1. Introduction
The development of flow processes has gained tremendous interest in recent years in organic chemistry due to numerous advantages of such types of processes over classic batch approaches [
1]. In addition to higher efficiency and safety advantages, the ability to ensure constant product quality has turned out to be a major driving force for the development of flow processes. In particular, for active pharmaceutical ingredients (APIs), this aspect is important, as the FDA along with the EPA, as the respective institutions in United States and Europe for approval of medicines, added aspects of continuous manufacturing to their guidelines [
2,
3], whereas for a long time the focus of flow processes was on “classic” chemical processes with, for example, hazardous and thermally labile substances and chemocatalytic reactions [
4,
5,
6]. Recently, biocatalysis in flow processes has become an emerging field with an increasing number of examples [
7,
8,
9]. An enzymatic reaction of particular interest for an application in flow mode due to its matured technology level is asymmetric ketone reduction, having a proven track record underlined by many successful examples demonstrated in the past for the batch mode [
10,
11,
12,
13]. Recently, significant process in the biocatalytic [
14,
15,
16,
17] reductive synthesis of alcohols starting from ketones, as well as syntheses by non-enzymatic means, have been reported [
18,
19,
20,
21]. In recent years, an increasing number of examples of biocatalysis in flow processes have been reported [
22,
23,
24,
25,
26,
27]. Although only being studied to a minor extent, some examples of ketone reductions with an alcohol dehydrogenase (ADH) in flow processes have been described [
28,
29,
30,
31,
32,
33,
34]. For example, Wandrey’s and Liese’s groups studied in detail the application of enzyme membrane reactors for enzymatic ketone reduction in a continuously running mode [
28,
29], and packed bed reactor setups were described by further groups using carrier-bound ADH from
Lactobacillus brevis (LB-ADH) in combination with a mobile aqueous phase [
30], or magnetic nanoparticle-bound ADH [
31]. In addition, Buehler et al. investigated intensively the use of ADH in aqueous/organic segmented flow systems. It was shown that emulsion formation could be eliminated, and enzymatic mass transfer-limited reactions can be enhanced [
32]. Furthermore, different means of stabilization of this reaction system were investigated [
33]. Very recently, our group published an improved downstream process by means of such a segmented flow process, illustrated using two different ADHs and substrates [
34].
In continuous processes using packed bed reactors (PBR), catalyst immobilization is a prerequisite. Also, downstream processes benefit from immobilization, as separation of the reaction mixture from the catalyst is easier, and the catalyst reusability is improved [
35]. Moreover, both, operational, as well as storage stability of the catalyst can potentially be significantly increased. However, often a significant loss of enzymatic activity during the immobilization process represents a drawback, and the reproducible production of immobilized catalyst may be a further challenge [
36]. Numerous techniques have been developed over the past years, which can be classified in the following three major methods: (a) binding to a solid support (carrier), (b) entrapment (encapsulation) in polymer networks, or (c) cross-linking in the form of cross-linked enzyme aggregates (CLEAs) or cross-linked enzyme crystals (CLECs) [
35]. Recently, much progress in the field of enzyme immobilization has been made [
37,
38,
39,
40,
41].
In this work, the option of entrapment of enzymes in polymer networks has been chosen. Often for this purpose, organic polymers containing cavities, sol-gel-processed particles, or membranes are used. A special case is the use of superabsorbent polymers to immobilize the entire aqueous phase. [
42,
43]. In this case, the enzyme is immobilized in its native aqueous environment, whereby undesired interactions of protein moieties with the support can be prevented. Recently, Gröger et al. applied a method of immobilizing ADHs with superabsorber efficiently for the synthesis of diols in enantiomerically pure form [
44]. In this example, the immobilized ADH from
Rhodococcus sp. was used in organic solvents without a major loss in activity. Advantages of this method is the low cost, the very fast immobilization procedure (<30 min), and the excellent catalyst reusability. Challenging, in particular with respect to an application in a fixed bed reactor, might be the gel-like consistency of the hydrogel and mass transfer limitations through phase interfaces in general.
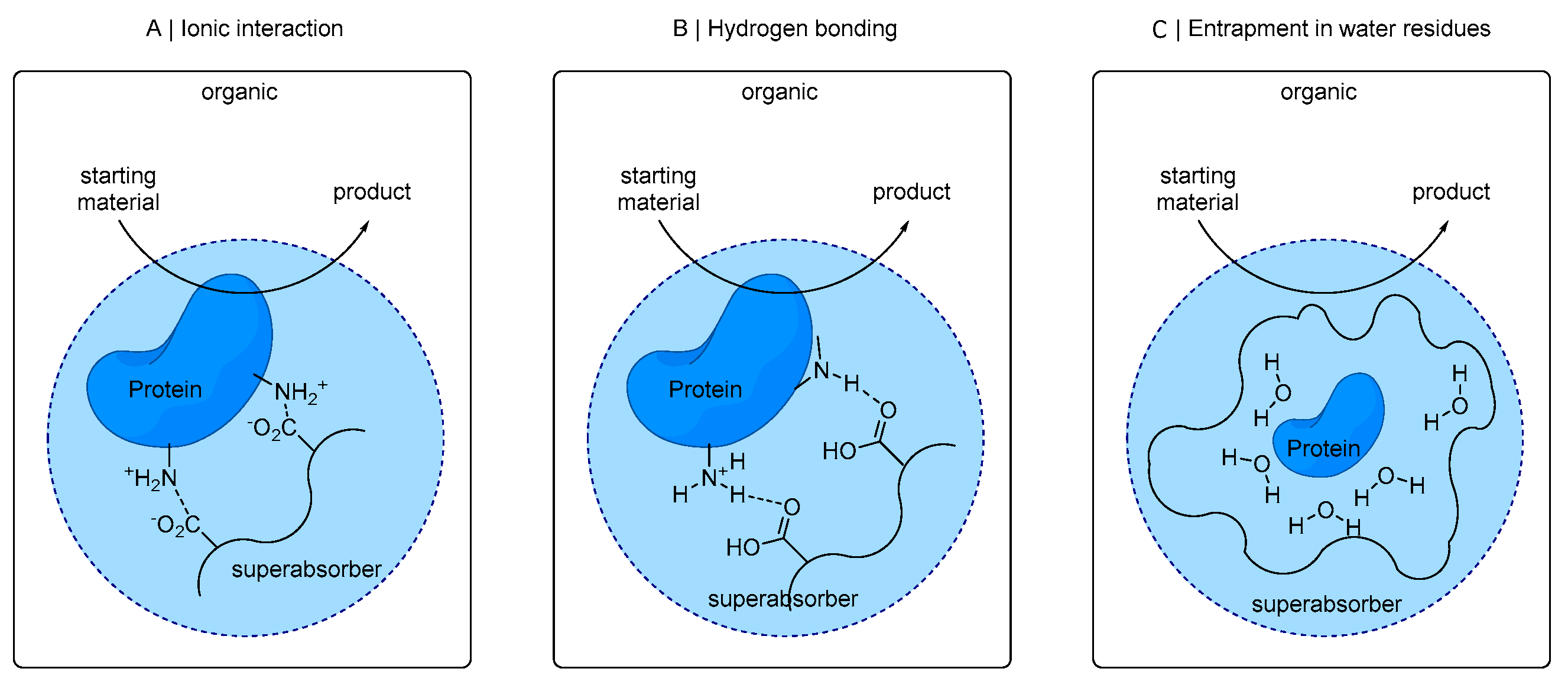
The absorbent polymer consists of cross-linked polyacrylic acid, which was partially neutralized [
45]. Proteins can be embedded in this polymer matrix and can be bound, for example, by ionic interactions of its acid moieties to ionic amino acid residues of the protein (see
Figure 1A). Also, hydrogen bridges from non-ionic protein residues with superabsorber functionalities (
Figure 1B) or entrapment of enzyme in superabsorber-bound water residues (
Figure 1C) can be a possible mode of interaction.
An (
R)-enantioselective ADH from
Lactobacillus brevis (LB-ADH) has been shown to be a versatile catalyst for the reduction of a variety of ketones, ranging from aliphatic ketones to benzylic or propargylic ketones [
46,
47]. It can be used as an isolated enzyme or as a whole-cell catalyst. In particular, its tolerance towards organic solvents is outstanding. The stability depends on several factors, such as the nature of the buffer or solvent additives [
46,
47].
In this contribution, the application of a superabsorber-immobilized ADH from L. brevis for an enzymatic reduction of acetophenone (1) in a flow process is described.
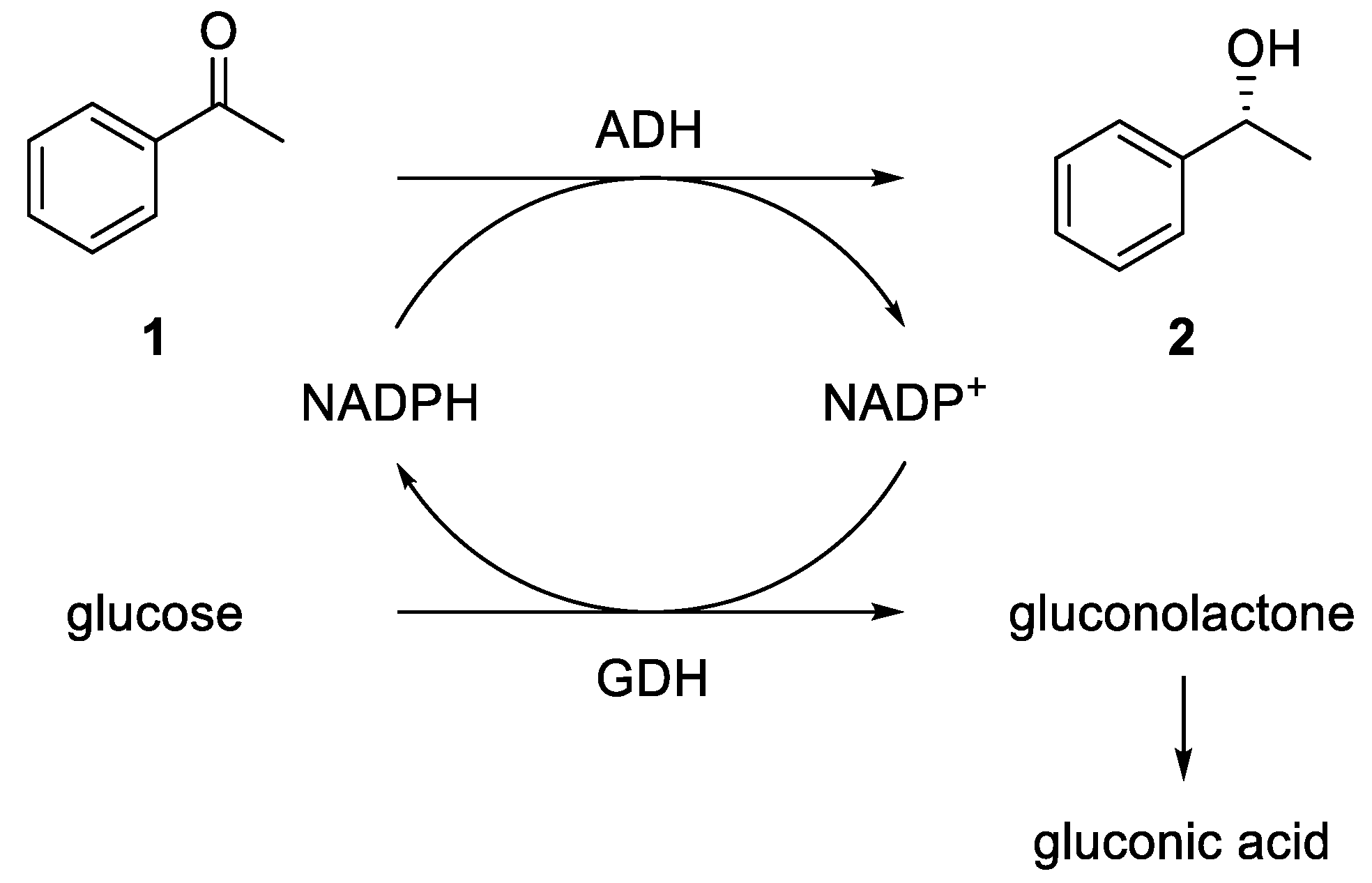
Because the ADHs are cofactor-dependent enzymes and the cofactor NADPH is very expensive, an efficient cofactor regeneration system is required. In the literature, different options are described [
48]. An elegant method is the use of isopropanol for an oxidation of the cofactor using the ADH itself. A further commonly used method is an enzyme cascade using an ADH and a glucose dehydrogenase (GDH). Here, the GDH reduces NADP
+ back to NADPH, consuming one equivalent of glucose, which is oxidized to gluconolactone and irreversibly opens to gluconic acid in an aqueous environment (see
Figure 2).
3. Results and Discussion
3.1. Studies on the Comparison of Enzymatic Acetophenone Reduction in Biphasic Buffer/Methyl tert-Butyl Ether (MTBE) System versus Superabsorber/MTBE System in Batch Mode
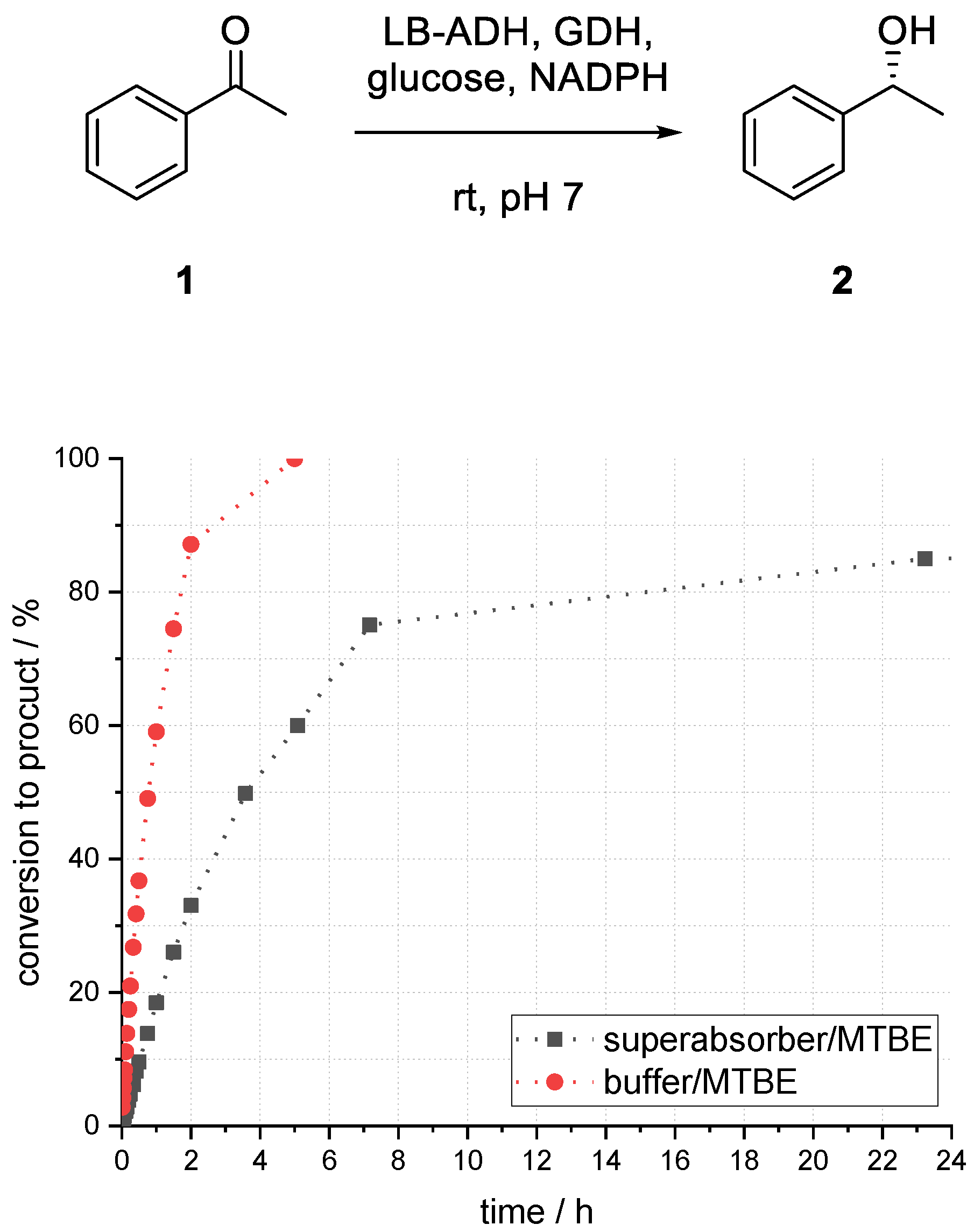
At first, the LB-ADH-catalyzed reduction of acetophenone (1) to (R)-1-phenylethanol (2) was investigated in a biphasic system of methyl tert-butyl ether (MTBE) and buffer to set a benchmark for the immobilized enzymes. Initial investigations on influences of different parameters and optimization thereof were carried out in batch mode.
Compared to the reaction in buffer, which provided 86% conversion to alcohol (
R)-
2 after 1.5 h, the biphasic reaction with a ratio of 1:1 (buffer/organic) provided only 45% (see
Figure 3). After 24 h, the conversion to product (
R)-
2 reached its maximum of 89%. In the case of a higher ratio of 1:5 (buffer/organic), a much faster conversion could be observed. After 1.5 h, 75% conversion to alcohol (
R)-
2 and after 5 h full conversion could be obtained. Emulsion formation and formation of protein aggregates, sticking on the glass surface, could be observed within a short reaction time in all experiments by utilizing a biphasic reaction system.
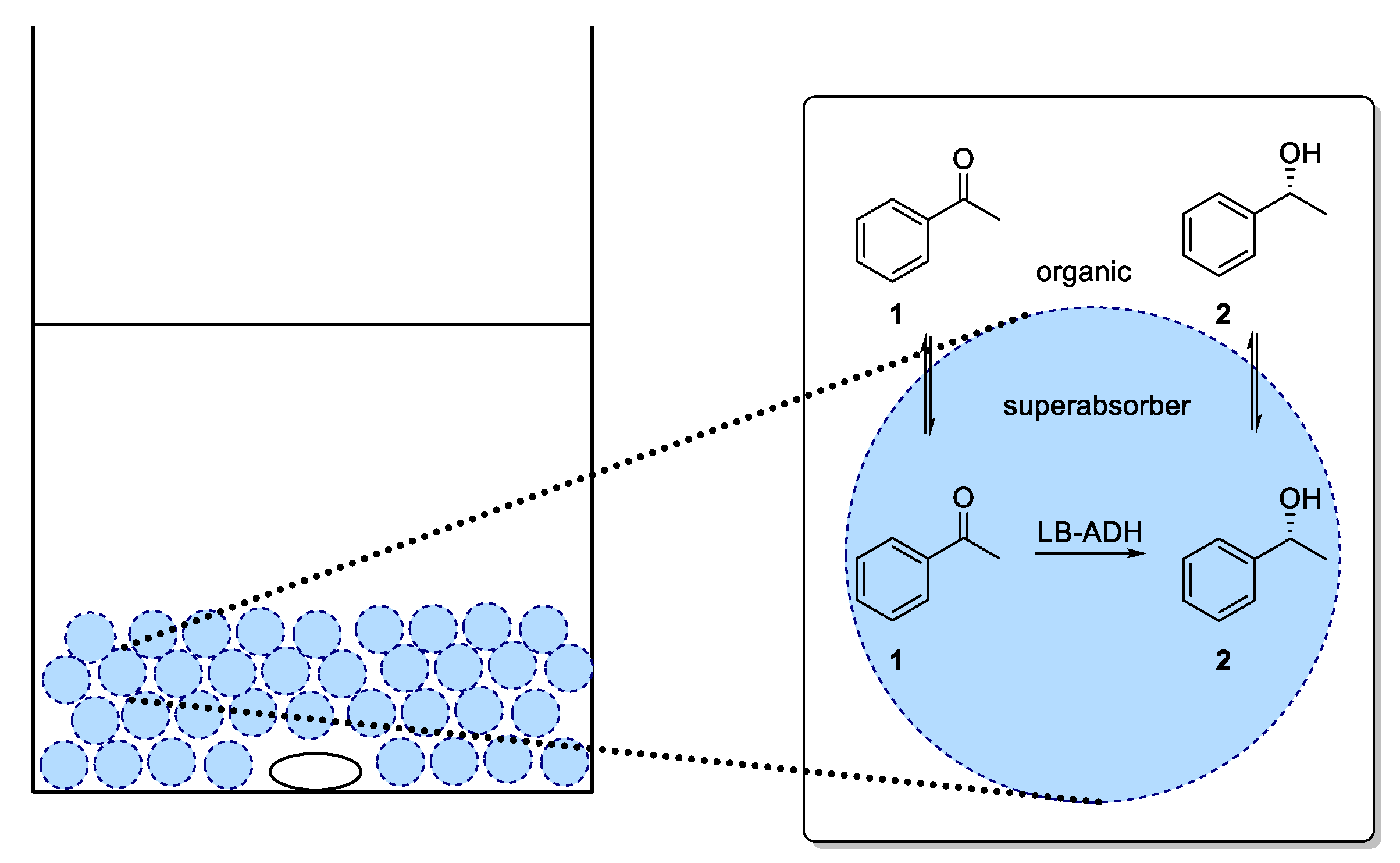
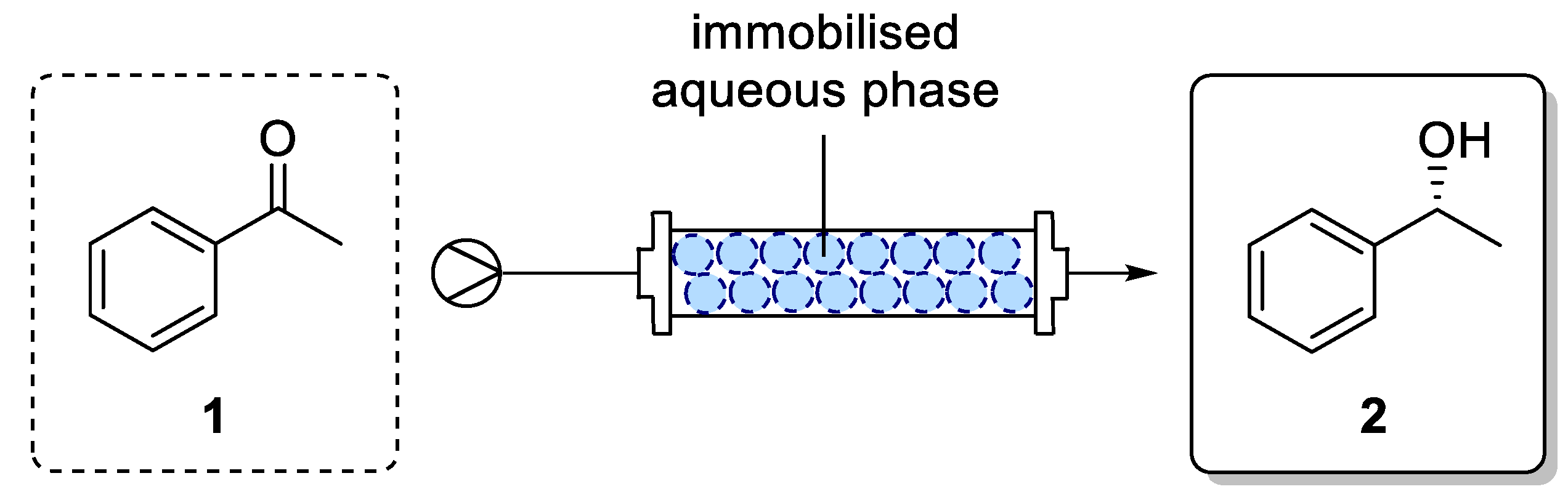
For an application of the ADH in a continuous process, immobilization of ADH utilizing a superabsorber was carried out. In this system, the enzymes are entrapped in an immobilized aqueous phase (as illustrated in
Figure 4) and therein also cofactor and cosubstrate are present. In our studies, we exclusively focused on co-immobilization of both GDH and ADH. As diffusion of cofactor is crucial for an efficient process, immobilization of enzymes in different compartments being separated from each other was expected to be difficult in terms of cofactor availability. By means of a co-immobilization, such an effect could be minimized and the influence of process parameters on the ADH-catalyzed reduction could be studied. In the mobile organic phase, the substrate was dissolved. Thus, this type of immobilization enabled the enzymes to operate in their native aqueous environment, which also might lead to a sufficient stability when being used in such a superabsorber immobilizate.
To gain a better understanding of this system, batch experiments screening different parameters were carried out. The conversion was monitored by gas chromatography (GC) analysis and the results after a reaction time of 25 h were compared. Different organic solvents, amounts of superabsorber, enzyme loadings, and buffer-to-organic solvent ratios were investigated. All aqueous solutions (ADH, GDH, NADPH, and glucose each in buffer) were mixed and immobilized by addition of superabsorber. After a short incubation of 5 min, a 100 mM acetophenone (
1) solution in MTBE was added. All reactions were carried out at 25 °C. The results were compared to a buffer/MTBE system without immobilization (
Figure 5).
Compared to a reduction using a biphasic buffer/MTBE system, the reaction in the superabsorber/MTBE system was much slower. In the biphasic system, full conversion to alcohol (
R)-
2 could be achieved after 5 h, whereas in the superabsorber system after 5 h only 60% conversion to (
R)-
2 was observed. A maximum conversion to product (
R)-
2 of 85% after 23 h was observed (
Figure 5). Thereafter, no further conversion could be observed.
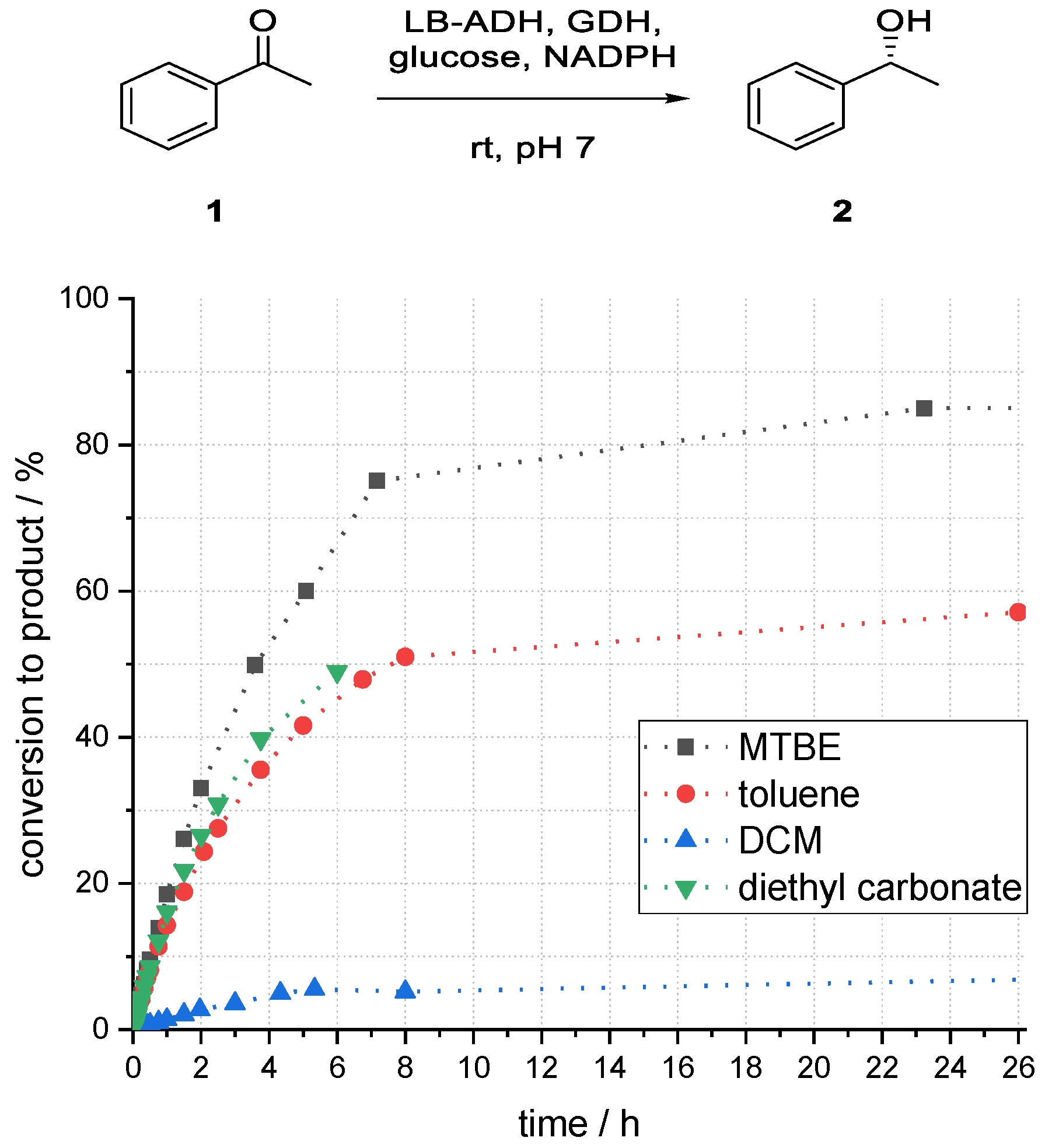
3.2. Studies on the Influence of Organic Solvents on the Reduction of Acetophenone with a Superabsorber-Immobilized Recombinant Alcohol Dehydrogenase from Lactobacillus brevis
To investigate the influence of different solvents, the organic phase was changed while other parameters were kept constant. A range of standard solvents with different properties were investigated. As dichloromethane (DCM) is usually not suitable for enzyme catalysis, it would be interesting to learn about the performance of the investigated system towards this solvent in comparison to other organic “standard solvents”. In addition, methyl tert-butyl ether (MTBE) and diethyl carbonate were chosen as greener solvent options.
As shown in
Figure 6, the highest conversion to product (
R)-
2 after 24 h was obtained for a system using MTBE as an organic solvent component (85%). A maximum conversion to the desired product of 55% for toluene could be observed after 26 h. Thereafter, no further product formation was observed. The result for diethyl carbonate was comparable to that of toluene. With DCM as solvent, the conversion of acetophenone to product (
R)-
2 was with 10% even after 48 h reaction time very low. By far the best tested solvent was MTBE, which was therefore used for further experiments.
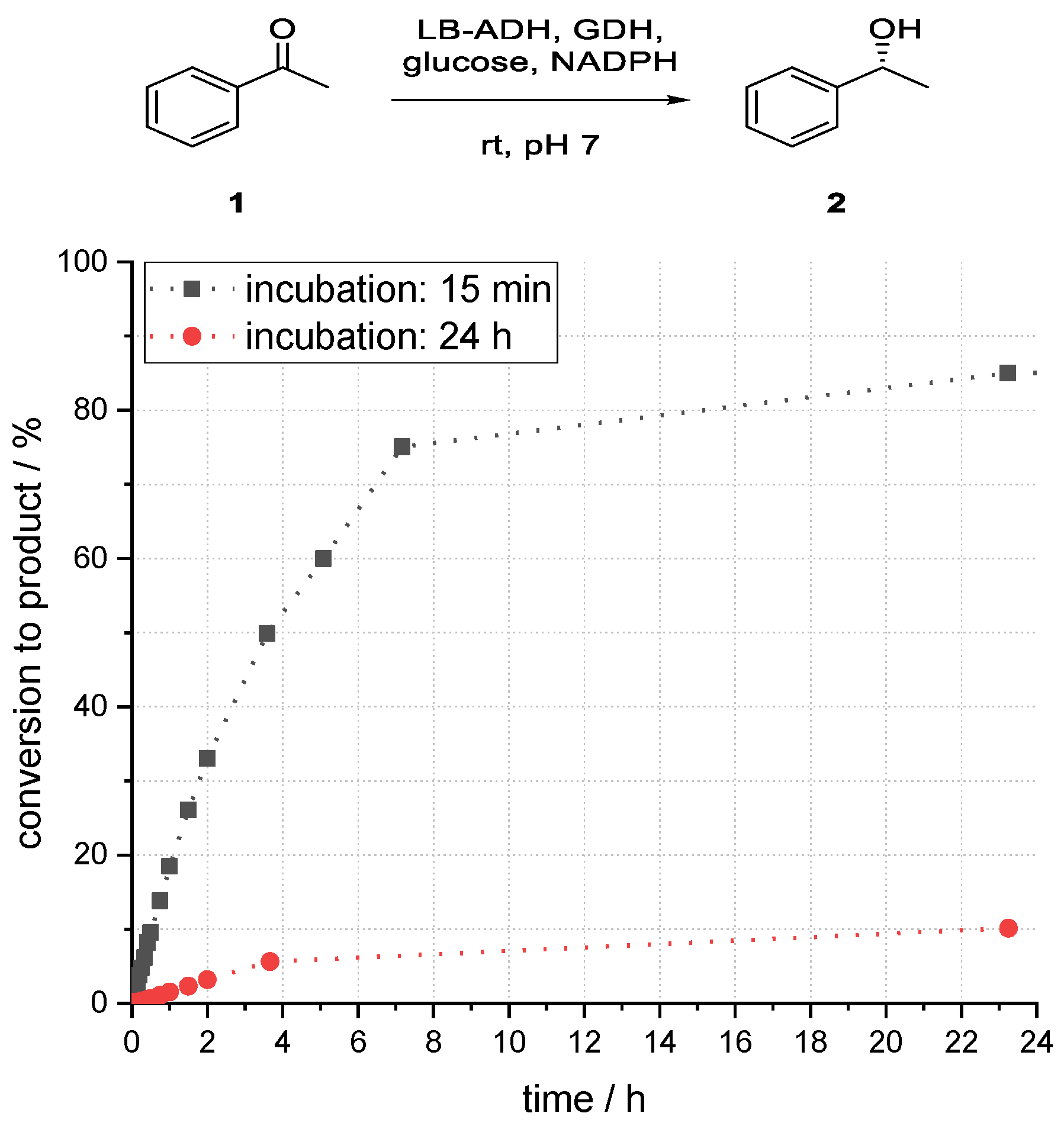
3.3. Studies on the Influence of Incubation Time on the Acetophenone Reduction with a Superabsorber-Immobilized Recombinant Alcohol Dehydrogenase from Lactobacillus brevis
A further investigation of the storage stability of the absorbed enzymes was carried out. For this purpose, the immobilized aqueous phase was incubated for 24 h at room temperature without organic solvent.
It was found that the immobilized enzymes were not stable over a longer period. After incubation of the immobilized aqueous phase for 24 h, the maximum conversion to alcohol (
R)-
2 after 23 h reaction time dropped from 85% to below 10%, as shown in
Figure 7. Therefore, the storage stability of the immobilized enzymes was not very high.
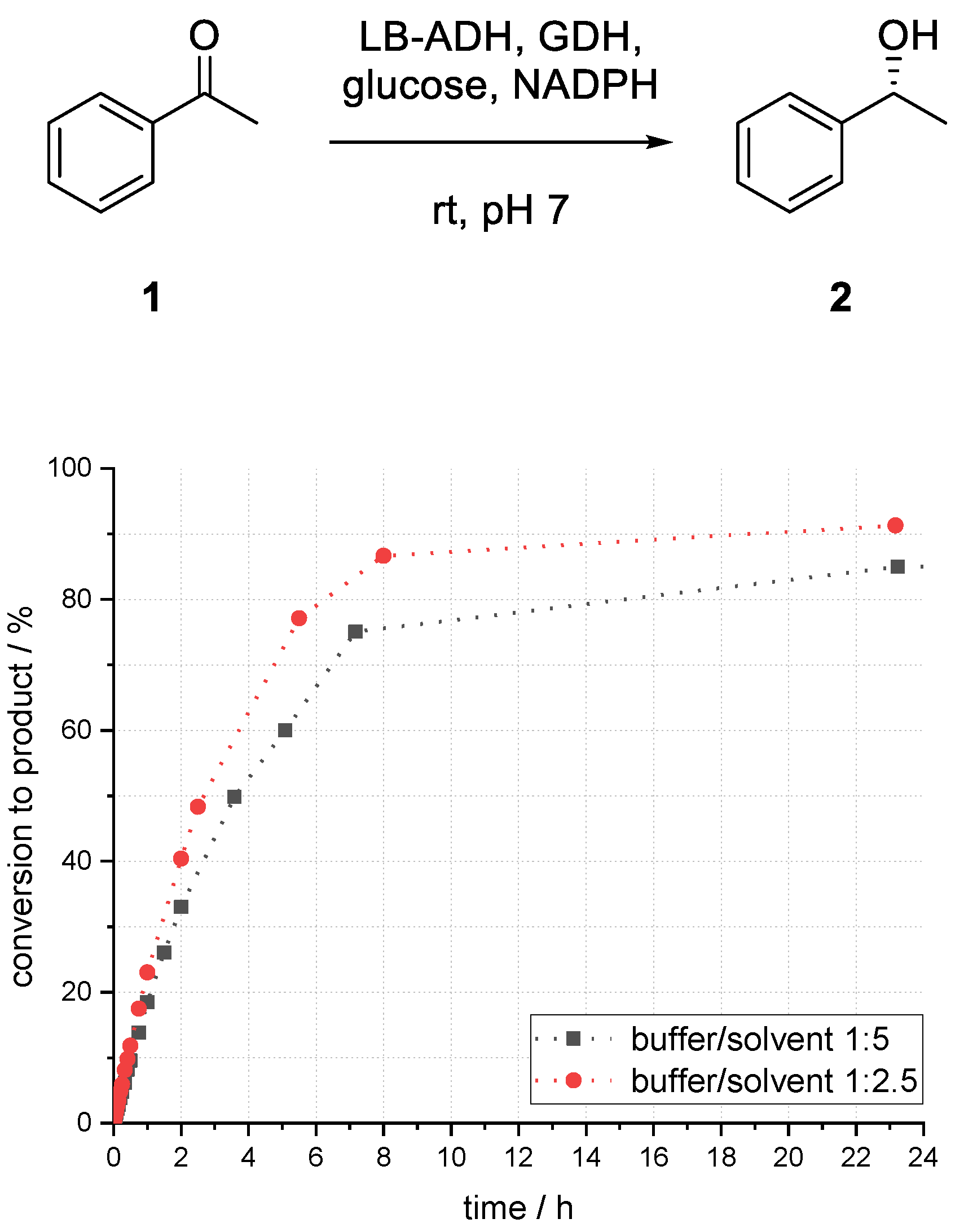
3.4. Studies on the Influence of the Ratio of Buffer to Solvent on the Acetophenone Reduction with a Superabsorber-Immobilized Recombinant Alcohol Dehydrogenase from Lactobacillus brevis
The influence of the ratio of the immobilized aqueous phase and organic phase was investigated. Change of phase interface area, distribution of compounds, as well as the enzyme environment can influence the performance of the system.
By changing the aqueous to organic phase ratio from 1:5 to 1:2.5, an increase of conversion to product (
R)-
2 after 23 h from 85% to 90% could be observed (
Figure 8). Higher enzyme stability due to less direct contact of enzyme and solvent, and more favorable compound distribution might be the reasons for higher product formation.
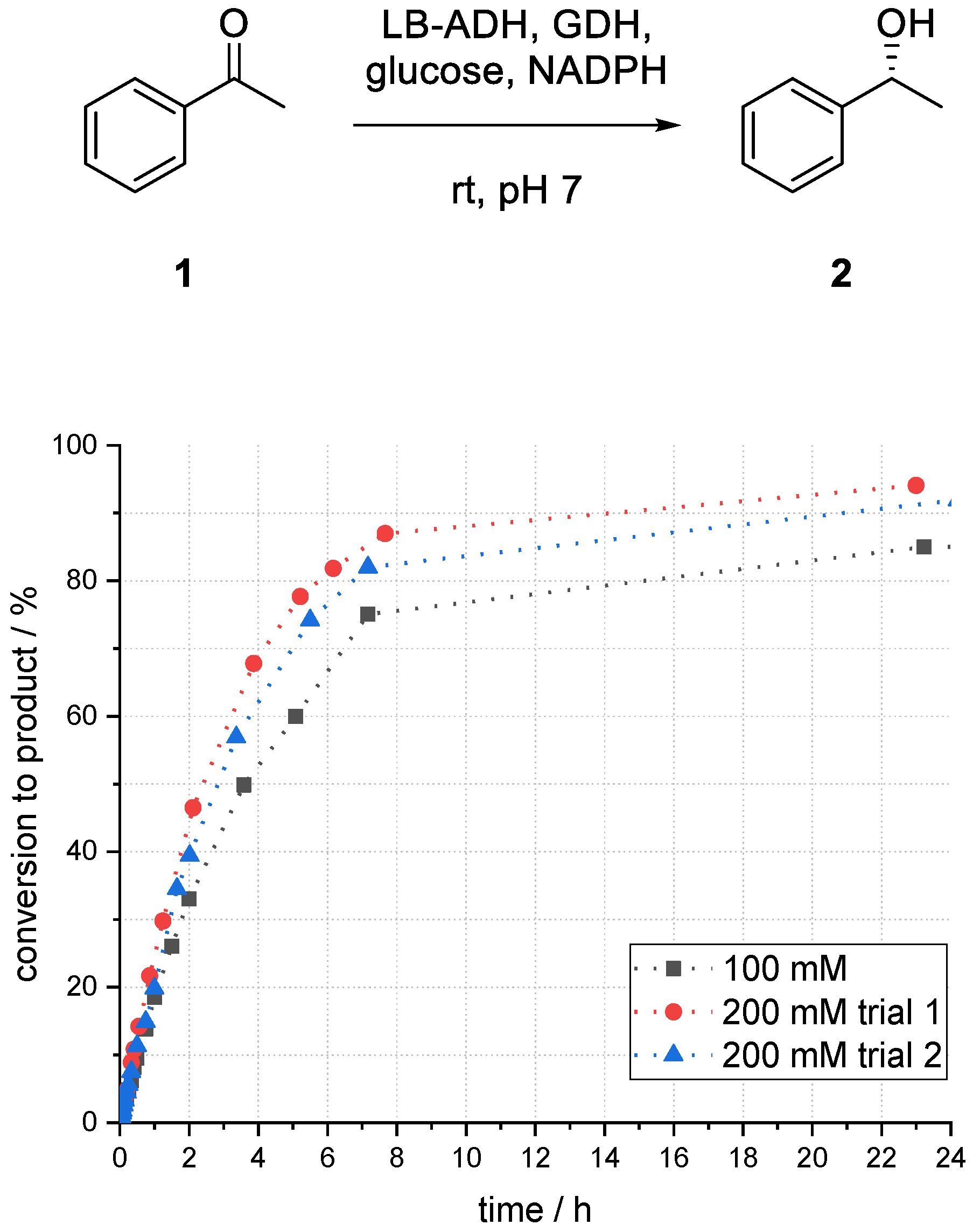
3.5. Studies on the Influence of the Buffer Concentration on the Acetophenone Reduction with a Superabsorber-Immobilized Recombinant Alcohol Dehydrogenase from Lactobacillus brevis
Also, the influence of potassium phosphate buffer (PPB) buffer concentration was investigated. Because the superabsorber itself acts as a buffer system and because stoichiometric amounts of acid were formed due to the GDH-based cofactor regeneration system, this system was difficult to quantify.
By increasing the PPB buffer concentration from 100 to 200 mM, the conversion to the desired product after 24 h increased from 85% to 94% (
Figure 9). The results have been confirmed in a second experiment (92%).
3.6. Studies on the Influence of the Superabsorber Particle Size on the Acetophenone Reduction with a Superabsorber-Immobilized Recombinant Alcohol Dehydrogenase from Lactobacillus brevis
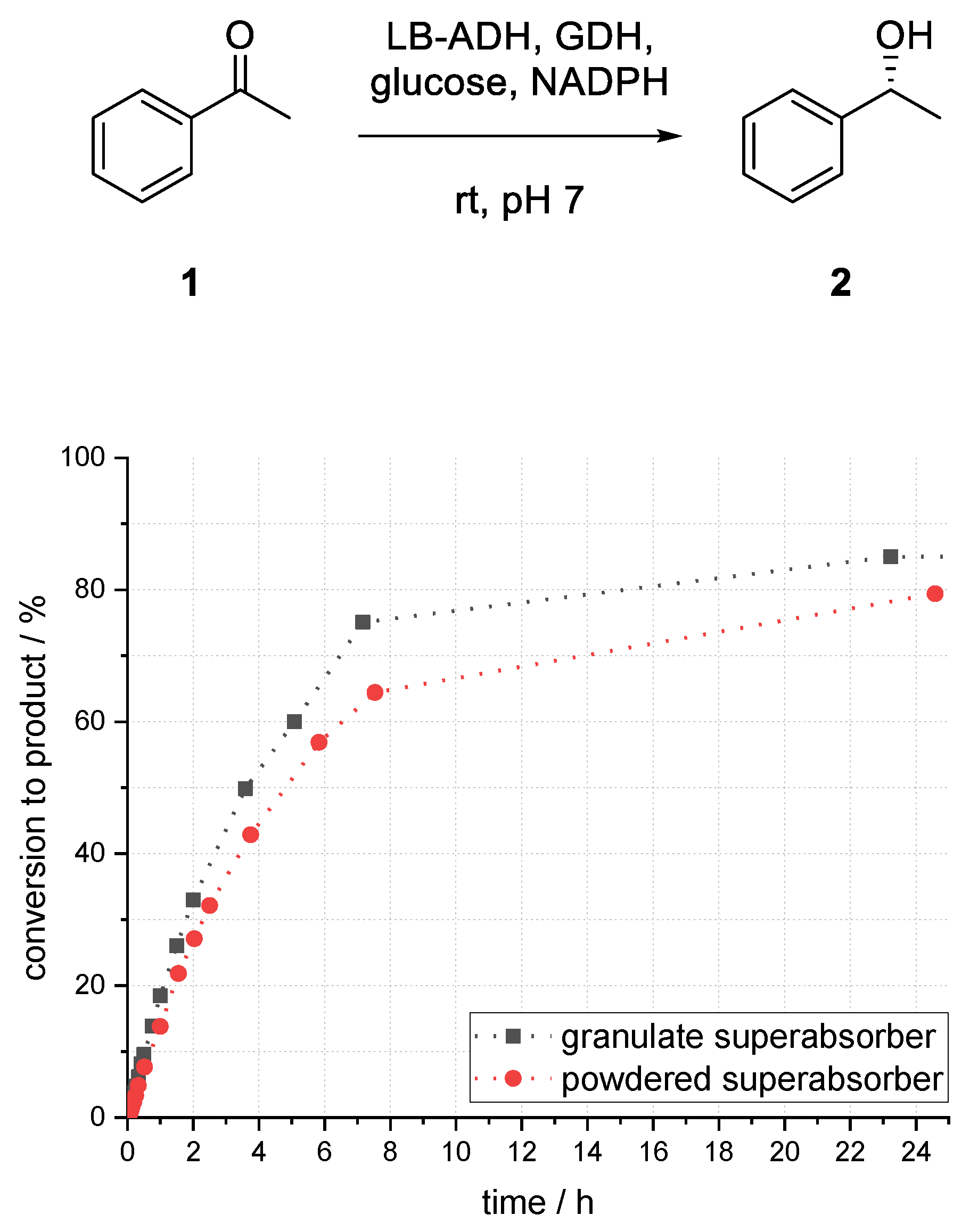
Another process parameter of potential relevance was the interface area of organic and aqueous phase. By powdering the superabsorber granulate, the interface area, and therefore the mass transfer phenomena, should be significantly increased. For the preparation of superabsorber powder (<100 µm), the superabsorber granulate was powdered by grinding in a mortar and sieving with a steel mesh.
As shown in
Figure 10, a smaller particle size led to lower conversion. After 25 h, 6% less product (
R)-
2 was formed compared to larger particle size after 23 h. Considering that the surface of the superabsorber particles was increased, phase transfer was not a limiting factor for this system. Due to smaller aqueous superabsorber particles, the solvent played a major role in deactivation of enzymes, as enzymes might be more often near to the surface.
3.7. Studies on the Influence of the Biocatalyst Loading on the Acetophenone Reduction with a Superabsorber-Immobilized Recombinant Alcohol Dehydrogenase from Lactobacillus brevis
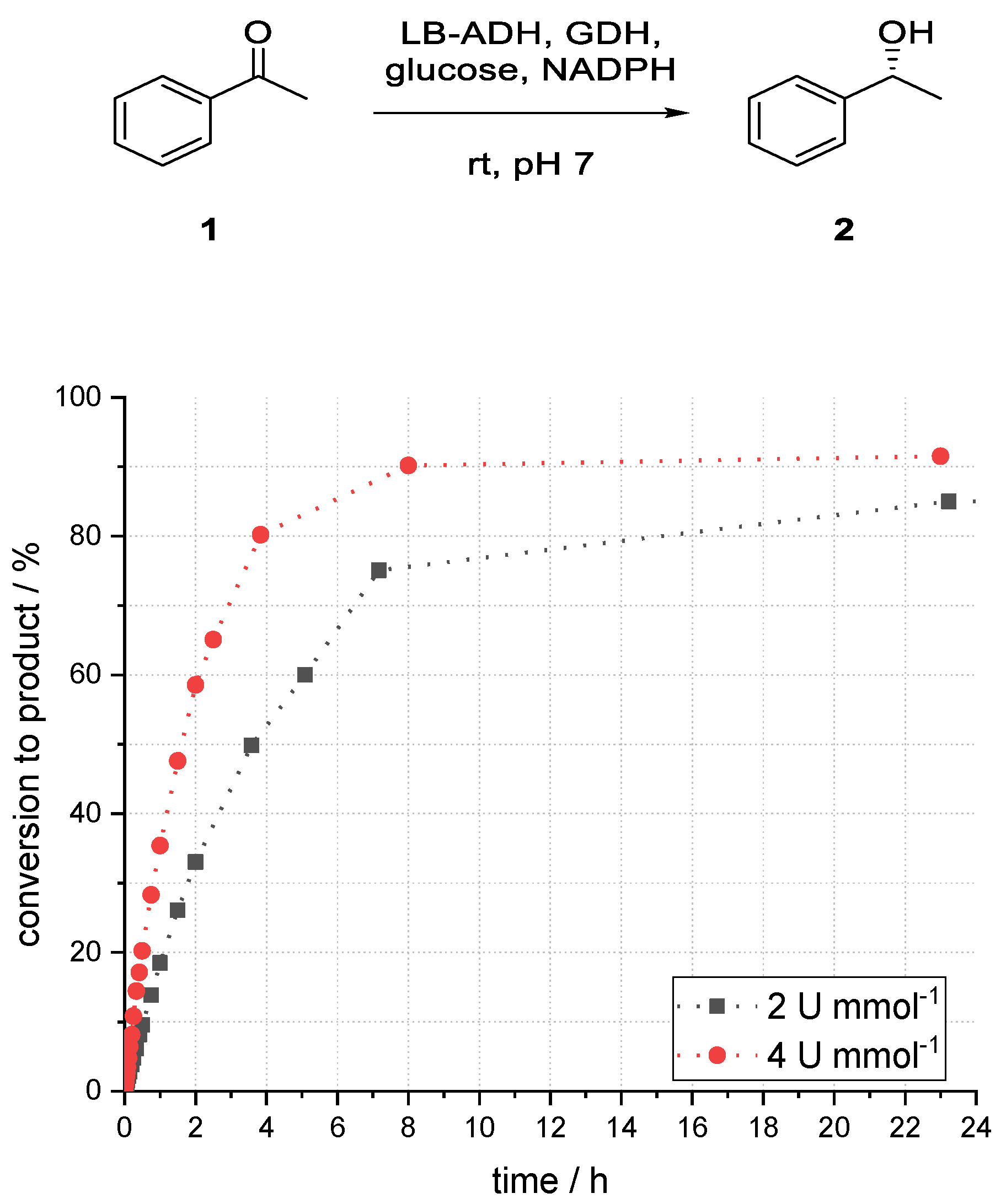
Finally, the ADH loading was increased form 2 U mL−1 to 4 U mL−1. The amount of cofactor regeneration system was adjusted accordingly.
As expected, the reaction using more catalyst performed better (85% vs. 92% after 23 h,
Figure 10). This result also supported the assumption that mass transfer was not a limiting factor in the reaction system, as the conversion can be increased using more catalysts. Enzyme deactivation, however, was likely to have played a major role in this reaction because at lower catalyst loading (2 U mL−1) prolonging the reaction time after 8 h, no significant further increase of conversion could be observed (
Figure 11).
As for the tested solvents, it could be shown that MTBE was the best performing one. As expected, solvents that have previously shown to be not tolerated by enzymes, such as DCM, turned out not to be suitable for the investigated reaction system. MTBE and diethyl carbonate have both shown high conversions, indicating deactivation of the enzyme to a lesser extent. Besides stability issues, according to the solvent-dependent partition coefficient, substrate distribution in the aqueous and organic phases might also have played a role here. In the case of an unfavored partition coefficient (caused by a very high solubility of the substrate in the organic phase), the concentration of substrate in the aqueous superabsorber system might have been below the KM value, thus slowing down the overall process.
3.8. Set-Up of a Packed Bed Reactor
Figure 12 shows a schematic flow process for an enzymatic reduction of acetophenone (
1) to phenylethanol (
R)-
2, coupled with a GDH-based cofactor regeneration system. The packed bed reactor was loaded with the immobilized aqueous phase, containing the enzymes, cofactor, and glucose. A total amount of 2 U ADH and a substrate concentration of 100 mM in MTBE were used for the experiments. For the setup, an in-house custom build glass tube reactor (ID 5 mm) was used (
Figure 13).
The reactor was fed with a solution of ketone 1 in MTBE using a syringe pump. A residence time of 1 hour was set and collected fractions of the resulting product solution were analyzed by GC. Because the catalyst was immobilized, no separation or quenching of the reaction mixture was required.
3.9. Studies on the Acetophenone Reduction in a Flow Process with a Superabsorber-Immobilized Recombinant Alcohol Dehydrogenase from Lactobacillus brevis
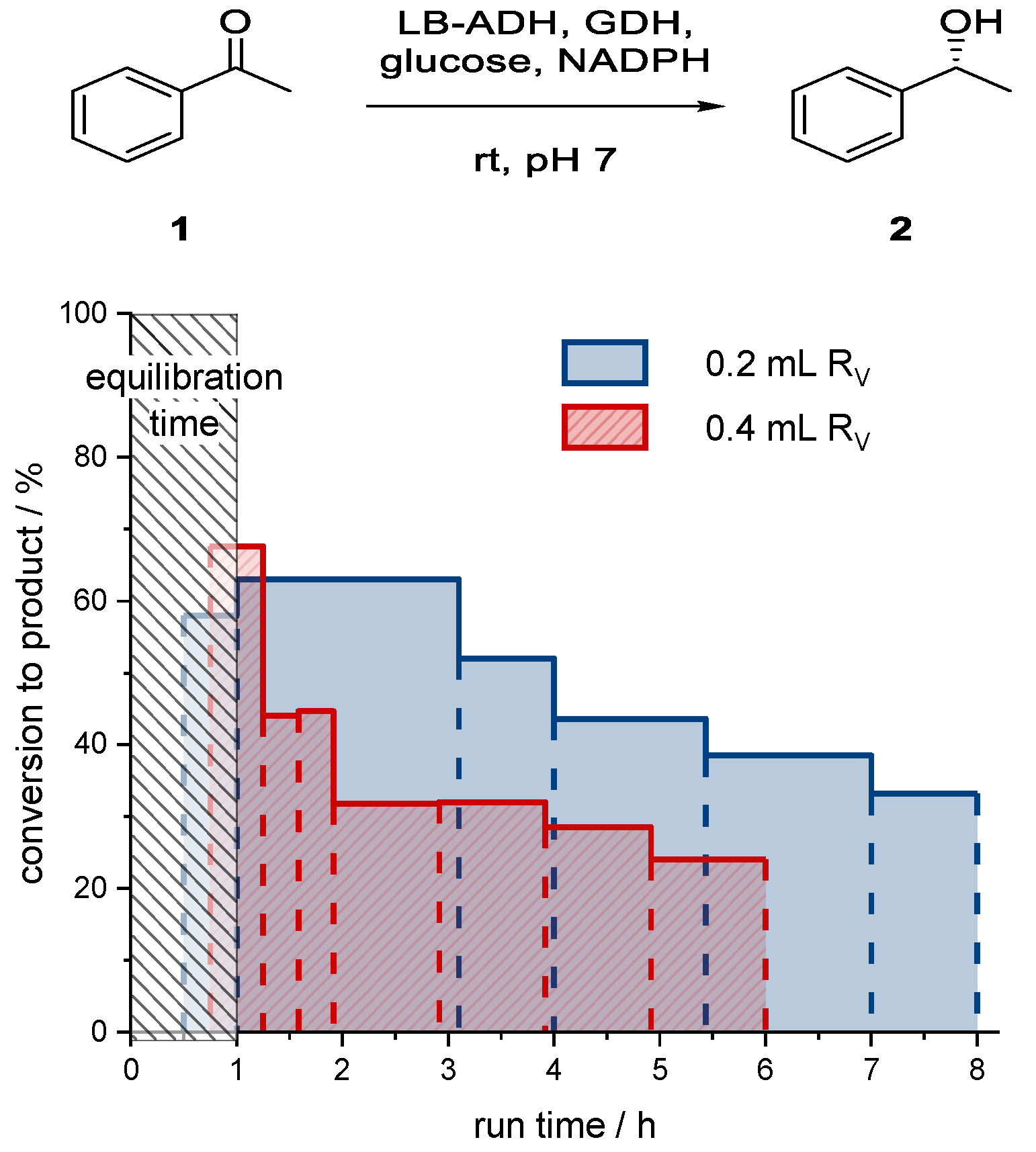
The reactor was charged with different volumes of immobilized aqueous phase. The amounts of superabsorber and catalytic system (ADH, GDH, glucose, NADPH) were kept constant, whereas different volumes of buffer were used to adjust the overall reactor bed volume (R
V). After charging of the reactor, the end pieces of the reactor were adjusted to the catalyst bed. The average conversions of collected fractions are shown in
Figure 14.
As for the larger reactor volume, the initial conversion to alcohol (R)-2 in the first hour was very high (67%). However, the conversion decreased very quickly to only 24% after 5 h. In the case of the smaller reactor bed volume, the conversion to alcohol (R)-2 for the first fractions collected from the start to the first hour was about 61%. However, the conversion was not constant over a longer period, and dropped to 33% after 7 h of reactor operating time (or seven residence times). Compared to the larger reactor volume, the system was more stable. The smaller system was probably more stable due to lower flow rates and therefore less organic solvent pumped through the reactor bed. In accordance with the batch experiments, the stability of the catalyst was a major issue.
Compared to batch processes, a much higher initial conversion within 1 h reaction time could be achieved. Due to the nature of a packed bed reactor, the catalyst could be used more efficiently, as the catalyst concentration in the reaction was much higher (a summary can be found in
Table 3).
It was successfully shown that a superabsorber-immobilisate can be applied in a packed bed reactor, thus offering an alternative to the standard approach using carrier-immobilized biocatalysts.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}













