
How Do Mechanics Guide Fibroblast Activity? Complex Disruptions during Emphysema Shape Cellular Responses and Limit Research


 , ,
, , {kind=link}
{kind=link}
Abstract
1. Introduction
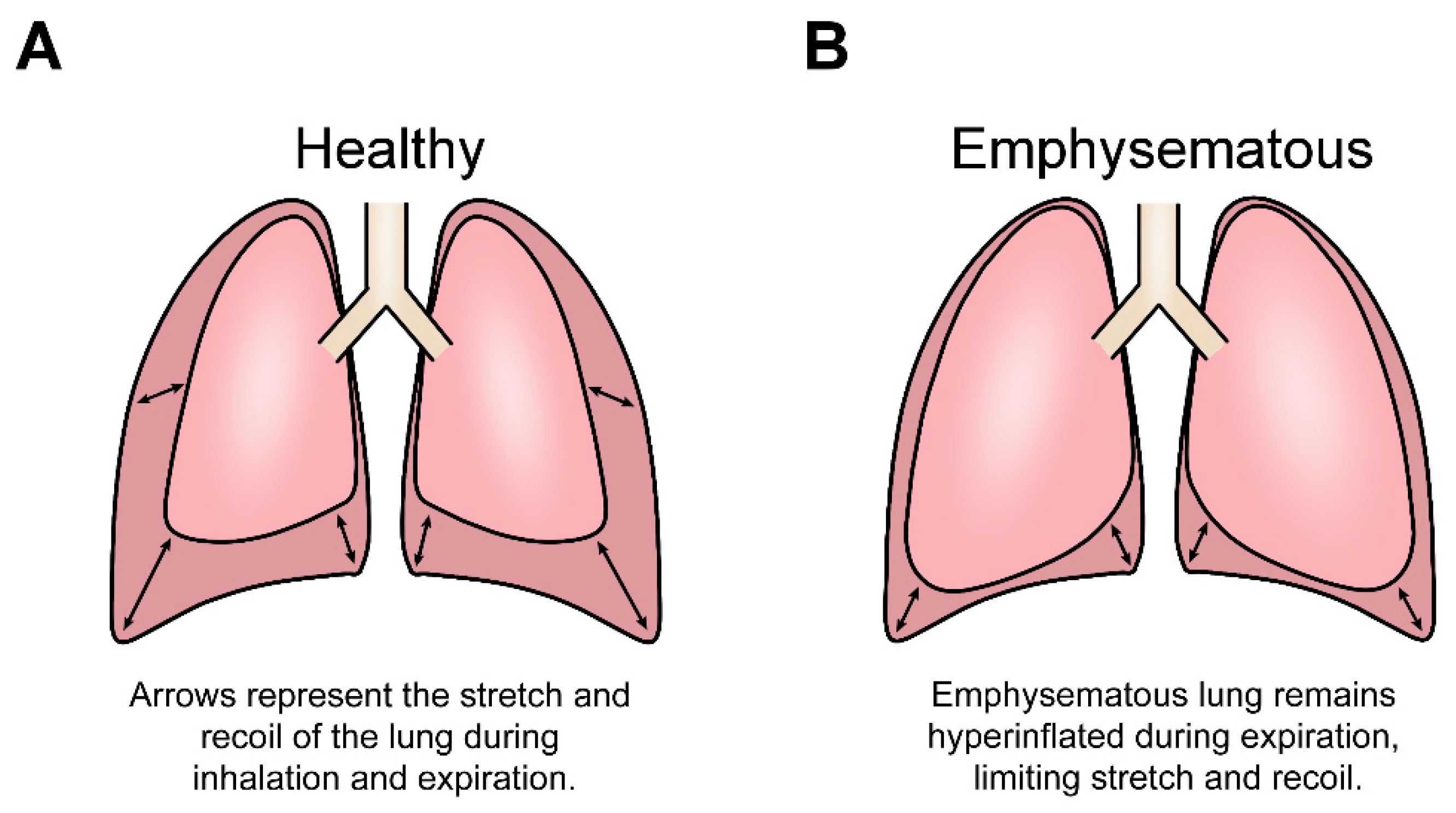
1.1. Lung Structure Allows for the Cyclical Stretch and Recoil of Inspiration and Expiration
1.2. Emphysema Disease Progression Alters Mechanical Cues by Destroying Lung Structure
1.3. Fibroblasts Maintain Their Mechanical Environment by Maintaining the ECM
2. Mechanical Cues Guide Fibroblast ECM Maintenance
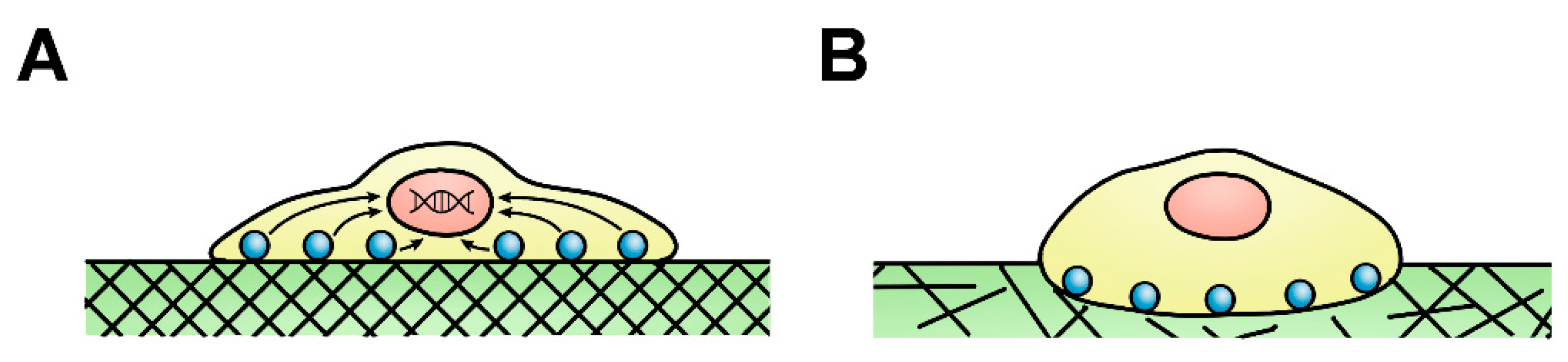
2.1. Mechanical Cues Dynamically Regulate Fibroblast Activity
2.2. Fibroblast Heterogeneity Demonstrates Mechanical Adaptations
2.3. A Healthy Mechanical Environment Is Essential to Preserve Lung Function and Prevent Lung Pathologies
3. The Emphysematous Fibroblast Phenotype Could Be Caused by Mechanical Cues
3.1. The Effects of Emphysematous Progression on Lung Fibroblasts Correlate with the Effects of Lessened Mechanical Cues
3.2. The Complexity of the Emphysematous Environment Limits Research into the Disease
4. The Mechanical Nature of Emphysema Limits the Study and Treatment of the Disease
4.1. In Vitro Models of Emphysema Rarely Replicate the Lung Mechanical Environment
4.2. Altered Emphysematous Lung Mechanobiology Is Inadequately Represented in In Vivo Models of Emphysema and Is Not Reversed by Current Treatments
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Burgstaller, G.; Oehrle, B.; Gerckens, M.; White, E.S.; Schiller, H.B.; Eickelberg, O. The Instructive Extracellular Matrix of the Lung: Basic Composition and Alterations in Chronic Lung Disease. Eur. Respir. J. 2017, 50. [Google Scholar] [CrossRef]
- Macklem, P.T. The Mechanics of Breathing. Am. J. Respir. Crit. Care Med. 1998, 157, S88–S94. [Google Scholar] [CrossRef] [PubMed]
- Rohrer, F. Physiologie der Atembewegung. In Handbuch der Normalen und Pathologischen Physiologie: Zweiter Band Atmung; Aufnahme und Abgabe Gasförmiger Stoffe; Bethe, A., Embden, G.V., Bergmann, G., Ellinger, A., Amersbach, K., Bayer, G., Brunner, A., Felix, W., Flury, F., Eds.; Springer: Berlin/Heidelberg, Germany, 1925; pp. 70–127. ISBN 978-3-642-91002-9. [Google Scholar]
- Sorokin, L. The Impact of the Extracellular Matrix on Inflammation. Nat. Rev. Immunol. 2010, 10, 712–723. [Google Scholar] [CrossRef] [PubMed]
- Herrera, J.; Henke, C.A.; Bitterman, P.B. Extracellular Matrix as a Driver of Progressive Fibrosis. J. Clin. Investig. 2018, 128, 45–53. [Google Scholar] [CrossRef]
- Humphrey, J.D.; Dufresne, E.R.; Schwartz, M.A. Mechanotransduction and Extracellular Matrix Homeostasis. Nat. Rev. Mol. Cell Biol. 2014, 15, 802–812. [Google Scholar] [CrossRef] [PubMed]
- Leung, D.Y.; Glagov, S.; Mathews, M.B. Cyclic Stretching Stimulates Synthesis of Matrix Components by Arterial Smooth Muscle Cells in Vitro. Science 1976, 191, 475–477. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, M.; Fung, Y.C.; Sobin, S.S. Collagen and Elastin Fibers in Human Pulmonary Alveolar Mouths and Ducts. J. Appl. Physiol. 1987, 63, 1185–1194. [Google Scholar] [CrossRef] [PubMed]
- Shelton, L.; Rada, J.S. Effects of Cyclic Mechanical Stretch on Extracellular Matrix Synthesis by Human Scleral Fibroblasts. Exp. Eye Res. 2007, 84, 314–322. [Google Scholar] [CrossRef] [PubMed]
- Faffe, D.S.; Zin, W.A. Lung Parenchymal Mechanics in Health and Disease. Physiol. Rev. 2009, 89, 759–775. [Google Scholar] [CrossRef] [PubMed]
- Suki, B.; Stamenovic, D.; Hubmayr, R. Lung Parenchymal Mechanics. Compr. Physiol. 2011, 1, 1317. [Google Scholar] [CrossRef]
- Chiquet, M.; Gelman, L.; Lutz, R.; Maier, S. From Mechanotransduction to Extracellular Matrix Gene Expression in Fibroblasts. Biochim. Biophys. Acta 2009, 1793, 911–920. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.H.-C.; Thampatty, B.P. An Introductory Review of Cell Mechanobiology. Biomech. Model. Mechanobiol. 2006, 5, 1–16. [Google Scholar] [CrossRef]
- Yue, B. Biology of the Extracellular Matrix: An Overview. J. Glaucoma 2014, 23, S20–S23. [Google Scholar] [CrossRef]
- Raghow, R. The Role of Extracellular Matrix in Postinflammatory Wound Healing and Fibrosis. FASEB J. 1994, 8, 823–831. [Google Scholar] [CrossRef]
- Rosmark, O.; Åhrman, E.; Müller, C.; Elowsson Rendin, L.; Eriksson, L.; Malmström, A.; Hallgren, O.; Larsson-Callerfelt, A.-K.; Westergren-Thorsson, G.; Malmström, J. Quantifying Extracellular Matrix Turnover in Human Lung Scaffold Cultures. Sci. Rep. 2018, 8, 5409. [Google Scholar] [CrossRef]
- White, E.S. Lung Extracellular Matrix and Fibroblast Function. Ann. Am. Thorac. Soc. 2015, 12, S30–S33. [Google Scholar] [CrossRef] [PubMed]
- Pryor, W.A.; Stone, K. Oxidants in Cigarette Smoke. Radicals, Hydrogen Peroxide, Peroxynitrate, and Peroxynitrite. Ann. N. Y. Acad. Sci. 1993, 686. [Google Scholar] [CrossRef] [PubMed]
- Decramer, M.; Janssens, W.; Miravitlles, M. Chronic Obstructive Pulmonary Disease. Lancet Lond. Engl. 2012, 379, 1341–1351. [Google Scholar] [CrossRef]
- Papandrinopoulou, D.; Tzouda, V.; Tsoukalas, G. Lung Compliance and Chronic Obstructive Pulmonary Disease. Pulm. Med. 2012, 2012. [Google Scholar] [CrossRef]
- Pandey, K.C.; De, S.; Mishra, P.K. Role of Proteases in Chronic Obstructive Pulmonary Disease. Front. Pharmacol. 2017, 8, 502. [Google Scholar] [CrossRef] [PubMed]
- Bates, J.H.T.; Davis, G.S.; Majumdar, A.; Butnor, K.J.; Suki, B. Linking Parenchymal Disease Progression to Changes in Lung Mechanical Function by Percolation. Am. J. Respir. Crit. Care Med. 2007, 176, 617–623. [Google Scholar] [CrossRef] [PubMed]
- Goldklang, M.; Stockley, R. Pathophysiology of Emphysema and Implications. Chronic Obstr. Pulm. Dis. 2016, 3, 454–458. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Suki, B.; Jesudason, R.; Sato, S.; Parameswaran, H.; Araujo, A.D.; Majumdar, A.; Allen, P.G.; Bartolák-Suki, E. Mechanical Failure, Stress Redistribution, Elastase Activity and Binding Site Availability on Elastin during the Progression of Emphysema. Pulm. Pharmacol. Ther. 2012, 25, 268–275. [Google Scholar] [CrossRef]
- Turino, G.M. Emphysema in COPD: Consequences and Causes. Thorax 2006, 61, 1031–1036. [Google Scholar] [CrossRef][Green Version]
- Yang, S.; Chida, A.; Bauter, M.; Shafiq, N.; Seweryniak, K.; Maggirwar, S.; Kilty, I.; Rahman, I. Cigarette Smoke Induces Proinflammatory Cytokine Release by Activation of NF-KappaB and Posttranslational Modifications of Histone Deacetylase in Macrophages. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 291. [Google Scholar] [CrossRef] [PubMed]
- Sharafkhaneh, A.; Hanania, N.A.; Kim, V. Pathogenesis of Emphysema: From the Bench to the Bedside. Proc. Am. Thorac. Soc. 2008, 5, 475. [Google Scholar] [CrossRef] [PubMed]
- Suki, B.; Lutchen, K.R.; Ingenito, E.P. On the Progressive Nature of Emphysema. Am. J. Respir. Crit. Care Med. 2003, 168, 516–521. [Google Scholar] [CrossRef]
- Bates, J.H.T.; Maksym, G.N.; Navajas, D.; Suki, B. Lung Tissue Rheology and 1/f Noise. Ann. Biomed. Eng. 1994, 22, 674–681. [Google Scholar] [CrossRef]
- De Ryk, J.; Thiesse, J.; Namati, E.; McLennan, G. Stress Distribution in a Three Dimensional, Geometric Alveolar Sac under Normal and Emphysematous Conditions. Int. J. Chronic Obstr. Pulm. Dis. 2007, 2, 81. [Google Scholar] [CrossRef]
- Togo, S.; Holz, O.; Liu, X.; Sugiura, H.; Kamio, K.; Wang, X.; Kawasaki, S.; Ahn, Y.; Fredriksson, K.; Skold, C.M.; et al. Lung Fibroblast Repair Functions in Patients with Chronic Obstructive Pulmonary Disease Are Altered by Multiple Mechanisms. Am. J. Respir. Crit. Care Med. 2008, 178, 248–260. [Google Scholar] [CrossRef]
- Hadjipanayi, E.; Mudera, V.; Brown, R.A. Close Dependence of Fibroblast Proliferation on Collagen Scaffold Matrix Stiffness. J. Tissue Eng. Regen. Med. 2009, 3, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Boudreault, F.; Tschumperlin, D.J. Stretch-Induced Mitogen-Activated Protein Kinase Activation in Lung Fibroblasts Is Independent of Receptor Tyrosine Kinases. Am. J. Respir. Cell Mol. Biol. 2010, 43, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Tschumperlin, D.J.; Ligresti, G.; Hilscher, M.B.; Shah, V.H. Mechanosensing and Fibrosis. J. Clin. Investig. 2018, 128, 74. [Google Scholar] [CrossRef]
- Breen, E.C. Mechanical Strain Increases Type I Collagen Expression in Pulmonary Fibroblasts in Vitro. J. Appl. Physiol. 2000, 88, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Lauffenburger, D.A.; Horwitz, A.F. Cell Migration: A Physically Integrated Molecular Process. Cell 1996, 84, 359–369. [Google Scholar] [CrossRef]
- Liu, F.; Lagares, D.; Choi, K.M.; Stopfer, L.; Marinković, A.; Vrbanac, V.; Probst, C.K.; Hiemer, S.E.; Sisson, T.H.; Horowitz, J.C.; et al. Mechanosignaling through YAP and TAZ Drives Fibroblast Activation and Fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 308, L344–L357. [Google Scholar] [CrossRef]
- Marinković, A.; Liu, F.; Tschumperlin, D.J. Matrices of Physiologic Stiffness Potently Inactivate Idiopathic Pulmonary Fibrosis Fibroblasts. Am. J. Respir. Cell Mol. Biol. 2012, 48, 422–430. [Google Scholar] [CrossRef]
- Liu, F.; Mih, J.D.; Shea, B.S.; Kho, A.T.; Sharif, A.S.; Tager, A.M.; Tschumperlin, D.J. Feedback Amplification of Fibrosis through Matrix Stiffening and COX-2 Suppression. J. Cell Biol. 2010, 190, 693. [Google Scholar] [CrossRef] [PubMed]
- Discher, D.E.; Janmey, P.; Wang, Y.-L. Tissue Cells Feel and Respond to the Stiffness of Their Substrate. Science 2005, 310, 1139–1143. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Zapata, A.M.; Heinz, A.; Kerkhof, M.H.; van de Westerlo-van Rijit, C.; Schmelzer, C.E.H.; Stoop, R.; Kluivers, K.B.; Oosterwijk, E. Extracellular Matrix Stiffness and Composition Regulate the Myofibroblast Differentiation of Vaginal Fibroblasts. Int. J. Mol. Sci. 2020, 21, 4762. [Google Scholar] [CrossRef]
- Tschumperlin, D.J.; Boudreault, F.; Liu, F. Recent Advances and New Opportunities in Lung Mechanobiology. J. Biomech. 2010, 43, 99–107. [Google Scholar] [CrossRef]
- Foote, A.G.; Wang, Z.; Kendziorski, C.; Thibeault, S.L. Tissue Specific Human Fibroblast Differential Expression Based on RNAsequencing Analysis. BMC Genom. 2019, 20. [Google Scholar] [CrossRef]
- Parker, M.W.; Rossi, D.; Peterson, M.; Smith, K.; Sikström, K.; White, E.S.; Connett, J.E.; Henke, C.A.; Larsson, O.; Bitterman, P.B. Fibrotic Extracellular Matrix Activates a Profibrotic Positive Feedback Loop. J. Clin. Investig. 2014, 124. [Google Scholar] [CrossRef]
- Booth, A.J.; Hadley, R.; Cornett, A.M.; Dreffs, A.A.; Matthes, S.A.; Tsui, J.L.; Weiss, K.; Horowitz, J.C.; Fiore, V.F.; Barker, T.H.; et al. Acellular Normal and Fibrotic Human Lung Matrices as a Culture System for In Vitro Investigation. Am. J. Respir. Crit. Care Med. 2012, 186, 866–876. [Google Scholar] [CrossRef]
- Kulkarni, T.; O’Reilly, P.; Antony, V.B.; Gaggar, A.; Thannickal, V.J. Matrix Remodeling in Pulmonary Fibrosis and Emphysema. Am. J. Respir. Cell Mol. Biol. 2016, 54, 751–760. [Google Scholar] [CrossRef] [PubMed]
- Suki, B.; Sato, S.; Parameswaran, H.; Szabari, M.V.; Takahashi, A.; Bartolák-Suki, E. Emphysema and Mechanical Stress-Induced Lung Remodeling. Physiology 2013, 28, 404–413. [Google Scholar] [CrossRef] [PubMed]
- Roy, B.; Yuan, L.; Lee, Y.; Bharti, A.; Mitra, A.; Shivashankar, G.V. Fibroblast Rejuvenation by Mechanical Reprogramming and Redifferentiation. Proc. Natl. Acad. Sci. USA 2020, 117, 10131–10141. [Google Scholar] [CrossRef] [PubMed]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A Framework for Advancing Our Understanding of Cancer-Associated Fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef]
- Wang, J.H.-C.; Thampatty, B.P.; Lin, J.-S.; Im, H.-J. Mechanoregulation of Gene Expression in Fibroblasts. Gene 2007, 391, 1. [Google Scholar] [CrossRef]
- Kono, T.; Tanii, T.; Furukawa, M.; Mizuno, N.; Kitajima, J.; Ishii, M.; Hamada, T.; Yoshizato, K. Cell Cycle Analysis of Human Dermal Fibroblasts Cultured on or in Hydrated Type I Collagen Lattices. Arch. Dermatol. Res. 1990, 282, 258–262. [Google Scholar] [CrossRef]
- Sarber, R.; Hull, B.; Merrill, C.; Sorrano, T.; Bell, E. Regulation of Proliferation of Fibroblasts of Low and High Population Doubling Levels Grown in Collagen Lattices. Mech. Ageing Dev. 1981, 17, 107–117. [Google Scholar] [CrossRef]
- Cross, V.L.; Zheng, Y.; Choi, N.W.; Verbridge, S.S.; Sutermaster, B.A.; Bonassar, L.J.; Fischbach, C.; Stroock, A.D. Dense Type I Collagen Matrices That Support Cellular Remodeling and Microfabrication for Studies of Tumor Angiogenesis and Vasculogenesis in Vitro. Biomaterials 2010, 31, 8596–8607. [Google Scholar] [CrossRef]
- Butcher, D.T.; Alliston, T.; Weaver, V.M. A Tense Situation: Forcing Tumour Progression. Nat. Rev. Cancer 2009, 9, 108–122. [Google Scholar] [CrossRef]
- Benham-Pyle, B.W.; Pruitt, B.L.; Nelson, W.J. Cell Adhesion. Mechanical Strain Induces E-Cadherin-Dependent Yap1 and β-Catenin Activation to Drive Cell Cycle Entry. Science 2015, 348, 1024–1027. [Google Scholar] [CrossRef]
- Desprat, N.; Supatto, W.; Pouille, P.-A.; Beaurepaire, E.; Farge, E. Tissue Deformation Modulates Twist Expression to Determine Anterior Midgut Differentiation in Drosophila Embryos. Dev. Cell 2008, 15, 470–477. [Google Scholar] [CrossRef]
- Engler, A.J.; Sen, S.; Sweeney, H.L.; Discher, D.E. Matrix Elasticity Directs Stem Cell Lineage Specification. Cell 2006, 126, 677–689. [Google Scholar] [CrossRef]
- Huang, S.; Chen, C.S.; Ingber, D.E. Control of Cyclin D1, P27(Kip1), and Cell Cycle Progression in Human Capillary Endothelial Cells by Cell Shape and Cytoskeletal Tension. Mol. Biol. Cell 1998, 9, 3179–3193. [Google Scholar] [CrossRef]
- Maître, J.-L.; Turlier, H.; Illukkumbura, R.; Eismann, B.; Niwayama, R.; Nédélec, F.; Hiiragi, T. Asymmetric Division of Contractile Domains Couples Cell Positioning and Fate Specification. Nature 2016, 536, 344–348. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Landmann, F.; Zahreddine, H.; Rodriguez, D.; Koch, M.; Labouesse, M. A Tension-Induced Mechanotransduction Pathway Promotes Epithelial Morphogenesis. Nature 2011, 471, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Harper, R.A.; Grove, G. Human Skin Fibroblasts Derived from Papillary and Reticular Dermis: Differences in Growth Potential in Vitro. Science 1979, 204, 526–527. [Google Scholar] [CrossRef] [PubMed]
- Muhl, L.; Genové, G.; Leptidis, S.; Liu, J.; He, L.; Mocci, G.; Sun, Y.; Gustafsson, S.; Buyandelger, B.; Chivukula, I.V.; et al. Single-Cell Analysis Uncovers Fibroblast Heterogeneity and Criteria for Fibroblast and Mural Cell Identification and Discrimination. Nat. Commun. 2020, 11, 3953. [Google Scholar] [CrossRef]
- Gurkan, U.A.; Akkus, O. The Mechanical Environment of Bone Marrow: A Review. Ann. Biomed. Eng. 2008, 36, 1978–1991. [Google Scholar] [CrossRef]
- Kasper, G.; Dankert, N.; Tuischer, J.; Hoeft, M.; Gaber, T.; Glaeser, J.D.; Zander, D.; Tschirschmann, M.; Thompson, M.; Matziolis, G.; et al. Mesenchymal Stem Cells Regulate Angiogenesis According to Their Mechanical Environment. STEM CELLS 2007, 25, 903–910. [Google Scholar] [CrossRef] [PubMed]
- D’Urso, M.; Kurniawan, N.A. Mechanical and Physical Regulation of Fibroblast–Myofibroblast Transition: From Cellular Mechanoresponse to Tissue Pathology. Front. Bioeng. Biotechnol. 2020, 8. [Google Scholar] [CrossRef]
- Mascharak, S.; desJardins-Park, H.E.; Longaker, M.T. Fibroblast Heterogeneity in Wound Healing: Hurdles to Clinical Translation. Trends Mol. Med. 2020, 26, 1101–1106. [Google Scholar] [CrossRef] [PubMed]
- Abercrombie, M.; Heaysman, J.E.; Pegrum, S.M. The Locomotion of Fibroblasts in Culture. II. “Ruffling”. Exp. Cell Res. 1970, 60, 437–444. [Google Scholar] [CrossRef]
- Hoon, J.-L.; Wong, W.-K.; Koh, C.-G. Functions and Regulation of Circular Dorsal Ruffles. Mol. Cell. Biol. 2012, 32, 4246–4257. [Google Scholar] [CrossRef]
- Deng, Z.; Fear, M.W.; Suk Choi, Y.; Wood, F.M.; Allahham, A.; Mutsaers, S.E.; Prêle, C.M. The Extracellular Matrix and Mechanotransduction in Pulmonary Fibrosis. Int. J. Biochem. Cell Biol. 2020, 126, 105802. [Google Scholar] [CrossRef]
- Ogawa, R. Keloid and Hypertrophic Scarring May Result from a Mechanoreceptor or Mechanosensitive Nociceptor Disorder. Med. Hypotheses 2008, 71. [Google Scholar] [CrossRef]
- Burgess, J.K.; Mauad, T.; Tjin, G.; Karlsson, J.C.; Westergren-Thorsson, G. The Extracellular Matrix—The under-Recognized Element in Lung Disease? J. Pathol. 2016, 240, 397–409. [Google Scholar] [CrossRef]
- Ito, S.; Ingenito, E.P.; Brewer, K.K.; Black, L.D.; Parameswaran, H.; Lutchen, K.R.; Suki, B. Mechanics, Nonlinearity, and Failure Strength of Lung Tissue in a Mouse Model of Emphysema: Possible Role of Collagen Remodeling. J. Appl. Physiol. 2005, 98, 503–511. [Google Scholar] [CrossRef]
- Jones, R.L.; Noble, P.B.; Elliot, J.G.; James, A.L. Airway Remodelling in COPD: It’s Not Asthma! Respirology 2016, 21, 1347–1356. [Google Scholar] [CrossRef]
- Holz, O.; Zühlke, I.; Jaksztat, E.; Müller, K.C.; Welker, L.; Nakashima, M.; Diemel, K.D.; Branscheid, D.; Magnussen, H.; Jörres, R.A. Lung Fibroblasts from Patients with Emphysema Show a Reduced Proliferation Rate in Culture. Eur. Respir. J. 2004, 24, 575–579. [Google Scholar] [CrossRef] [PubMed]
- Phillip, J.M.; Aifuwa, I.; Walston, J.; Wirtz, D. The Mechanobiology of Aging. Annu. Rev. Biomed. Eng. 2015, 17, 113–141. [Google Scholar] [CrossRef]
- Mitzner, W. Emphysema: A Disease of Small Airways or Lung Parenchyma? N. Engl. J. Med. 2011, 365, 1637–1639. [Google Scholar] [CrossRef]
- Dagouassat, M.; Gagliolo, J.; Chrusciel, S.; Bourin, M.; Duprez, C.; Caramelle, P.; Boyer, L.; Hue, S.; Stern, J.; Validire, P.; et al. The Cyclooxygenase-2-Prostaglandin E2 Pathway Maintains Senescence of Chronic Obstructive Pulmonary Disease Fibroblasts. Am. J. Respir. Crit. Care Med. 2013, 187. [Google Scholar] [CrossRef]
- Noordhoek, J.A.; Postma, D.S.; Chong, L.L.; Vos, J.T.W.M.; Kauffman, H.F.; Timens, W.; Van Straaten, J.F.M. Different Proliferative Capacity of Lung Fibroblasts Obtained from Control Subjects and Patients with Emphysema. Exp. Lung Res. 2003, 29, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Isik, B.; Ceylan, A.; Isik, R. Oxidative Stress in Smokers and Non-Smokers. Inhal. Toxicol. 2007, 19, 767–769. [Google Scholar] [CrossRef]
- Eickelberg, O.; Köhler, E.; Reichenberger, F.; Bertschin, S.; Woodtli, T.; Erne, P.; Perruchoud, A.P.; Roth, M. Extracellular Matrix Deposition by Primary Human Lung Fibroblasts in Response to TGF-Β1 and TGF-Β3. Am. J. Physiol. Lung Cell. Mol. Physiol. 1999, 276, L814–L824. [Google Scholar] [CrossRef]
- Plantier, L.; Boczkowski, J.; Crestani, B. Defect of Alveolar Regeneration in Pulmonary Emphysema: Role of Lung Fibroblasts. Int. J. Chronic Obstr. Pulm. Dis. 2007, 2, 463–469. [Google Scholar]
- Sanchez-Esteban, J.; Cicchiello, L.A.; Wang, Y.; Tsai, S.-W.; Williams, L.K.; Torday, J.S.; Rubin, L.P. Mechanical Stretch Promotes Alveolar Epithelial Type II Cell Differentiation. J. Appl. Physiol. 2001, 91, 589–595. [Google Scholar] [CrossRef]
- O’Donnell, D.E.; Parker, C.M. COPD Exacerbations. 3: Pathophysiology. Thorax 2006, 61, 354–361. [Google Scholar] [CrossRef] [PubMed]
- de Hilster, R.H.J.; Sharma, P.K.; Jonker, M.R.; White, E.S.; Gercama, E.A.; Roobeek, M.; Timens, W.; Harmsen, M.C.; Hylkema, M.N.; Burgess, J.K. Human Lung Extracellular Matrix Hydrogels Resemble the Stiffness and Viscoelasticity of Native Lung Tissue. Am. J. Physiol. Lung Cell. Mol. Physiol. 2020, 318, L698–L704. [Google Scholar] [CrossRef]
- Kleinman, H.K.; Martin, G.R. Matrigel: Basement Membrane Matrix with Biological Activity. Semin. Cancer Biol. 2005, 15, 378–386. [Google Scholar] [CrossRef]
- Zhang, Y.; He, Y.; Bharadwaj, S.; Hammam, N.; Carnagey, K.; Myers, R.; Atala, A.; Van Dyke, M. Tissue-Specific Extracellular Matrix Coatings for the Promotion of Cell Proliferation and Maintenance of Cell Phenotype. Biomaterials 2009, 30, 4021–4028. [Google Scholar] [CrossRef]
- Doryab, A.; Tas, S.; Taskin, M.B.; Yang, L.; Hilgendorff, A.; Groll, J.; Wagner, D.E.; Schmid, O. Evolution of Bioengineered Lung Models: Recent Advances and Challenges in Tissue Mimicry for Studying the Role of Mechanical Forces in Cell Biology. Adv. Funct. Mater. 2019, 29, 1903114. [Google Scholar] [CrossRef]
- Blaauboer, M.E.; Smit, T.H.; Hanemaaijer, R.; Stoop, R.; Everts, V. Cyclic Mechanical Stretch Reduces Myofibroblast Differentiation of Primary Lung Fibroblasts. Biochem. Biophys. Res. Commun. 2011, 404, 23–27. [Google Scholar] [CrossRef]
- Huh, D.; Kim, H.J.; Fraser, J.P.; Shea, D.E.; Khan, M.; Bahinski, A.; Hamilton, G.A.; Ingber, D.E. Microfabrication of Human Organs-on-Chips. Nat. Protoc. 2013, 8, 2135–2157. [Google Scholar] [CrossRef] [PubMed]
- Bonakdar, N.; Schilling, A.; Lennert, P.; Spörrer, M.; Gerum, R.C.; Alonso, J.L.; Goldmann, W.H. Measuring Mechanical Properties in Cells: Three Easy Methods for Biologists. Cell Biol. Int. 2014, 38, 1227–1232. [Google Scholar] [CrossRef] [PubMed]
- Hart, K.C.; Sim, J.Y.; Hopcroft, M.A.; Cohen, D.J.; Tan, J.; Nelson, W.J.; Pruitt, B.L. An Easy-to-Fabricate Cell Stretcher Reveals Density-Dependent Mechanical Regulation of Collective Cell Movements in Epithelia. bioRxiv 2021. [Google Scholar] [CrossRef]
- Kamble, H.; Barton, M.J.; Jun, M.; Park, S.; Nguyen, N.-T. Cell Stretching Devices as Research Tools: Engineering and Biological Considerations. Lab. Chip 2016, 16, 3193–3203. [Google Scholar] [CrossRef]
- Seriani, S.; Del Favero, G.; Mahaffey, J.; Marko, D.; Gallina, P.; Long, C.S.; Mestroni, L.; Sbaizero, O. The Cell-Stretcher: A Novel Device for the Mechanical Stimulation of Cell Populations. Rev. Sci. Instrum. 2016, 87, 084301. [Google Scholar] [CrossRef]
- Gould, R.A.; Chin, K.; Santisakultarm, T.P.; Dropkin, A.; Richards, J.M.; Schaffer, C.B.; Butcher, J.T. Cyclic Strain Anisotropy Regulates Valvular Interstitial Cell Phenotype and Tissue Remodeling in Three-Dimensional Culture. Acta Biomater. 2012, 8, 1710–1719. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.-Y.; Liu, Y.-C.; Wang, M.-X.; Hu, J.-J. Fibroblast-Seeded Collagen Gels in Response to Dynamic Equibiaxial Mechanical Stimuli: A Biomechanical Study. J. Biomech. 2018, 78, 134–142. [Google Scholar] [CrossRef]
- John, J.; Throm Quinlan, A.; Silvestri, C.; Billiar, K. Boundary Stiffness Regulates Fibroblast Behavior in Collagen Gels. Ann. Biomed. Eng. 2010, 38, 658–673. [Google Scholar] [CrossRef]
- Liang, G.-B.; He, Z.-H. Animal Models of Emphysema. Chin. Med. J. 2019, 132, 2465–2475. [Google Scholar] [CrossRef]
- Baron, R.M.; Choi, A.J.S.; Owen, C.A.; Choi, A.M.K. Genetically Manipulated Mouse Models of Lung Disease: Potential and Pitfalls. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 302, L485–L497. [Google Scholar] [CrossRef] [PubMed]
- Hautamaki, R.D.; Kobayashi, D.K.; Senior, R.M.; Shapiro, S.D. Requirement for Macrophage Elastase for Cigarette Smoke-Induced Emphysema in Mice. Science 1997, 277, 2002–2004. [Google Scholar] [CrossRef] [PubMed]
- Busch, R.H.; Lauhala, K.E.; Loscutoff, S.M.; McDonald, K.E. Experimental Pulmonary Emphysema Induced in the Rat by Intratracheally Administered Elastase: Morphogenesis. Environ. Res. 1984, 33, 497–513. [Google Scholar] [CrossRef]
- Tanner, L.; Single, A.B. Animal Models Reflecting Chronic Obstructive Pulmonary Disease and Related Respiratory Disorders: Translating Pre-Clinical Data into Clinical Relevance. J. Innate Immun. 2020, 12, 203–225. [Google Scholar] [CrossRef] [PubMed]
- Winkler, T.; Suki, B. Emergent Structure–Function Relations in Emphysema and Asthma. Crit. Rev. Biomed. Eng. 2011, 39, 263. [Google Scholar] [CrossRef]
- Cagle, P.T.; Thurlbeck, W.M. Postpneumonectomy Compensatory Lung Growth. Am. Rev. Respir. Dis. 1988, 138, 1314–1326. [Google Scholar] [CrossRef] [PubMed]
- Massaro, D.; Alexander, E.; Reiland, K.; Hoffman, E.P.; Massaro, G.D.; Clerch, L.B. Rapid Onset of Gene Expression in Lung, Supportive of Formation of Alveolar Septa, Induced by Refeeding Mice after Calorie Restriction. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 292. [Google Scholar] [CrossRef] [PubMed]
- Chu, X.; Chen, C.; Chen, C.; Zhang, J.-S.; Bellusci, S.; Li, X. Evidence for Lung Repair and Regeneration in Humans: Key Stem Cells and Therapeutic Functions of Fibroblast Growth Factors. Front. Med. 2020, 14, 262–272. [Google Scholar] [CrossRef]
- Zepp, J.A.; Zacharias, W.J.; Frank, D.B.; Cavanaugh, C.A.; Zhou, S.; Morley, M.P.; Morrisey, E.E. Distinct Mesenchymal Lineages and Niches Promote Epithelial Self-Renewal and Myofibrogenesis in the Lung. Cell 2017, 170, 1134. [Google Scholar] [CrossRef] [PubMed]
- Celli, B.; MacNee, W. Standards for the Diagnosis and Treatment of Patients with COPD: A Summary of the ATS/ERS Position Paper. Eur. Respir. J. 2004, 23. [Google Scholar] [CrossRef] [PubMed]
- Camp, P.C.; Sugarbaker, D.J. Surgical Interventions for Emphysema. Semin. Thorac. Cardiovasc. Surg. 2007, 19, 157–171. [Google Scholar] [CrossRef]
- Fishman, A.; Martinez, F.; Naunheim, K.; Piantadosi, S.; Wise, R.; Ries, A.; Weinmann, G.; Wood, D. A Randomized Trial Comparing Lung-Volume-Reduction Surgery with Medical Therapy for Severe Emphysema. N. Engl. J. Med. 2003, 348. [Google Scholar] [CrossRef]
- Wan, I.Y.P.; Toma, T.P.; Geddes, D.M.; Snell, G.; Williams, T.; Venuta, F.; Yim, A.P.C. Bronchoscopic Lung Volume Reduction for End-Stage Emphysema: Report on the First 98 Patients. Chest 2006, 129, 518–526. [Google Scholar] [CrossRef]
- Shah, P.L.; Herth, F.J.; van Geffen, W.H.; Deslee, G.; Slebos, D.-J. Lung Volume Reduction for Emphysema. Lancet Respir. Med. 2017, 5, 147–156. [Google Scholar] [CrossRef]
- Hopkinson, N.S.; Toma, T.P.; Hansell, D.M.; Goldstraw, P.; Moxham, J.; Geddes, D.M.; Polkey, M.I. Effect of Bronchoscopic Lung Volume Reduction on Dynamic Hyperinflation and Exercise in Emphysema. Am. J. Respir. Crit. Care Med. 2005, 171, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Gurtner, G.; Dauskardt, R.; Wong, V.; Bhatt, K.A.; Wu, K.; Vial, I.N.; Padois, K.; Korman, J.M.; Longaker, M.T. Improving Cutaneous Scar Formation by Controlling the Mechanical Environment: Large Animal and Phase I Studies. Ann. Surg. 2011, 254. [Google Scholar] [CrossRef] [PubMed]
- Müller, K.-C.; Paasch, K.; Feindt, B.; Welker, L.; Watz, H.; Weise, M.; Schmid, R.A.; Nakashima, M.; Branscheid, D.; Magnussen, H.; et al. In Contrast to Lung Fibroblasts—No Signs of Senescence in Skin Fibroblasts of Patients with Emphysema. Exp. Gerontol. 2008, 43, 623–628. [Google Scholar] [CrossRef] [PubMed][Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leslie, M.N.; Chou, J.; Young, P.M.; Traini, D.; Bradbury, P.; Ong, H.X. How Do Mechanics Guide Fibroblast Activity? Complex Disruptions during Emphysema Shape Cellular Responses and Limit Research. Bioengineering 2021, 8, 110. https://doi.org/10.3390/bioengineering8080110
Leslie MN, Chou J, Young PM, Traini D, Bradbury P, Ong HX. How Do Mechanics Guide Fibroblast Activity? Complex Disruptions during Emphysema Shape Cellular Responses and Limit Research. Bioengineering. 2021; 8(8):110. https://doi.org/10.3390/bioengineering8080110
Chicago/Turabian StyleLeslie, Mathew N., Joshua Chou, Paul M. Young, Daniela Traini, Peta Bradbury, and Hui Xin Ong. 2021. "How Do Mechanics Guide Fibroblast Activity? Complex Disruptions during Emphysema Shape Cellular Responses and Limit Research" Bioengineering 8, no. 8: 110. https://doi.org/10.3390/bioengineering8080110
APA StyleLeslie, M. N., Chou, J., Young, P. M., Traini, D., Bradbury, P., & Ong, H. X. (2021). How Do Mechanics Guide Fibroblast Activity? Complex Disruptions during Emphysema Shape Cellular Responses and Limit Research. Bioengineering, 8(8), 110. https://doi.org/10.3390/bioengineering8080110