
Microbiome Analysis via OTU and ASV-Based Pipelines—A Comparative Interpretation of Ecological Data in WWTP Systems


Abstract
:
1. Introduction
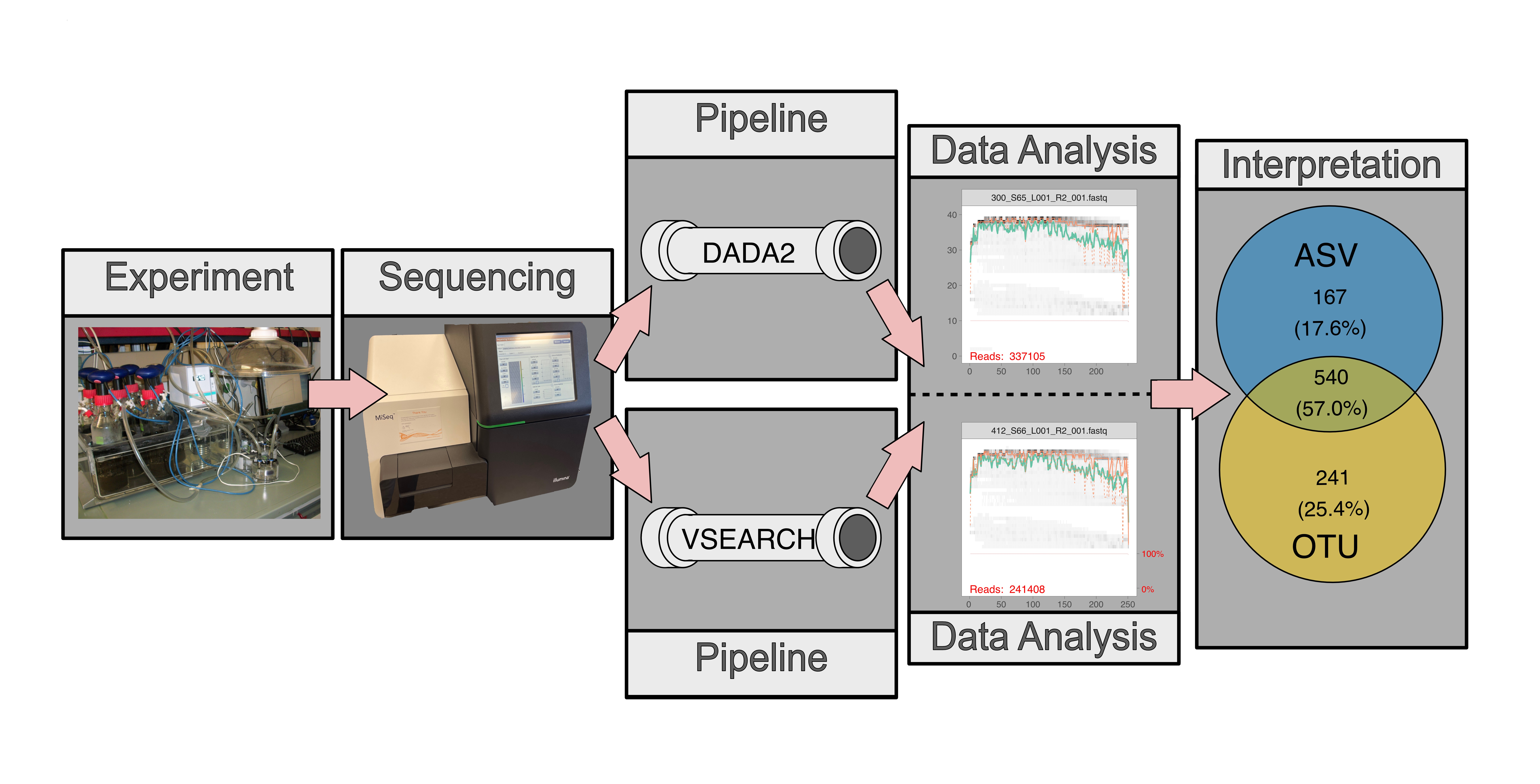
2. Materials and Methods
2.1. Sampling and Experimental Setup for Simulation of Anaerobic Co-Digestion Co-AD
2.2. Analytical Procedures
2.3. DNA Extraction and 16S rRNA Gene Amplicon Sequencing
2.4. Sequence Analysis OTU
2.5. Sequence Analysis ASV
2.6. Data Handling
3. Results
3.1. Operational Data and Reactor Performance
3.2. Sequencing Pipeline Data Overview
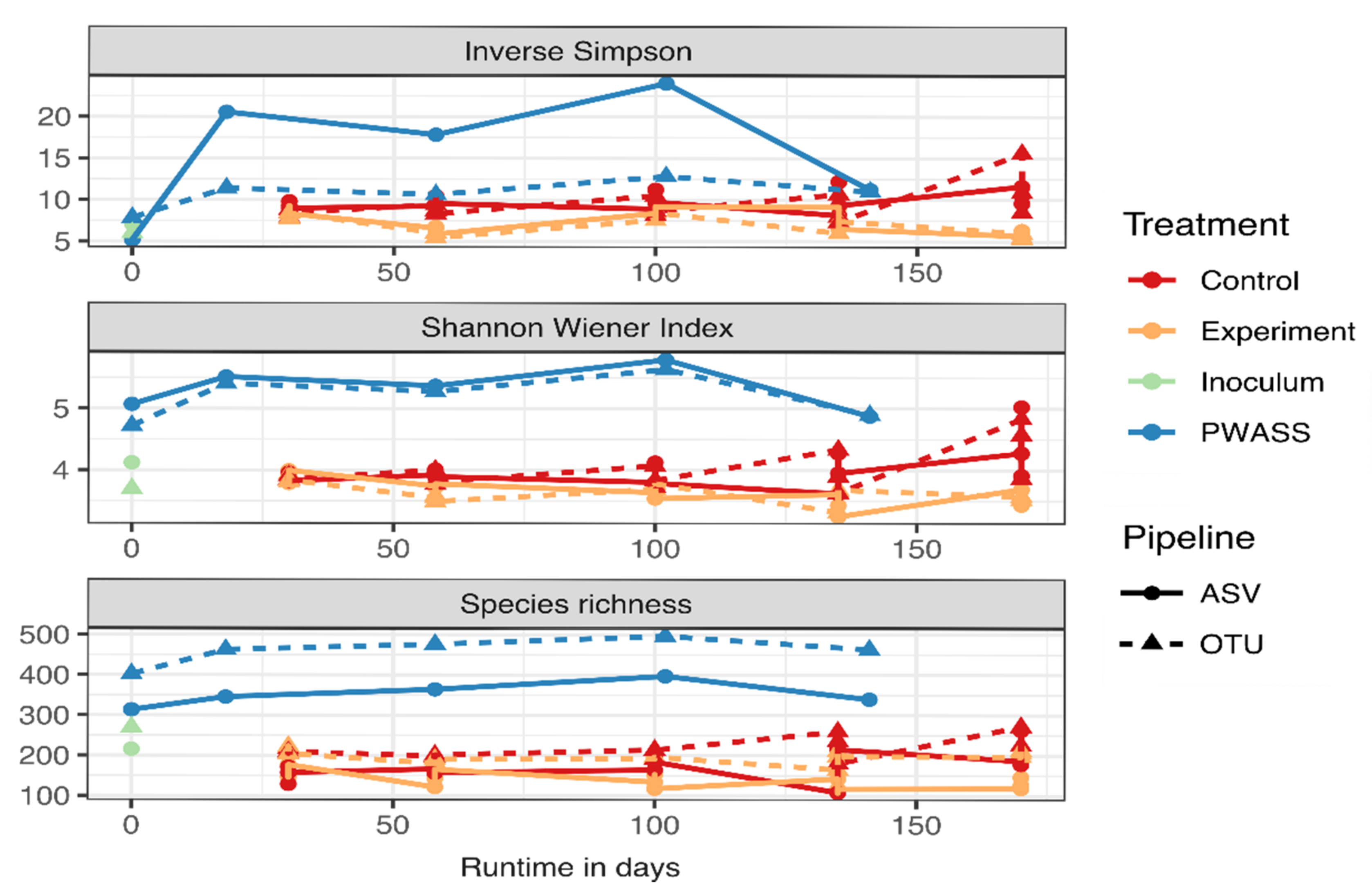
3.3. α-Diversity Comparison of Pipeline Outcomes
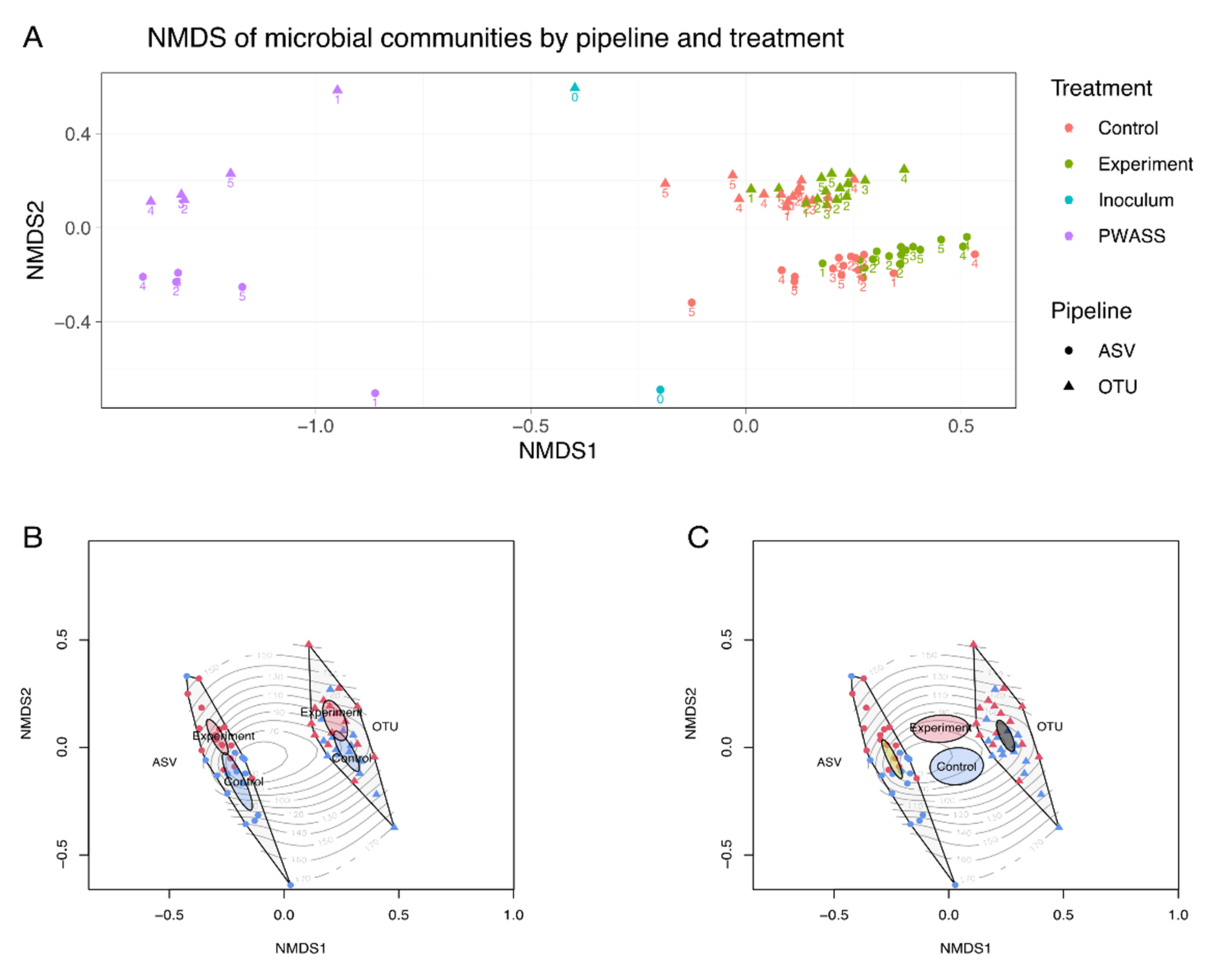
3.4. β-Diversity Comparison of Pipeline Outcomes—Prokaryotic Community Composition on Phylum Level
3.5. 𝛽-Diversity Comparison of Pipeline Outcomes—Prokaryotic Community Composition on Genus Level
3.6. Statistical Analysis of Pipeline Outcomes in the Light of Ecological Data
4. Discussion
4.1. Minimizing Sequencing Data Bias during Microbial Community Analysis
4.2. Equalizing Sequencing Data Bias during Microbial Community Analysis
4.3. Complex Relationships: Dissimilar or Similar
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Biogas in mL Day−1 Reactor−1,b | CH4 in % Day−1 Reactor−1 | |||
|---|---|---|---|---|
| Timepointc | Control d | Experiment d | Control | Experiment |
| 1 | 413.8 ± 18.9 | 349.6 ± 2.6 | 69.8 ± 2.8 | 68.2 ± 3.2 |
| 2 | 476.5 ± 10.9 | 507.7 ± 15.1 | 69.8 ± 5.0 | 69.9 ± 1.2 |
| 3 | 520.0 ± 69.0 | 587.7 ± 12.0 | 71.4 ± 6.1 | 69.2 ± 2.0 |
| 4 | 265.8 ± 88.1 | 434.6 ± 50.8 | 73.6 ± 8.8 | 66.5 ± 2.3 |
| 5 | 367.4 ± 50.1 | 721.8 ± 11.2 | 74.3 ± 8.7 | 70.8 ± 1.9 |
References
- Tang, K.; Zhang, Y.; Lin, D.; Han, Y.; Chen, C.T.A.; Wang, D.; Lin, Y.S.; Sun, J.; Zheng, Q.; Jiao, N. Cultivation-independent and cultivation-dependent analysis of microbes in the shallow-sea hydrothermal system off Kueishantao Island, Taiwan: Unmasking heterotrophic bacterial diversity and functional capacity. Front. Microbiol. 2018, 9, 279. [Google Scholar] [CrossRef]
- Gutknecht, J.L.M.; Goodman, R.M.; Balser, T.C. Linking soil process and microbial ecology in freshwater wetland ecosystems. Plant. Soil 2006, 289, 17–34. [Google Scholar] [CrossRef]
- Herlemann, D.P.R.; Labrenz, M.; Jürgens, K.; Bertilsson, S.; Waniek, J.J.; Andersson, A.F. Transitions in bacterial communities along the 2000 km salinity gradient of the Baltic Sea. ISME J. 2011, 5, 1571–1579. [Google Scholar] [CrossRef] [Green Version]
- Johnson, J.S.; Spakowicz, D.J.; Hong, B.-Y.; Petersen, L.M.; Demkowicz, P.; Chen, L.; Leopold, S.R.; Hanson, B.M.; Agresta, H.O.; Gerstein, M.; et al. Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis. Nat. Commun. 2019, 10, 5029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yilmaz, P.; Parfrey, L.W.; Yarza, P.; Gerken, J.; Pruesse, E.; Quast, C.; Schweer, T.; Peplies, J.; Ludwig, W.; Glöckner, F.O. The SILVA and “All-species living tree project (LTP)” taxonomic frameworks. Nucleic Acids Res. 2014, 42, D643–D648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ludwig, W.; Viver, T.; Westram, R.; Gago, J.F.; Bustos-Caparros, E.; Knittel, K.; Amann, R.; Rossello-Mora, R. Release LTP_12_2020, featuring a new ARB alignment and improved 16S rRNA tree for prokaryotic type strains. Syst. Appl. Microbiol. 2021, 44, 126218. [Google Scholar] [CrossRef]
- Woese, C.R.; Fox, G.E. Phylogenetic structure of the prokaryotic domain: The primary kingdoms. Proc. Natl. Acad. Sci. USA 1977, 74, 5088–5090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woese, C.R.; Kandler, O.; Wheelis, M.L. Towards a natural system of organisms: Proposal for the domains Archaea, Bacteria, and Eucarya. Proc. Natl. Acad. Sci. USA 1990, 87, 4576–4579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinclair, L.; Osman, O.A.; Bertilsson, S.; Eiler, A. Microbial community composition and diversity via 16S rRNA gene amplicons: Evaluating the illumina platform. PLoS ONE 2015, 10, e0116955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aird, D.; Ross, M.G.; Chen, W.-S.; Danielsson, M.; Fennell, T.; Russ, C.; Jaffe, D.B.; Nusbaum, C.; Gnirke, A. Analyzing and minimizing PCR amplification bias in Illumina sequencing libraries. Genome Biol. 2011, 12, R18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoler, N.; Nekrutenko, A. Sequencing error profiles of illumina sequencing instruments. NAR Genom. Bioinform. 2021, 3, lqab019. [Google Scholar] [CrossRef] [PubMed]
- Bukin, Y.u.S.; Galachyants, Y.u.P.; Morozov, I.V.; Bukin, S.V.; Zakharenko, A.S.; Zemskaya, T.I. The effect of 16S rRNA region choice on bacterial community metabarcoding results. Sci. Data 2019, 6, 190007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fadeev, E.; Cardozo-Mino, M.G.; Rapp, J.Z.; Bienhold, C.; Salter, I.; Salman-Carvalho, V.; Molari, M.; Tegetmeyer, H.E.; Buttigieg, P.L.; Boetius, A. Comparison of two 16S rRNA primers (V3–V4 and V4–V5) for studies of arctic microbial communities. Front. Microbiol. 2021, 12, 637526. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Tomita, J.; Nishioka, K.; Hisada, T.; Nishijima, M. Development of a Prokaryotic Universal Primer for Simultaneous Analysis of Bacteria and Archaea Using Next-Generation Sequencing. PLoS ONE 2014, 9, e105592. [Google Scholar] [CrossRef] [Green Version]
- Abellan-Schneyder, I.; Matchado, M.S.; Reitmeier, S.; Sommer, A.; Sewald, Z.; Baumbach, J.; List, M.; Neuhaus, K. Primer, Pipelines, Parameters: Issues in 16S rRNA Gene Sequencing. mSphere 2021, 6, e01202–e01220. [Google Scholar] [CrossRef]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [Green Version]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef] [Green Version]
- Callahan, B.J.; Mcmurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nearing, J.T.; Douglas, G.M.; Comeau, A.M.; Langille, M.G. Denoising the Denoisers: An independent evaluation of microbiome sequence error-correction approaches. PeerJ 2018, 6, e5364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiime2 Forum. ASV Naming: Reviewer Reply. 2022. Available online: https://forum.qiime2.org/t/asv-naming-reviewer-reply/19795 (accessed on 7 March 2022).
- Qiime2 Forum. ASV IDs in Publication. 2022. Available online: https://forum.qiime2.org/t/asv-ids-in-publication/10646 (accessed on 7 March 2022).
- Jain, M.; Olsen, H.E.; Paten, B.; Akeson, M. The Oxford Nanopore MinION: Delivery of nanopore sequencing to the genomics community. Genome Biol. 2016, 17, 239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.; Dennehy, C.; Lawlor, P.G.; Hu, Z.; McCabe, M.; Cormican, P.; Zhan, X.; Gardiner, G.E. Exploring the roles of and interactions among microbes in dry co-digestion of food waste and pig manure using high-throughput 16S rRNA gene amplicon sequencing. Biotechnol. Biofuels 2019, 12, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, A.W.; Naphtali, J.; Schellhorn, H.E. High-throughput DNA sequencing technologies for water and wastewater analysis. Sci. Prog. 2019, 102, 351–376. [Google Scholar] [CrossRef] [PubMed]
- De Celis, M.; Belda, I.; Ortiz-Álvarez, R.; Arregui, L.; Marquina, D.; Serrano, S.; Santos, A. Tuning up microbiome analysis to monitor WWTPs’ biological reactors functioning. Sci. Rep. 2020, 10, 4079. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, A.; Zan, F.; Siddiqui, M.A.; Nizamuddin, S.; Chen, G. Integrated treatment of food waste with wastewater and sewage sludge: Energy and carbon footprint analysis with economic implications. Sci. Total Environ. 2022, 825, 154052. [Google Scholar] [CrossRef] [PubMed]
- Baloch, M.Y.J.; Talpur, S.A.; Iqbal, J.; Munir, M. Process Design for Biohydrogen Production from Waste Materials and Its Application. SE 2022, 7, 47. [Google Scholar] [CrossRef]
- Vergara-Araya, M.; Hilgenfeldt, V.; Peng, D.; Steinmetz, H.; Wiese, J. Modelling to Lower Energy Consumption in a Large WWTP in China While Optimising Nitrogen Removal. Energies 2021, 14, 5826. [Google Scholar] [CrossRef]
- Jeske, J.T.; Gallert, C. Mechanisms driving microbial community composition in anaerobic co-digestion of waste-activated sewage sludge. Bioengineering 2021, 8, 197. [Google Scholar] [CrossRef]
- Uhlenhut, F.; Schlüter, K.; Gallert, C. Wet biowaste digestion: ADM1 model improvement by implementation of known genera and activity of propionate oxidizing bacteria. Water Res. 2018, 129, 384–393. [Google Scholar] [CrossRef]
- 2540 SOLIDS. Standard Methods For the Examination of Water and Wastewater; American Public Health Association: Washington, DC, USA, 2018. [Google Scholar]
- Dyksma, S.; Jansen, L.; Gallert, C. Syntrophic acetate oxidation replaces acetoclastic methanogenesis during thermophilic digestion of biowaste. Microbiome 2020, 8, 105. [Google Scholar] [CrossRef] [PubMed]
- A DADA2 Workflow for Big Data: Paired-End (1.4 or Later). Available online: https://benjjneb.github.io/dada2/bigdata_paired.html (accessed on 10 January 2022).
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021. [Google Scholar]
- McMurdie, P.J.; Holmes, S. Phyloseq: An r package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oksanen, J. Vegan: An introduction to ordination. Management 2008, 1, 1–10. [Google Scholar]
- Oksanen, J.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’hara, R.B.; Simpson, G.L.; Solymos, P.; Stevens, M.H.H.; Wagner, H. Vegan: Community Ecology Package; R Package Version 2. 2020. Available online: https://rdrr.io/rforge/vegan/ (accessed on 7 March 2022).
- Lathi, L.; Shetty, S. Tools for microbiome analysis in R Version (2019). Available online: http://microbiome.github.com/microbiome (accessed on 7 March 2022).
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Wilke, C.O. (2020) cowplot: Streamlined Plot Theme and Plot Annotations for ‘ggplot2′. Available online: https://CRAN.R-project.org/package=cowplot (accessed on 6 March 2022).
- Cameron, E.S.; Schmidt, P.J.; Tremblay, B.J.-M.; Emelko, M.B.; Müller, K.M. Enhancing diversity analysis by repeatedly rarefying next generation sequencing data describing microbial communities. Sci. Rep. 2021, 11, 22302. [Google Scholar] [CrossRef]
- McMurdie, P.; Holmes, S. Waste Not, Want Not: Why Rarefying Microbiome Data Is Inadmissible. PLoS Comput. Biol. 2014, 10, e1003531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. UNOISE2: Improved error-correction for Illumina 16S and ITS amplicon sequencing. Bioinformatics 2016, 081257. [Google Scholar]
- Oren, A.; Arahal, D.R.; Rosselló-Móra, R.; Sutcliffe, I.C.; Moore, E.R. Preparing a revision of the International Code of Nomenclature of Prokaryotes. Int. J. Syst. Evol. Microbiol. 2021, 71, 004598. [Google Scholar] [CrossRef]
- Prodan, A.; Tremaroli, V.; Brolin, H.; Zwinderman, A.H.; Nieuwdorp, M.; Levin, E. Comparing bioinformatic pipelines for microbial 16S rRNA amplicon sequencing. PLoS ONE 2020, 15, e0227434. [Google Scholar] [CrossRef] [Green Version]
- D’Argenio, V.; Casaburi, G.; Precone, V.; Salvatore, F. Comparative Metagenomic Analysis of Human Gut Microbiome Composition Using Two Different Bioinformatic Pipelines. BioMed Res. Int. 2014, 2014, 325340. [Google Scholar] [CrossRef]
- Barnes, C.J.; Rasmussen, L.; Asplund, M.; Knudsen, S.W.; Clausen, M.-L.; Agner, T.; Hansen, A.J. Comparing DADA2 and OTU clustering approaches in studying the bacterial communities of atopic dermatitis. J. Med. Microbiol. 2020, 69, 1293–1302. [Google Scholar] [CrossRef]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef] [PubMed]
- Reitmeier, S.; Hitch, T.C.A.; Treichel, N.; Fikas, N.; Hausmann, B.; Ramer-Tait, A.E.; Neuhaus, K.; Berry, D.; Haller, D.; Lagkouvardos, I.; et al. Handling of spurious sequences affects the outcome of high-throughput 16S rRNA gene amplicon profiling. ISME Commun. 2021, 1, 31. [Google Scholar] [CrossRef]
- Edgar, R.C.; Flyvbjerg, H. Error filtering, pair assembly and error correction for next-generation sequencing reads. Bioinformatics 2015, 31, 3476–3482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cameron, E.S.; Schmidt, P.J.; Tremblay, B.J.M.; Emelko, M.B.; Müller, K.M. To rarefy or not to rarefy: Enhancing diversity analysis of microbial communities through next-generation sequencing and rarefying repeatedly. Bioinformatics. 2020, 11, 22302. [Google Scholar]
- Zhang, B.; Yu, Q.; Yan, G.; Zhu, H.; Zhu, L. Seasonal bacterial community succession in four typical wastewater treatment plants: Correlations between core microbes and process performance. Sci. Rep. 2018, 8, 4566. [Google Scholar] [CrossRef] [PubMed]
- LaMartina, E.L.; Mohaimani, A.A.; Newton, R.J. Urban wastewater bacterial communities assemble into seasonal steady states. Microbiome 2021, 9, 116. [Google Scholar] [CrossRef] [PubMed]
- Calusinska, M.; Goux, X.; Fossépré, M.; Muller, E.; Wilmes, P.; Delfosse, P. A year of monitoring 20 mesophilic full-scale bioreactors reveals the existence of stable but different core microbiomes in bio-waste and wastewater anaerobic digestion systems. Biotechnol. Biofuels 2018, 11, 196. [Google Scholar] [CrossRef]
- Campanaro, S.; Treu, L.; Rodriguez-R, L.M.; Kovalovszki, A.; Ziels, R.M.; Maus, I.; Zhu, X.; Kougias, P.G.; Basile, A.; Luo, G.; et al. New insights from the biogas microbiome by comprehensive genome-resolved metagenomics of nearly 1600 species originating from multiple anaerobic digesters. Biotechnol. Biofuels 2020, 13, 25. [Google Scholar] [CrossRef] [Green Version]
- Campanaro, S.; Treu, L.; Kougias, P.G.; Luo, G.; Angelidaki, I. Metagenomic binning reveals the functional roles of core abundant microorganisms in twelve full-scale biogas plants. Water Res. 2018, 140, 123–134. [Google Scholar] [CrossRef]
- Mondal, S.K.; Kundu, S.; Das, R.; Roy, S. Analysis of phylogeny and codon usage bias and relationship of GC content, amino acid composition with expression of the structural nif genes. J. Biomol. Struct. Dyn. 2016, 34, 1649–1666. [Google Scholar] [CrossRef]
- Hu, E.-Z.; Lan, X.-R.; Liu, Z.-L.; Gao, J.; Niu, D.-K. A positive correlation between GC content and growth temperature in prokaryotes. BMC Genom. 2022, 23, 110. [Google Scholar] [CrossRef] [PubMed]
- Musto, H.; Naya, H.; Zavala, A.; Romero, H.; Alvarez-Valı́n, F.; Bernardi, G. Correlations between genomic GC levels and optimal growth temperatures in prokaryotes. FEBS Lett. 2004, 573, 73–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mongad, D.S.; Chavan, N.S.; Narwade, N.P.; Dixit, K.; Shouche, Y.S.; Dhotre, D.P. MicFunPred: A conserved approach to predict functional profiles from 16S rRNA gene sequence data. Genomics 2021, 113, 3635–3643. [Google Scholar] [CrossRef] [PubMed]


| Number of Taxa | ||||
|---|---|---|---|---|
| Threshold | OTU Based 1 | OTU Based 2 | ASV Based | Merged 3 |
| full Dataset | 67,015 | 50,065 | 8005 | |
| >0.001% relative Abundance | 1,204 | 1,201 | 1,089 | |
| Genus level | 910 | 877 | 781 | 1053 |
| Genus level >0.001% relative Abundance | 285 | 283 | 279 | 348 |
| Phylum | Treatment | ASV | OTU | ∆ of Total Community | Fold Deviation |
|---|---|---|---|---|---|
| Acidobacteriota | Control | 1.12% | <0.01% | 1.11% | 839 |
| Armatimonadota | 0.40% | <0.01% | 0.40% | 510 | |
| Coprothermobacterota | 16.46% | 18.45% | 2.00% | 1.12 | |
| Firmicutes | 21.10% | 18.82% | 2.28% | 1.12 | |
| Hydrothermae | 1.47% | ND | 1.47% | ∞ | |
| Patescibacteria | 0.41% | 0.01% | 0.40% | 32.6 | |
| Synergistota | 13.62% | 15.59% | 1.97% | 1.14 | |
| Thermotogota | 18.30% | 19.48% | 1.18% | 1.06 | |
| Acidobacteriota | Experiment | 0.64% | <0.01% | 0.64% | 271 |
| Armatimonadota | 0.31% | <0.01% | 0.31% | 519 | |
| Chloroflexota | 0.48% | 0.35% | 0.13% | 1.36 | |
| Coprothermobacterota | 17.44% | 18.94% | 1.50% | 1.09 | |
| Firmicutes | 13.99% | 11.91% | 2.08% | 1.17 | |
| Hydrothermae | 0.71% | ND | 0.71% | ∞ | |
| Synergistota | 12.90% | 14.28% | 1.38% | 1.11 | |
| Thermotogota | 30.40% | 31.43% | 1.03% | 1.03 | |
| Chloroflexota | Inoculum | 1.24% | 0.94% | 0.30% | 1.32 |
| Coprothermobacterota | 19.72% | 25.31% | 5.58% | 1.28 | |
| Patescibacteria | 3.22% | 0.12% | 3.10% | 26.9 | |
| Verrucomicrobiota | 0.29% | 0.39% | 0.09% | 1.32 | |
| Acidobacteriota | PWASS | 1.22% | 0.29% | 0.93% | 4.22 |
| Caldatribacteriota | 0.32% | 0.26% | 0.07% | 1.26 | |
| Caldisericota | 0.43% | 0.58% | 0.15% | 1.36 | |
| Halobacterota | 1.43% | 1.83% | 0.40% | 1.28 | |
| Patescibacteria | 1.00% | 0.02% | 0.98% | 46.2 | |
| Proteobacteria | 34.55% | 36.33% | 1.78% | 1.05 | |
| Synergistota | 2.06% | 2.47% | 0.41% | 1.20 |
| AD Reactors Only | AD and PWASS | |||
|---|---|---|---|---|
| Explanatory Variable | Without Pipeline Interaction | Including pipeline | Without Pipeline Interaction | Including Pipeline |
| Pipeline | *** | NA | . | NA |
| Treatment | *** | … | *** | .. |
| Timepoint | *** | … | * | … |
| Timepoint * Treatment | *** | … | * | … |
| Timepoint * Treatment * Temperature | NA | NA | *** | * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeske, J.T.; Gallert, C. Microbiome Analysis via OTU and ASV-Based Pipelines—A Comparative Interpretation of Ecological Data in WWTP Systems. Bioengineering 2022, 9, 146. https://doi.org/10.3390/bioengineering9040146
Jeske JT, Gallert C. Microbiome Analysis via OTU and ASV-Based Pipelines—A Comparative Interpretation of Ecological Data in WWTP Systems. Bioengineering. 2022; 9(4):146. https://doi.org/10.3390/bioengineering9040146
Chicago/Turabian StyleJeske, Jan Torsten, and Claudia Gallert. 2022. "Microbiome Analysis via OTU and ASV-Based Pipelines—A Comparative Interpretation of Ecological Data in WWTP Systems" Bioengineering 9, no. 4: 146. https://doi.org/10.3390/bioengineering9040146
APA StyleJeske, J. T., & Gallert, C. (2022). Microbiome Analysis via OTU and ASV-Based Pipelines—A Comparative Interpretation of Ecological Data in WWTP Systems. Bioengineering, 9(4), 146. https://doi.org/10.3390/bioengineering9040146