.; Coelho, A.I.; Rubio-Gozalbo, M.E. Current and future treatments for classic galactosemia. J. Pers. Med. 2021, 11, 75. [Google Scholar] [CrossRef]
- Balakrishnan, B.; An, D.; Nguyen, V.; DeAntonis, C.; Martini, P.G.V.; Lai, K. Novel mRNA-based therapy reduces toxic galactose metabolites and overcomes galactose sensitivity in a mouse model of classic galactosemia. Mol. Ther. 2020, 28, 304–312. [Google Scholar] [CrossRef]
- Coelho, A.I.; Lourenço, S.; Trabuco, M.; Silva, M.J.; Oliveira, A.; Gaspar, A.; Diogo, L.; de Almeida, I.T.; Vicente, J.B.; Rivera, I. Functional correction by antisense therapy of a splicing mutation in the GALT gene. Eur. J. Hum. Genet. 2015, 23, 500–506. [Google Scholar] [CrossRef][Green Version]
- Kauppinen, R. Porphyrias. Lancet 2005, 365, 241–252. [Google Scholar] [CrossRef]
- Puy, H.; Gouya, L.; Deybach, J.-C. Porphyrias. Lancet 2010, 375, 924–937. [Google Scholar] [CrossRef]
- Meyer, U.A.; Strand, L.J.; Doss, M.; Rees, A.C.; Marver, H.S. Intermittent acute porphyria—Demonstration of a genetic defect in porphobilinogen metabolism. N. Engl. J. Med. 1972, 286, 1277–1282. [Google Scholar] [CrossRef] [PubMed]
- Elder, G.; Harper, P.; Badminton, M.; Sandberg, S.; Deybach, J.-C. The incidence of inherited porphyrias in Europe. J. Inherit. Metab. Dis. Off. J. Soc. Study Inborn Errors Metab. 2013, 36, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Badminton, M.; Elder, G. Molecular mechanisms of dominant expression in porphyria. J. Inherit. Metab. Dis. 2005, 28, 277–286. [Google Scholar] [CrossRef]
- Lissing, M.; Nowak, G.; Adam, R.; Karam, V.; Boyd, A.; Gouya, L.; Meersseman, W.; Melum, E.; Ołdakowska-Jedynak, U.; Reiter, F.P.; et al. Liver transplantation for acute intermittent porphyria. Liver Transplant. 2021, 27, 491–501. [Google Scholar] [CrossRef]
- Jiang, L.; Berraondo, P.; Jericó, D.; Guey, L.T.; Sampedro, A.; Frassetto, A.; Benenato, K.E.; Burke, K.; Santamaría, E.; Alegre, M.; et al. Systemic messenger RNA as an etiological treatment for acute intermittent porphyria. Nat. Med. 2018, 24, 1899–1909. [Google Scholar] [CrossRef]
- Yasuda, M.; Gan, L.; Chen, B.; Kadirvel, S.; Yu, C.; Phillips, J.D.; New, M.I.; Liebow, A.; Fitzgerald, K.; Querbes, W.; et al. RNAi-mediated silencing of hepatic Alas1 effectively prevents and treats the induced acute attacks in acute intermittent porphyria mice. Proc. Natl. Acad. Sci. USA 2014, 111, 7777–7782. [Google Scholar] [CrossRef]
- D’Avola, D.; López-Franco, E.; Sangro, B.; Pañeda, A.; Grossios, N.; Gil-Farina, I.; Benito, A.; Twisk, J.; Paz, M.; Ruiz, J.; et al. Phase I open label liver-directed gene therapy clinical trial for acute intermittent porphyria. J. Hepatol. 2016, 65, 776–783. [Google Scholar] [CrossRef]
- Chan, A.; Liebow, A.; Yasuda, M.; Gan, L.; Racie, T.; Maier, M.; Kuchimanchi, S.; Foster, D.; Milstein, S.; Charisse, K.; et al. Preclinical development of a subcutaneous ALAS1 RNAi therapeutic for treatment of hepatic porphyrias using circulating RNA quantification. Mol. Ther.-Nucleic Acids 2015, 4, e263. [Google Scholar] [CrossRef] [PubMed]
- Córdoba, K.M.; Serrano-Mendioroz, I.; Jericó, D.; Merino, M.; Jiang, L.; Sampedro, A.; Alegre, M.; Corrales, F.; Garrido, M.J.; Martini, P.G.V.; et al. Recombinant porphobilinogen deaminase targeted to the liver corrects enzymopenia in a mouse model of acute intermittent porphyria. Sci. Transl. Med. 2022, 14, eabc0700. [Google Scholar] [CrossRef] [PubMed]
- Berntorp, E.; Shapiro, A.D. Modern haemophilia care. Lancet 2012, 379, 1447–1456. [Google Scholar] [CrossRef]
- Hooiveld, M.; Roosendaal, G.; Vianen, M.; Berg, M.V.D.; Bijlsma, J.; Lafeber, F. Blood-induced joint damage: Longterm effects in vitro and in vivo. J. Rheumatol. 2003, 30, 339–344. [Google Scholar]
- van Vulpen, L.F.D.; Schutgens, R.E.G.; Coeleveld, K.; Alsema, E.C.; Roosendaal, G.; Mastbergen, S.C.; Lafeber, F.P.J.G. IL-1β, in contrast to TNFα, is pivotal in blood-induced cartilage damage and is a potential target for therapy. Blood J. Am. Soc. Hematol. 2015, 126, 2239–2246. [Google Scholar] [CrossRef]
- Balkaransingh, P.; Young, G. Novel therapies and current clinical progress in hemophilia A. Ther. Adv. Hematol. 2018, 9, 49–61. [Google Scholar] [CrossRef]
- Fischer, K.; Ljung, R.; Platokouki, H.; Liesner, R.; Claeyssens, S.; Smink, E.; Berg, H.M.V.D. Prospective observational cohort studies for studying rare diseases: The European PedNet Haemophilia Registry. Haemophilia 2014, 20, e280–e286. [Google Scholar] [CrossRef]
- Ar, M.C.; Balkan, C.; Kavaklı, K. Extended half-life coagulation factors: A new era in the management of hemophilia patients. Turk. J. Hematol. 2019, 36, 141. [Google Scholar] [CrossRef]
- Patel, S.R.; Lundgren, T.S.; Spencer, H.T.; Doering, C.B. The immune response to the fVIII gene therapy in preclinical models. Front. Immunol. 2020, 11, 494. [Google Scholar] [CrossRef] [PubMed]
- George, L.A.; Sullivan, S.K.; Giermasz, A.; Rasko, J.E.; Samelson-Jones, B.J.; Ducore, J.; Cuker, A.; Sullivan, L.M.; Majumdar, S.; Teitel, J.; et al. Hemophilia B gene therapy with a high-specific-activity factor IX variant. N. Engl. J. Med. 2017, 377, 2215–2227. [Google Scholar] [CrossRef] [PubMed]
- Rangarajan, S.; Walsh, L.; Lester, W.; Perry, D.; Madan, B.; Laffan, M.; Yu, H.; Vettermann, C.; Pierce, G.F.; Wong, W.Y.; et al. AAV5—Factor VIII gene transfer in severe hemophilia A. N. Engl. J. Med. 2017, 377, 2519–2530. [Google Scholar] [CrossRef] [PubMed]
- Pasi, K.J.; Rangarajan, S.; Georgiev, P.; Mant, T.; Creagh, M.D.; Lissitchkov, T.; Bevan, D.; Austin, S.; Hay, C.R.; Hegemann, I.; et al. Targeting of antithrombin in hemophilia A or B with RNAi therapy. N. Engl. J. Med. 2017, 377, 819–828. [Google Scholar] [CrossRef]
- Machin, N.; Ragni, M.V. An investigational RNAi therapeutic targeting antithrombin for the treatment of hemophilia A and B. J. Blood Med. 2018, 9, 135. [Google Scholar] [CrossRef] [PubMed]
- Shima, M.; Hanabusa, H.; Taki, M.; Matsushita, T.; Sato, T.; Fukutake, K.; Fukazawa, N.; Yoneyama, K.; Yoshida, H.; Nogami, K. Factor VIII—Mimetic function of humanized bispecific antibody in hemophilia A. N. Engl. J. Med. 2016, 374, 2044–2053. [Google Scholar] [CrossRef]
- Han, J.P.; Kim, M.; Choi, B.S.; Lee, J.H.; Lee, G.S.; Jeong, M.; Lee, Y.; Kim, E.-A.; Oh, H.-K.; Go, N.; et al. In vivo delivery of CRISPR-Cas9 using lipid nanoparticles enables antithrombin gene editing for sustainable hemophilia A and B therapy. Sci. Adv. 2022, 8, eabj6901. [Google Scholar] [CrossRef]
- George, L.A.; Ragni, M.V.; Samelson-Jones, B.J.; Cuker, A.; Runoski, A.R.; Cole, G.; Wright, F.; Chen, Y.; Hui, D.J.; Wachtel, K.; et al. Spk-8011: Preliminary results from a phase 1/2 dose escalation trial of an investigational AAV-mediated gene therapy for hemophilia A. Blood Cells Mol. Dis. 2017, 130, 604. [Google Scholar]
- Nathwani, A.C.; Reiss, U.M.; Tuddenham, E.G.; Rosales, C.; Chowdary, P.; McIntosh, J.; Della Peruta, M.; Lheriteau, E.; Patel, N.; Raj, D.; et al. Long-term safety and efficacy of factor IX gene therapy in hemophilia B. N. Engl. J. Med. 2014, 371, 1994–2004. [Google Scholar] [CrossRef]
- Huang, H.-R.; Moroski-Erkul, C.; Bialek, P.; Wang, C.; Gong, G.; Hartfort, S.; Sattler, R.; White, D.; Lai, K.; Chalothorn, D.; et al. CRISPR/Cas9-mediated targeted insertion of human F9 achieves therapeutic circulating protein levels in mice and non-human primates. Mol. Ther. 2019, 27. [Google Scholar]
- Brown, B.D.; Cantore, A.; Annoni, A.; Sergi, L.S.; Lombardo, A.; Della Valle, P.; D’Angelo, A.; Naldini, L. A microRNA-regulated lentiviral vector mediates stable correction of hemophilia B mice. Blood J. Am. Soc. Hematol. 2007, 110, 4144–4152. [Google Scholar] [CrossRef]
- Gao, J.; Bergmann, T.; Zhang, W.; Schiwon, M.; Ehrke-Schulz, E.; Ehrhardt, A. Viral vector-based delivery of CRISPR/Cas9 and donor DNA for homology-directed repair in an in vitro model for canine hemophilia B. Mol. Ther.-Nucleic Acids 2019, 14, 364–376. [Google Scholar] [CrossRef]
- Chowdary, P.; Shapiro, S.; Makris, M.; Evans, G.; Boyce, S.; Talks, K.; Dolan, G.; Reiss, U.; Phillips, M.; Riddell, A.; et al. A novel adeno associated virus (AAV) gene therapy (FLT180a) achieves normal FIX activity levels in severe hemophilia B (HB) patients (B-AMAZE Study). Res. Pract. Thromb. Haemost. 2020, 4 (Suppl. 2), 17. [Google Scholar]
- Blau, N.; van Spronsen, F.J.; Levy, H.L. Phenylketonuria. Lancet 2010, 376, 1417–1427. [Google Scholar] [CrossRef]
- Matalon, R.; Michals, K. Phenylketonuria: Screening, treatment and maternal PKU. Clin. Biochem. 1991, 24, 337–342. [Google Scholar] [CrossRef]
- Vockley, J.; Andersson, H.C.; Antshel, K.M.; Braverman, N.E.; Burton, B.K.; Frazier, D.M.; Mitchell, J.; Smith, W.E.; Thompson, B.H.; Berry, S.A. Phenylalanine hydroxylase deficiency: Diagnosis and management guideline. Genet. Med. 2014, 16, 188–200. [Google Scholar] [CrossRef] [PubMed]
- Jahja, R.; Huijbregts, S.; de Sonneville, L.; van der Meere, J.J.; Bosch, A.M.; Hollak, C.E.; Rubio-Gozalbo, M.E.; Brouwers, M.C.; Hofstede, F.C.; de Vries, M.C.; et al. Mental health and social functioning in early treated Phenylketonuria: The PKU-COBESO study. Mol. Genet. Metab. 2013, 110, S57–S61. [Google Scholar] [CrossRef]
- Kono, K.; Okano, Y.; Nakayama, K.; Hase, Y.; Minamikawa, S.; Ozawa, N.; Yokote, H.; Inoue, Y. Diffusion-weighted MR imaging in patients with phenylketonuria: Relationship between serum phenylalanine levels and ADC values in cerebral white matter. Radiology 2005, 236, 630–636. [Google Scholar] [CrossRef] [PubMed]
- Soltys, K.A.; Setoyama, K.; Tafaleng, E.N.; Gutiérrez, A.S.; Fong, J.; Fukumitsu, K.; Nishikawa, T.; Nagaya, M.; Sada, R.; Haberman, K.; et al. Host conditioning and rejection monitoring in hepatocyte transplantation in humans. J. Hepatol. 2017, 66, 987–1000. [Google Scholar] [CrossRef]
- Eisensmith, R.; Woo, S. Gene therapy for phenylketonuria. Acta Paediatr. 1994, 83, 124–129. [Google Scholar] [CrossRef] [PubMed]
- Villiger, L.; Grisch-Chan, H.M.; Lindsay, H.; Ringnalda, F.; Pogliano, C.B.; Allegri, G.; Fingerhut, R.; Häberle, J.; Matos, J.; Robinson, M.D.; et al. Treatment of a metabolic liver disease by in vivo genome base editing in adult mice. Nat. Med. 2018, 24, 1519–1525. [Google Scholar] [CrossRef] [PubMed]
- Richards, D.Y.; Winn, S.R.; Dudley, S.; Nygaard, S.; Mighell, T.L.; Grompe, M.; Harding, C.O. AAV-mediated CRISPR/Cas9 gene editing in murine phenylketonuria. Mol. Ther.-Methods Clin. Dev. 2020, 17, 234–245. [Google Scholar] [CrossRef] [PubMed]
- Tao, R.; Xiao, L.; Zhou, L.; Zheng, Z.; Long, J.; Zhou, L.; Tang, M.; Dong, B.; Yao, S. Long-term metabolic correction of phenylketonuria by AAV-delivered phenylalanine amino lyase. Mol. Ther.-Methods Clin. Dev. 2020, 19, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, R.A.; Carlson, D.F.; Allen, K.L.; Webster, D.A.; VanLith, C.J.; Nicolas, C.T.; Hillin, L.G.; Yu, Y.; Kaiser, C.W.; Wahoff, W.R.; et al. Development of a porcine model of phenylketonuria with a humanized R408W mutation for gene editing. PLoS ONE 2021, 16, e0245831. [Google Scholar] [CrossRef] [PubMed]
- Perez-Garcia, C.G.; Diaz-Trelles, R.; Vega, J.B.; Bao, Y.; Sablad, M.; Limphong, P.; Chikamatsu, S.; Yu, H.; Taylor, W.; Karmali, P.P.; et al. Development of an mRNA replacement therapy for phenylketonuria. Mol. Ther.-Nucleic Acids 2022, 28, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Strauss, K.A.; Puffenberger, E.G.; Carson, V.J. Maple syrup urine disease. GeneReviews 2020. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1319 (accessed on 21 June 2022).
- Therrell, B.L., Jr.; Lloyd-Puryear, M.A.; Camp, K.M.; Mann, M.Y. Inborn errors of metabolism identified via newborn screening: Ten-year incidence data and costs of nutritional interventions for research agenda planning. Mol. Genet. Metab. 2014, 113, 14–26. [Google Scholar] [CrossRef]
- Muelly, E.R.; Moore, G.J.; Bunce, S.C.; Mack, J.; Bigler, D.C.; Morton, D.H.; Strauss, K.A. Biochemical correlates of neuropsychiatric illness in maple syrup urine disease. J. Clin. Investig. 2013, 123, 1809–1820. [Google Scholar] [CrossRef]
- Strauss, K.; Puffenberger, E.; Morton, D. Maple syrup urine disease. GeneReviews 2016. [Google Scholar]
- Mazariegos, G.V.; Morton, D.H.; Sindhi, R.; Soltys, K.; Nayyar, N.; Bond, G.; Shellmer, D.; Shneider, B.; Vockley, J.; Strauss, K.A. Liver transplantation for classical maple syrup urine disease: Long-term follow-up in 37 patients and comparative United Network for Organ Sharing experience. J. Pediatr. 2012, 160, 116–121.e1. [Google Scholar] [CrossRef] [PubMed]
- Greig, J.A.; Jennis, M.; Dandekar, A.; Chorazeczewski, J.K.; Smith, M.K.; Ashley, S.N.; Yan, H.; Wilson, J.M. Muscle-directed AAV gene therapy rescues the maple syrup urine disease phenotype in a mouse model. Mol. Genet. Metab. 2021, 134, 139–146. [Google Scholar] [CrossRef]
- Van Mil, S.; Houwen, R.H.J.; Klomp, L.W.J. Genetics of familial intrahepatic cholestasis syndromes. J. Med. Genet. 2005, 42, 449–463. [Google Scholar] [CrossRef]
- Jacquemin, E. Progressive familial intrahepatic cholestasis: Genetic basis and treatment. Clin. Liver Dis. 2000, 4, 753–763. [Google Scholar] [CrossRef]
- Gonzales, E.; Spraul, A.; Jacquemin, E. Clinical utility gene card for: Progressive familial intrahepatic cholestasis type 2. Eur. J. Hum. Genet. 2014, 22, 572. [Google Scholar]
- Davit-Spraul, A.; Gonzales, E.; Baussan, C.; Jacquemin, E. Progressive familial intrahepatic cholestasis. Orphanet J. Rare Dis. 2009, 4, 1. [Google Scholar] [CrossRef]
- Stapelbroek, J.M.; van Erpecum, K.J.; Klomp, L.W.; Houwen, R.H. Liver disease associated with canalicular transport defects: Current and future therapies. J. Hepatol. 2010, 52, 258–271. [Google Scholar] [CrossRef]
- Strautnieks, S.S.; Byrne, J.A.; Pawlikowska, L.; Cebecauerová, D.; Rayner, A.; Dutton, L.; Meier, Y.; Antoniou, A.; Stieger, B.; Arnell, H.; et al. Severe bile salt export pump deficiency: 82 different ABCB11 mutations in 109 families. Gastroenterology 2008, 134, 1203–1214.e8. [Google Scholar] [CrossRef]
- Davit-Spraul, A.; Gonzales, E.; Baussan, C.; Jacquemin, E. The spectrum of liver diseases related to ABCB4 gene mutations: Pathophysiology and clinical aspects. In Seminars in Liver Disease; Thieme Medical Publishers: New York, NY, USA, 2010. [Google Scholar]
- Jacquemin, E.; Bernard, O.; Hadchouel, M.; Cresteil, D.; De Vree, J.L.; Paul, M.; Elferink, R.P.; Bosma, P.J.; Sokal, E.; Sturm, E.; et al. The wide spectrum of multidrug resistance 3 deficiency: From neonatal cholestasis to cirrhosis of adulthood. Gastroenterology 2001, 120, 1448–1458. [Google Scholar] [CrossRef]
- Davit-Spraul, A.; Fabre, M.; Branchereau, S.; Baussan, C.; Gonzales, E.; Stieger, B.; Bernard, O.; Jacquemin, E. ATP8B1 and ABCB11 analysis in 62 children with normal gamma-glutamyl transferase progressive familial intrahepatic cholestasis (PFIC): Phenotypic differences between PFIC1 and PFIC2 and natural history. Hepatology 2010, 51, 1645–1655. [Google Scholar] [CrossRef]
- Bosma, P.J.; Wits, M.; Oude-Elferink, R.P.J. Gene therapy for progressive familial intrahepatic cholestasis: Current progress and future prospects. Int. J. Mol. Sci. 2020, 22, 273. [Google Scholar] [CrossRef]
- Aronson, S.J.; Bakker, R.S.; Shi, X.; Duijst, S.; Bloemendaal, L.T.; de Waart, D.R.; Verheij, J.; Ronzitti, G.; Elferink, R.P.O.; Beuers, U.; et al. Liver-directed gene therapy results in long-term correction of progressive familial intrahepatic cholestasis type 3 in mice. J. Hepatol. 2019, 71, 153–162. [Google Scholar] [CrossRef]
- Weiss, K.H.; Stremmel, W. Evolving perspectives in Wilson disease: Diagnosis, treatment and monitoring. Curr. Gastroenterol. Rep. 2012, 14, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Weiss, K.H.; Stremmel, W. Clinical considerations for an effective medical therapy in Wilson’s disease. Ann. N. Y. Acad. Sci. 2014, 1315, 81–85. [Google Scholar] [CrossRef]
- Bandmann, O.; Weiss, K.H.; Kaler, S.G. Wilson’s disease and other neurological copper disorders. Lancet Neurol. 2015, 14, 103–113. [Google Scholar] [CrossRef]
- Gitlin, J.D. Wilson disease. Gastroenterology 2003, 125, 1868–1877. [Google Scholar] [CrossRef]
- Stremmel, W.; Meyerrose, K.-W.; Niederau, C.; Hefter, H.; Kreuzpaintner, G.; Strohmeyer, G. Wilson disease: Clinical presentation, treatment, and survival. Ann. Intern. Med. 1991, 115, 720–726. [Google Scholar] [CrossRef]
- Weiss, K.H.; Schafer, M.; Gotthardt, D.N.; Angerer, A.; Mogler, C.; Schirmacher, P.; Schemmer, P.; Stremmel, W.; Sauer, P. Outcome and development of symptoms after orthotopic liver transplantation for Wilson disease. Clin. Transplant. 2013, 27, 914–922. [Google Scholar] [CrossRef]
- Członkowska, A.; Litwin, T. Wilson disease—Currently used anticopper therapy. Handb. Clin. Neurol. 2017, 142, 181–191. [Google Scholar]
- Członkowska, A.; Litwin, T.; Dusek, P.; Ferenci, P.; Lutsenko, S.; Medici, V.; Rybakowski, J.K.; Weiss, K.H.; Schilsky, M.L. Wilson disease. Nat. Rev. Dis. Primers 2018, 4, 21. [Google Scholar] [CrossRef] [PubMed]
- Mohr, I.; Weiss, K.H. Current anti-copper therapies in management of Wilson disease. Ann. Transl. Med. 2019, 7 (Suppl. 2), S69. [Google Scholar] [CrossRef] [PubMed]
- Buiakova, O.I.; Xu, J.; Lutsenko, S.; Zeitlin, S.; Das, K.; Das, S.; Ross, B.M.; Mekios, C.; Scheinberg, I.H.; Gilliam, T.C. Null mutation of the murine ATP7B (Wilson disease) gene results in intracellular copper accumulation and late-onset hepatic nodular transformation. Hum. Mol. Genet. 1999, 8, 1665–1671. [Google Scholar] [CrossRef] [PubMed]
- Lutsenko, S. Atp7b−/− mice as a model for studies of Wilson’s disease. Biochem. Soc. Trans. 2008, 36, 1233–1238. [Google Scholar] [CrossRef] [PubMed]
- Murillo, O.; Luqui, D.M.; Gazquez, C.; Martinez-Espartosa, D.; Navarro-Blasco, I.; Monreal, J.I.; Guembe, L.; Moreno-Cermeno, A.; Corrales, F.J.; Prieto, J.; et al. Long-term metabolic correction of Wilson’s disease in a murine model by gene therapy. J. Hepatol. 2016, 64, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.-K.; Hatanaka, F.; Araoka, T.; Reddy, P.; Wu, M.-Z.; Sui, Y.; Yamauchi, T.; Sakurai, M.; O’Keefe, D.D.; Núñez-Delicado, E.; et al. In vivo target gene activation via CRISPR/Cas9-mediated trans-epigenetic modulation. Cell 2017, 171, 1495–1507.e15. [Google Scholar] [CrossRef]
- Gramignoli, R.; Vosough, M.; Kannisto, K.; Srinivasan, R.C.; Strom, S.C. Clinical hepatocyte transplantation: Practical limits and possible solutions. Eur. Surg. Res. 2015, 54, 162–177. [Google Scholar] [CrossRef]
- Pöhler, M.; Guttmann, S.; Nadzemova, O.; Lenders, M.; Brand, E.; Zibert, A.; Schmidt, H.H.; Sandfort, V. CRISPR/Cas9-mediated correction of mutated copper transporter ATP7B. PLoS ONE 2020, 15, e0239411. [Google Scholar] [CrossRef]
- Roach, P.J. Glycogen and its metabolism. Curr. Mol. Med. 2002, 2, 101–120. [Google Scholar] [CrossRef]
- Kanungo, S.; Wells, K.; Tribett, T.; El-Gharbawy, A. Glycogen metabolism and glycogen storage disorders. Ann. Transl. Med. 2018, 6, 474. [Google Scholar] [CrossRef]
- Adeva-Andany, M.M.; González-Lucán, M.; Donapetry-García, C.; Fernández-Fernández, C.; Ameneiros-Rodríguez, E. Glycogen metabolism in humans. BBA Clin. 2016, 5, 85–100. [Google Scholar] [CrossRef]
- Davis, M.K.; Weinstein, D.A. Liver transplantation in children with glycogen storage disease: Controversies and evaluation of the risk/benefit of this procedure. Pediatr. Transplant. 2008, 12, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Koeberl, D.D.; Sun, B.D.; Damodaran, T.V.; Brown, T.; Millington, D.S.; Benjamin, D.K.; Bird, A.; Schneider, A.; Hillman, S.; Jackson, M.; et al. Early, sustained efficacy of adeno-associated virus vector-mediated gene therapy in glycogen storage disease type Ia. Gene Ther. 2006, 13, 1281–1289. [Google Scholar] [CrossRef] [PubMed]
- Perez-Pinera, P.; Ousterout, D.G.; Brown, M.T.; Gersbach, C.A. Gene targeting to the ROSA26 locus directed by engineered zinc finger nucleases. Nucleic Acids Res. 2012, 40, 3741–3752. [Google Scholar] [CrossRef]
- Miller, D.G.; Petek, L.M.; Russell, D.W. Adeno-associated virus vectors integrate at chromosome breakage sites. Nat. Genet. 2004, 36, 767–773. [Google Scholar] [CrossRef] [PubMed]
- Landau, D.J.; Brooks, E.; Perez-Pinera, P.; Amarasekara, H.; Mefferd, A.; Li, S.; Bird, A.; Gersbach, C.A.; Koeberl, D.D. In vivo zinc finger nuclease-mediated targeted integration of a glucose-6-phosphatase transgene promotes survival in mice with glycogen storage disease type IA. Mol. Ther. 2016, 24, 697–706. [Google Scholar] [CrossRef] [PubMed]
- Sentner, C.P.; Hoogeveen, I.J.; Weinstein, D.A.; Santer, R.; Murphy, E.; McKiernan, P.J.; Steuerwald, U.; Beauchamp, N.J.; Taybert, J.; Laforêt, P.; et al. Glycogen storage disease type III: Diagnosis, genotype, management, clinical course and outcome. J. Inherit. Metab. Dis. 2016, 39, 697–704. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.-A.; Choi, S.J.; Gao, F.; Kishnani, P.S.; Sun, B. A novel gene therapy approach for GSD III using an AAV vector encoding a bacterial glycogen debranching enzyme. Mol. Ther.-Methods Clin. Dev. 2020, 18, 240–249. [Google Scholar] [CrossRef]
- Pursell, N.; Gierut, J.; Zhou, W.; Dills, M.; Diwanji, R.; Gjorgjieva, M.; Saxena, U.; Yang, J.-S.; Shah, A.; Venkat, N.; et al. Inhibition of glycogen synthase II with RNAi prevents liver injury in mouse models of glycogen storage diseases. Mol. Ther. 2018, 26, 1771–1782. [Google Scholar] [CrossRef]
- Li, S.-C.; Chen, C.-M.; Goldstein, J.L.; Wu, J.-Y.; Lemyre, E.; Burrow, T.A.; Kang, P.B.; Chen, Y.-T.; Bali, D.S. Glycogen storage disease type IV: Novel mutations and molecular characterization of a heterogeneous disorder. J. Inherit. Metab. Dis. 2010, 33, 83–90. [Google Scholar] [CrossRef]
- Kakhlon, O.; Vaknin, H.; Mishra, K.; D’Souza, J.; Marisat, M.; Sprecher, U.; Wald-Altman, S.; Dukhovny, A.; Raviv, Y.; Da’Adoosh, B.; et al. Alleviation of a polyglucosan storage disorder by enhancement of autophagic glycogen catabolism. EMBO Mol. Med. 2021, 13, e14554. [Google Scholar] [CrossRef]
- Yi, H.; Zhang, Q.; Brooks, E.; Yang, C.; Thurberg, B.L.; Kishnani, P.S.; Sun, B. Systemic correction of murine glycogen storage disease type IV by an AAV-mediated gene therapy. Hum. Gene Ther. 2017, 28, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Bhandari, J.; Thada, P.K.; Yadav, D. Crigler Najjar Syndrome. In StatPearls; StatPearls Publishing: Tampa, FL, USA, 2021. [Google Scholar]
- Ebrahimi, A.; Rahim, F. Crigler-Najjar syndrome: Current perspectives and the application of clinical genetics. Endocr. Metab. Immune Disord.-Drug Targets 2018, 18, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Vandamme, C.; Adjali, O.; Mingozzi, F. Unraveling the complex story of immune responses to AAV vectors trial after trial. Hum. Gene Ther. 2017, 28, 1061–1074. [Google Scholar] [CrossRef]
- Greig, J.A.; Nordin, J.; Draper, C.; Bell, P.; Wilson, J. AAV8 gene therapy rescues the newborn phenotype of a mouse model of Crigler–Najjar. Hum. Gene Ther. 2018, 29, 763–770. [Google Scholar] [CrossRef] [PubMed]
- Ronzitti, G.; Bortolussi, G.; van Dijk, R.; Collaud, F.; Charles, S.; Leborgne, C.; Vidal, P.; Martin, S.; Gjata, B.; Sola, M.S.; et al. A translationally optimized AAV-UGT1A1 vector drives safe and long-lasting correction of Crigler-Najjar syndrome. Mol. Ther.-Methods Clin. Dev. 2016, 3, 16049. [Google Scholar] [CrossRef] [PubMed]



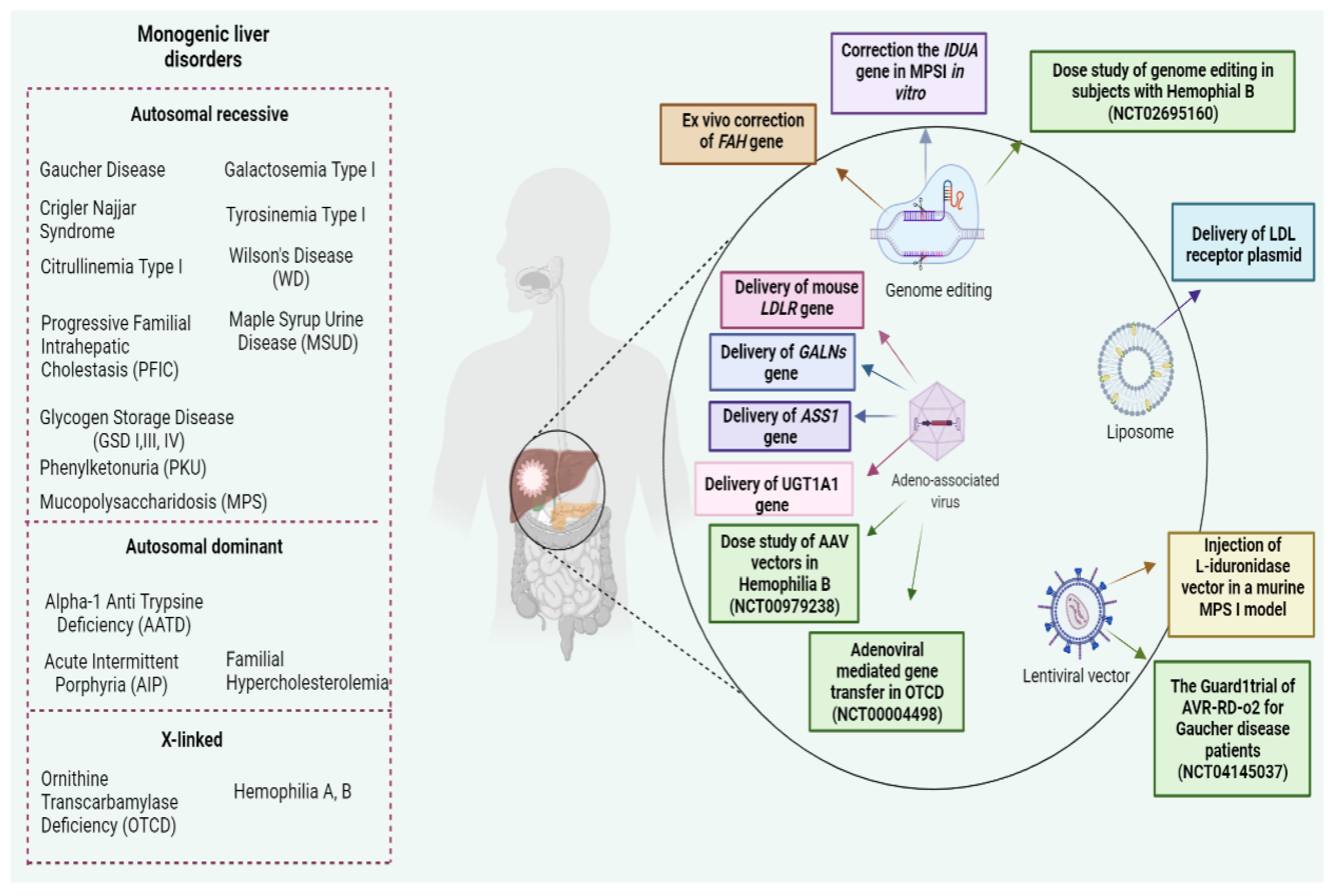{kind=link}
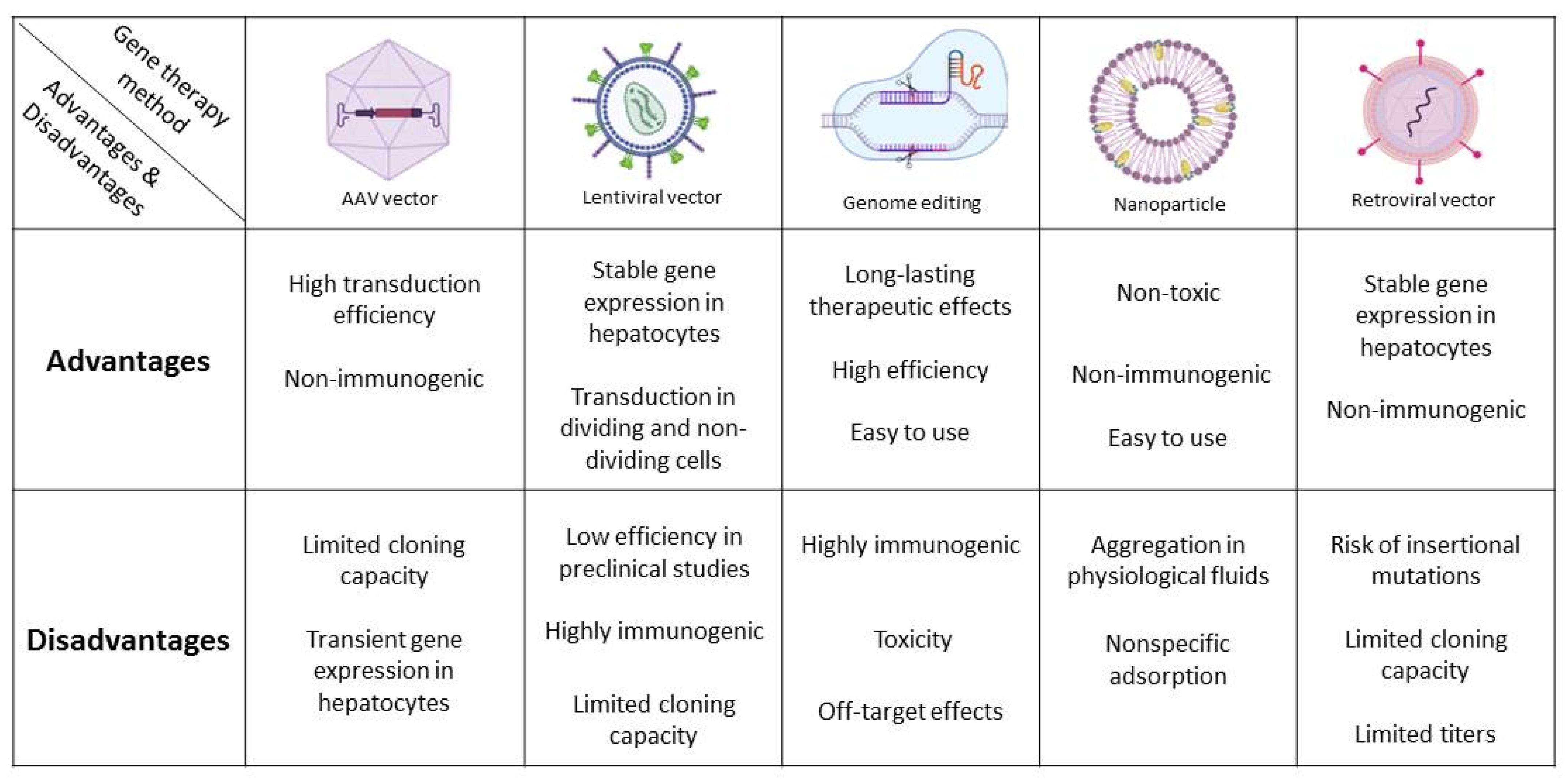{kind=link}
| Genetic Disorders | Liver Transplant | Hepatocyte Transplant | Gene Therapy |
|---|---|---|---|
| 1. Familial Hypercholesterolemia | Clinical study, W. Bilheimer et al. [8] | Clinical study, Grossman et al. [9] | Clinical study, (NCT02651675) |
| 2. Gaucher Disease | Clinical study, M. Ayto et al. [10] | N.A. | Clinical study, (NCT05139316) |
| 3. Mucopolysaccharidosis | Preclinical study, Toyama et al. [11] | N.A. | Clinical study, (NCT04201405) |
| 4. Urea cycle defects | |||
| a. OTC Deficiency | Clinical study, A. Busuttil et al. [12] | Clinical study, Stéphenne et al. [13] | Clinical study, (NCT05092685) |
| b. Citrullinemia type I | Clinical study, Yuan et al. [14] | Clinical study, Meyburg et al. [15] | Preclinical study, Chandler et al. [16] |
| 5. Alpha-1-anti Trypsin Deficiency | Clinical study, Hood et al. [17] | Preclinical study, Kay et al. [18] | Clinical study, (NCT04474197) |
| 6. Tyrosinemia Type I | Clinical study, Freese et al. [19] | Clinical study, Ribes-Koninckx et al. [20] | Preclinical study, VanLith et al. [21] |
| 7. Galactosemia | Clinical study, Otto et al. [22] | N.A. | Preclinical study, Rasmussen et al. [23] |
| 8. Acute Intermittent Porphyria | Clinical study, F. Soonawalla et al. [24] | N.A. | Clinical study, (NCT02082860) |
| 9. Hemophilia | Clinical study, Kurian et al. [25] | Clinical study, Kohei et al. [26] | Clinical study, Ozelo et al. [27] |
| 10. Phenylketonuria | Clinical study, Vajro et al. [28] | Clinical study, Stéphenne et al. [29] | Clinical study, (NCT04480567) |
| 11. Maple Syrup Urine Disease | Clinical study, Wendel et al. [30] | Preclinical study, Skvorak et al. [31] | Clinical study, (NCT03173521) |
| 12. Progressive familial intrahepatic cholestasis | Clinical study, Aydogdu et al. [32] | Preclinical study, De Vree et al. [33] | Preclinical study, Weber et al. [34] |
| 13. Wilson Disease | Clinical study, Bellary et al. [35] | Preclinical study, Allen et al. [36] | Clinical study, (NCT04884815) |
| 14. Glycogen Storage Diseases | Clinical study, Li et al. [37] | Preclinical study, Malhi et al. [38] | Clinical study, (NCT00976352) |
| 15. Crigler–Najjar Syndrome | Clinical study, Rela et al. [39] | Clinical study, Ambrosino et al. [40] | Clinical study, (NCT03466463) |
| Hereditary Disease (Monogenic Liver Disorder) | Gene Therapy Approach | Status | Phase | Outcome of Intervention | NCT Number |
|---|---|---|---|---|---|
| Ornithine Transcarbamylase Deficiency (OTCD) | single dose of recombinant adenovirus infused into the liver under fluoroscopic guidance | Terminated | Phase 1 | Not Provided | NCT00004386 |
| Intravascular adenoviral vector mediated gene transfer into the live | Terminated | Phase 1 | Not Provided | NCT00004498 | |
| HORACE 1 (AAVLK03hOTC); specifically targets the liver | Not yet recruiting | Phase 1/2 | Efficacy and safety outcomes | NCT05092685 | |
| AAV serotype 8 (AAV8)-Mediated Gene Transfer | Recruiting | Phase 3 | Change in plasma ammonia (AUC0-24) from baseline to week 64 for all participants | NCT05345171 | |
| single IV infusion of DTX301 (scAAV8OTC) | Completed | Phase 1/2 | Change in baseline in ureagenesis rate | NCT02991144 | |
| Maple Syrup Urine Disease (MSUD) | AAV8 for the delivery of the human ARSB gene (AAV2/8.TBG.hARSB 2) to liver | Active, not recruiting | Phase 1/2 | Efficacy outcome | NCT03173521 |
| Phenylketonoria (PKU) | single I.V. administration AAVHSC15 vector containing a functional copy of the human PAH gene (HMI-102) | Recruiting | Phase 1/2 | Change in plasma Phe concentration from baseline | NCT03952156 |
| AAV-mediated gene transfer of BMN 307 | Active, not recruiting | Phase 1/2 | Change from baseline in mean plasma Phe levels | NCT04480567 | |
| IV administration of HMI-103 AAVHSC15 vector containing a functional copy of the human PAH gene | Active, not recruiting | Phase 1 | Change from baseline in natural and total protein intake (g/day) at each timepoint post-administration of HMI-103 | NCT05222178 | |
| Alpha-1-anti Trypsin Deficiency (AATD) | Oral administration of VX-864 iRNA | Completed | Phase 2 | Change in plasma antigenic AAT levels | NCT04474197 |
| Administration of a serotype rh.10 replication deficient AAV expressing the human alpha-1 antitrypsin cDNA (ADVM-043) | Completed | Phase 1/2 | Change in therapeutic serum and alveolar epithelial lining fluid levels of a1AT as a preliminary measure of efficacy | NCT02168686 | |
| rAAV2-CB-hAAT gene Vector | Completed | Early Phase 1 | Human AAT levels and phenotype in the blood | NCT00377416 | |
| rAAV1-CB-hAAT | Completed | Phase 1 | Human AAT levels and phenotype in the blood | NCT00430768 | |
| Acute Intermittent Porphyria (AIP) | rAAV2/5-PBGD | Completed | Phase 1 | Health-related quality of life of AIP patients | NCT02082860 |
| Gene therapy rAAV2/5-PBGD for the treatment of acute intermittent porphyria | Completed | Phase 1 | Effect of the treatment on porphobilinogen (PBG) and delta-aminolevulinic acid (ALA) urinary level. | NCT02082860 | |
| Hemophilia B | IV infusion of SPK-9001 3 | Completed | Phase 2 | Change from baseline in FIX:C Antigen Level at Steady State | NCT02484092 |
| Genome editing by zinc finger nuclease therapeutic SB-FIX | Terminated | Phase 1 | Effect of SB-FIX on presence and shedding in AAV2/6 vector DNA | NCT02695160 | |
| AAV-mediated gene transfer of scAAV2/8-LP1-hFIXco | Active | Phase 1 | Not Provided | NCT00979238 | |
| Using a Single-Stranded, Adeno-Associated Pseudotype 8 Viral Vector (AAV8-hFIX19) | Terminated | Phase 1 | Factor IX activity and antigen; PT; and aPTT. | NCT01620801 | |
| AAV vector containing Factor IX gene named FLT180a | Terminated | Phase 1/2 | Change from baseline in FIX concentrate consumption and annualized bleeding rate | NCT03369444 | |
| AAV containing BBM-H901 4 | Active, not recruiting | Not applicable | Vector-derived FIX:C and FIX antigen levels. | NCT04135300 | |
| Hemophilia A | single IV infusion of ASC618 5 | Not yet recruiting | Phase 1/2 | Changes in FVIII activity levels from baseline | NCT04676048 |
| novel AAV vector (with a stronger attraction to the human liver) to deliver the human factor VIII (hFVIII) named SPK-8011 | Recruiting | Phase 1/2 | Increased FVIII:C levels to prevent spontaneous bleeding | NCT03003533 | |
| AAV-based gene therapy (Valoctocogene roxaparvovec 6) | Active, not recruiting | Phase 1/2 | Frequency of FVIII replacement therapy during the study | NCT02576795 | |
| Infusion of AAV2/8-HLP-FVIII-V3 | Recruiting | Phase 1 | Plasma hFVIII activity | NCT03001830 | |
| Mucopolysaccharidosis | Autologous CD34+ cells transduced with a lentiviral vector containing the human N-Sulfoglucosamine Sulfohydrolase (SGSH) gene | Active, not recruiting | Phase 1/2 | change in ng/mL glycosaminoglycans in CSF from baseline following IMP administration | NCT04201405 |
| Retroviral-mediated gene transfer of Lymphocyte gene | Completed | Phase 1/2 | Not Provided | NCT00004454 | |
| Genome editing by zinc finger nuclease for SB-318 | Terminated | Phase 1/2 | Effect of SB-318 on leukocyte IDUA activity | NCT02702115 | |
| Fabry Disease Lysosomal Storage Diseases | Single-ascending dose study of a novel AAV containing FLT190 | Recruiting | Phase 1/2 | Frequency of treatment-emergent adverse events (AEs) | NCT04040049 |
| Single dose of investigational product, ST-920 7 | Recruiting | Phase 1/2 | Incidence of treatment-emergent adverse events (TEAEs) | NCT04046224 | |
| Wilson Disease | AAV-mediated gene transfer using infusion of UX701 | Recruiting | Phase 1/2 | Change in Liver Copper Concentration | NCT04884815 |
| Recombinant AAV-mediated gene transfer of VTX-801 | Recruiting | Phase 1/2 | Serum ceruloplasmin activity (enzymatic assay) | NCT04537377 | |
| Familial Hypercholesterolemia | Low Density Lipoprotein Receptor mRNA Exosomes | Not yet recruiting | Phase 1 | Changes in Stability of Carotid Artery Plaques | NCT05043181 |
| Recombinant retroviral vector (ex-vivo liver directed gene therapy) | Completed | Phase 1 | Not Provided | NCT00004809 | |
| AAV directed hlDLR gene therapy | Completed | Phase 1/2 | Percent change in LDL-C compared to baseline | NCT02651675 | |
| Gaucher disease | Lentiviral-mediated gene transfer of AVR-RD-02 | Recruiting | Phase 1/2 | Change from Baseline in plasma Chitotriosidase activity levels | NCT04145037 |
| Retroviral-mediated gene transfer containing human glucocerebrosidase cDNA (ex vivo) | Completed | Phase 1 | Not Provided | NCT00001234 | |
| Glycogen Storage Diseases | AAV8-mediated gene transfer of DTX401 | Recruiting | Phase 3 | Change from Baseline to Week 48 in Time to Hypoglycemia | NCT05139316 |
| Recombinant AAV1-mediated gene transfer of rAAV1-CMV-GAA | Completed | Phase 1/2 | Change in AAV antibody level; change in Alglucosidase alpha (GAA) Antibody level; maximal inspiratory pressure | NCT00976352 | |
| AAV8-mediated gene transfer of AT845 | Recruiting | Phase 1/2 | Change from baseline in thigh fat fraction | NCT04174105 | |
| Crigler–Najjar Syndrome | AAV-mediated gene transfer of GNT0003 | Recruiting | Phase 1/2 | Decrease in total Serum bilirubin level | NCT03466463 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghasemzad, M.; Hashemi, M.; Lavasani, Z.M.; Hossein-khannazer, N.; Bakhshandeh, H.; Gramignoli, R.; Keshavarz Alikhani, H.; Najimi, M.; Nikeghbalian, S.; Vosough, M. Novel Gene-Correction-Based Therapeutic Modalities for Monogenic Liver Disorders. Bioengineering 2022, 9, 392. https://doi.org/10.3390/bioengineering9080392
Ghasemzad M, Hashemi M, Lavasani ZM, Hossein-khannazer N, Bakhshandeh H, Gramignoli R, Keshavarz Alikhani H, Najimi M, Nikeghbalian S, Vosough M. Novel Gene-Correction-Based Therapeutic Modalities for Monogenic Liver Disorders. Bioengineering. 2022; 9(8):392. https://doi.org/10.3390/bioengineering9080392
Chicago/Turabian StyleGhasemzad, Mahsa, Mahdieh Hashemi, Zohre Miri Lavasani, Nikoo Hossein-khannazer, Haleh Bakhshandeh, Roberto Gramignoli, Hani Keshavarz Alikhani, Mustapha Najimi, Saman Nikeghbalian, and Massoud Vosough. 2022. "Novel Gene-Correction-Based Therapeutic Modalities for Monogenic Liver Disorders" Bioengineering 9, no. 8: 392. https://doi.org/10.3390/bioengineering9080392
APA StyleGhasemzad, M., Hashemi, M., Lavasani, Z. M., Hossein-khannazer, N., Bakhshandeh, H., Gramignoli, R., Keshavarz Alikhani, H., Najimi, M., Nikeghbalian, S., & Vosough, M. (2022). Novel Gene-Correction-Based Therapeutic Modalities for Monogenic Liver Disorders. Bioengineering, 9(8), 392. https://doi.org/10.3390/bioengineering9080392