Thermoplastic PCL-b-PEG-b-PCL and HDI Polyurethanes for Extrusion-Based 3D-Printing of Tough Hydrogels

Abstract

1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
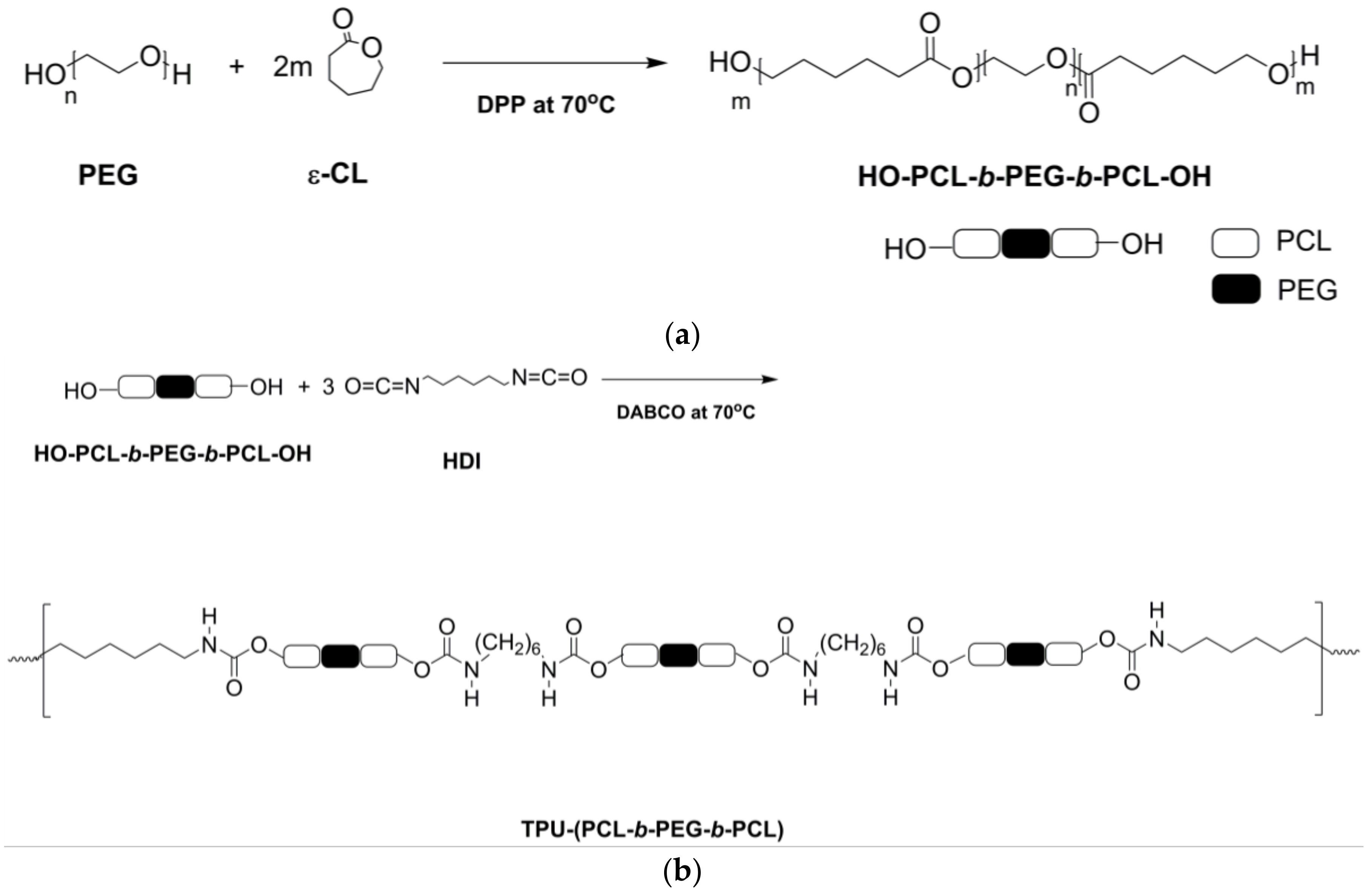
2.2.1. Synthesis of PCL-b-PEG-b-PCL and TPU-(PCL-b-PEG-b-PCL) Polymers
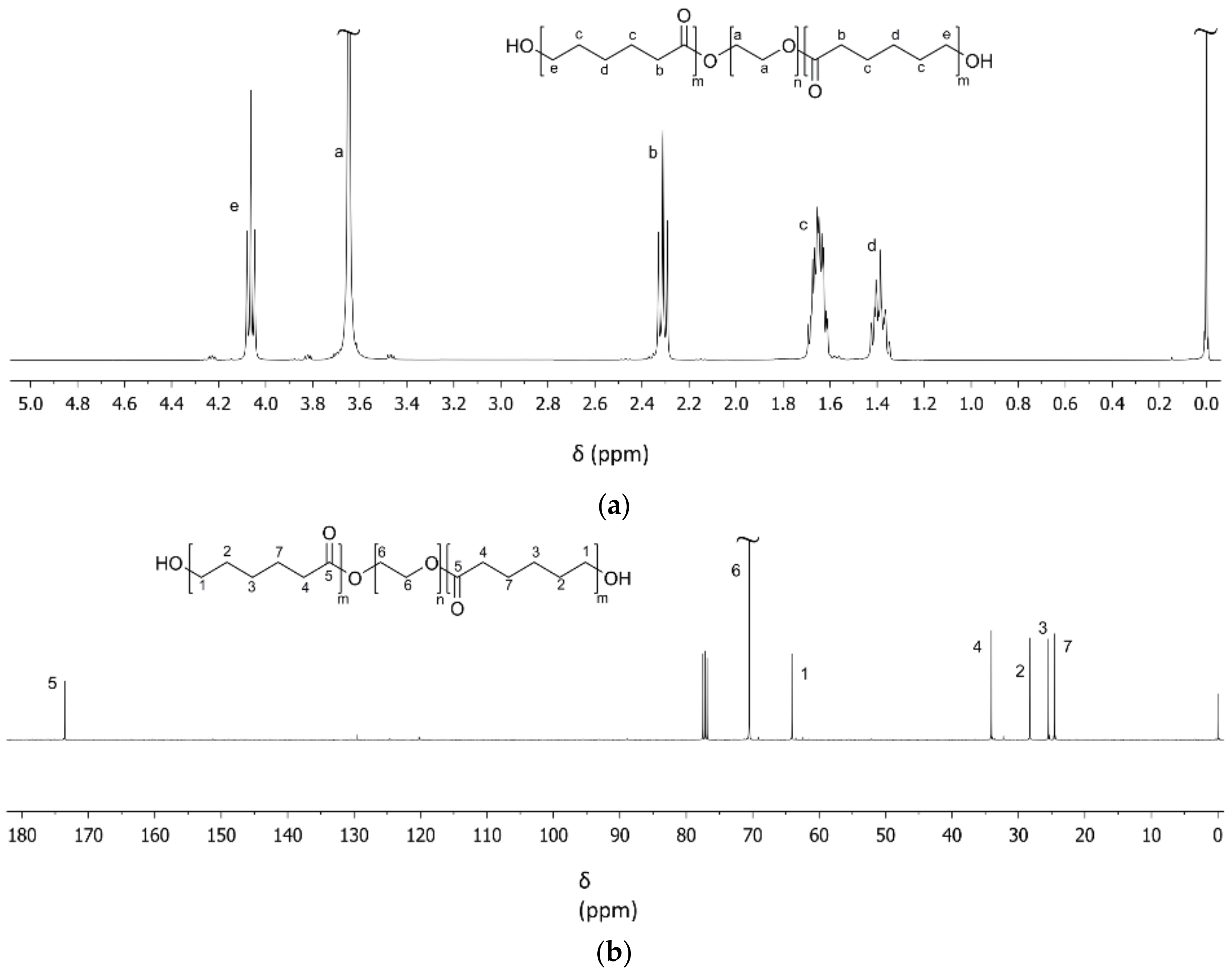
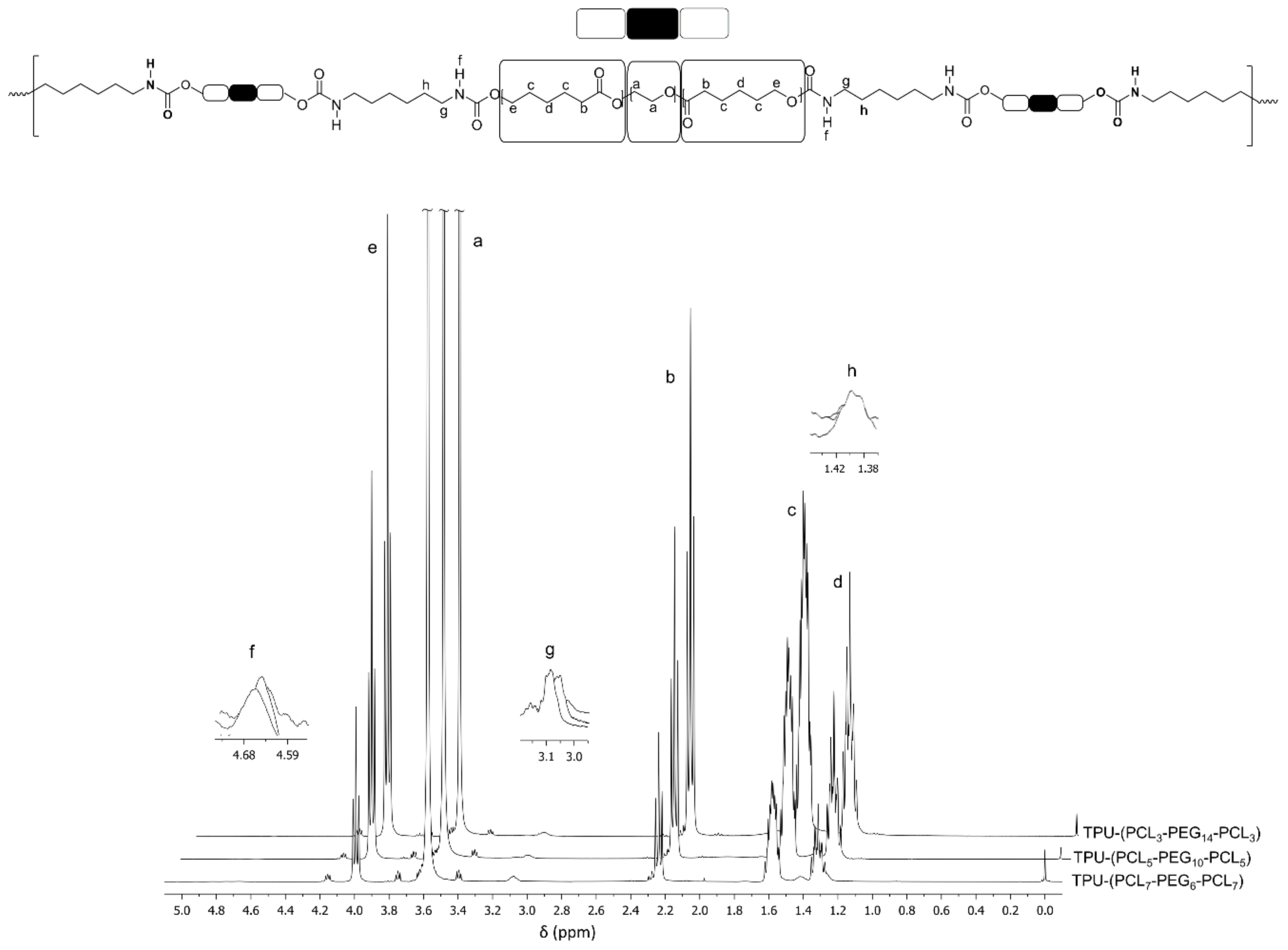
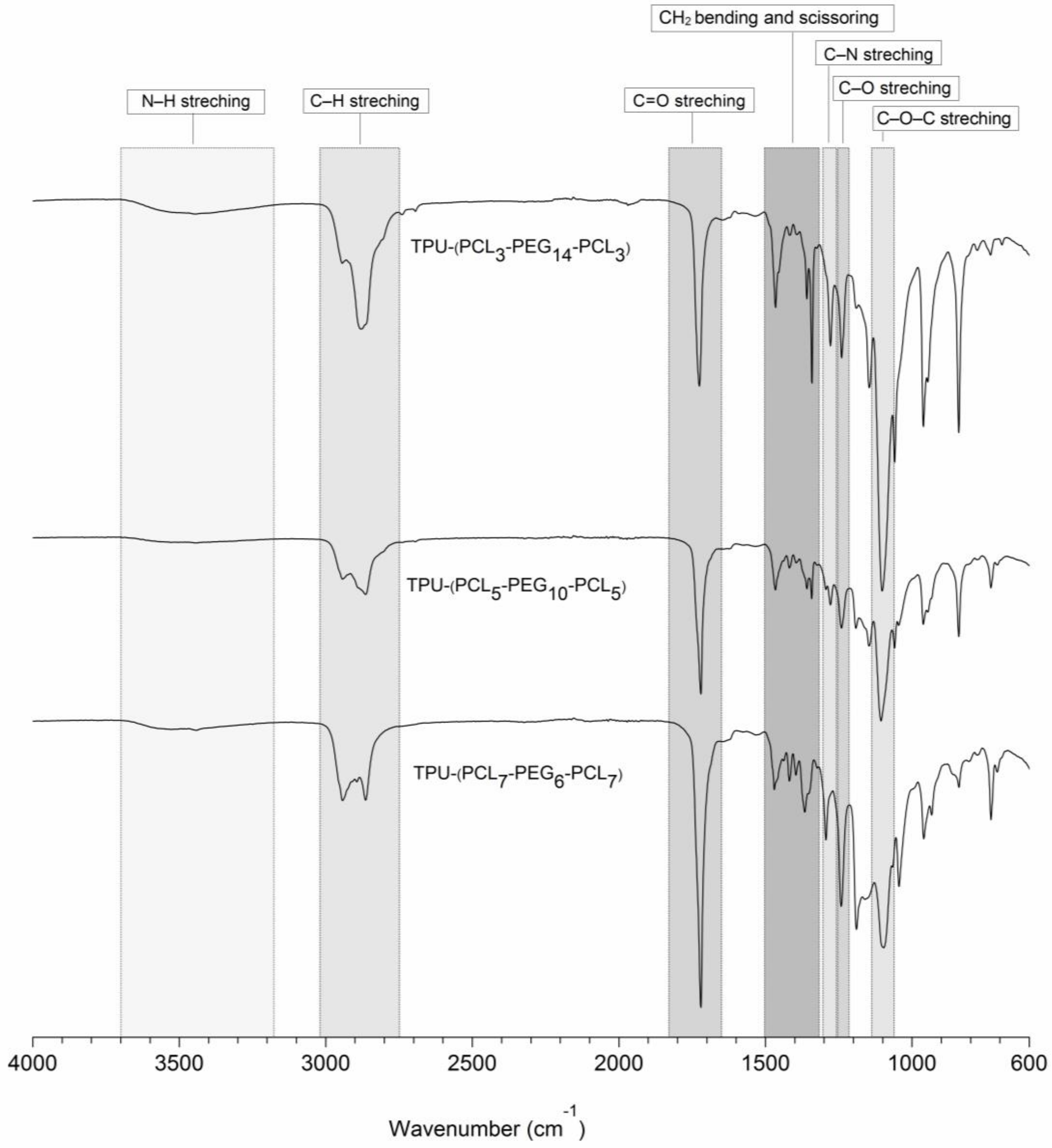
2.2.2. Characterization of PCL-b-PEG-b-PCL and TPU-(PCL-b-PEG-b-PCL) Polymers
2.2.3. Physical and Mechanical Properties of PCL-b-PEG-b-PCL and TPU-(PCL-b-PEG-b-PCL) Polymers
2.2.4. Fabrication of Designed Structures by Fused Deposition Modelling (FDM)
3. Results
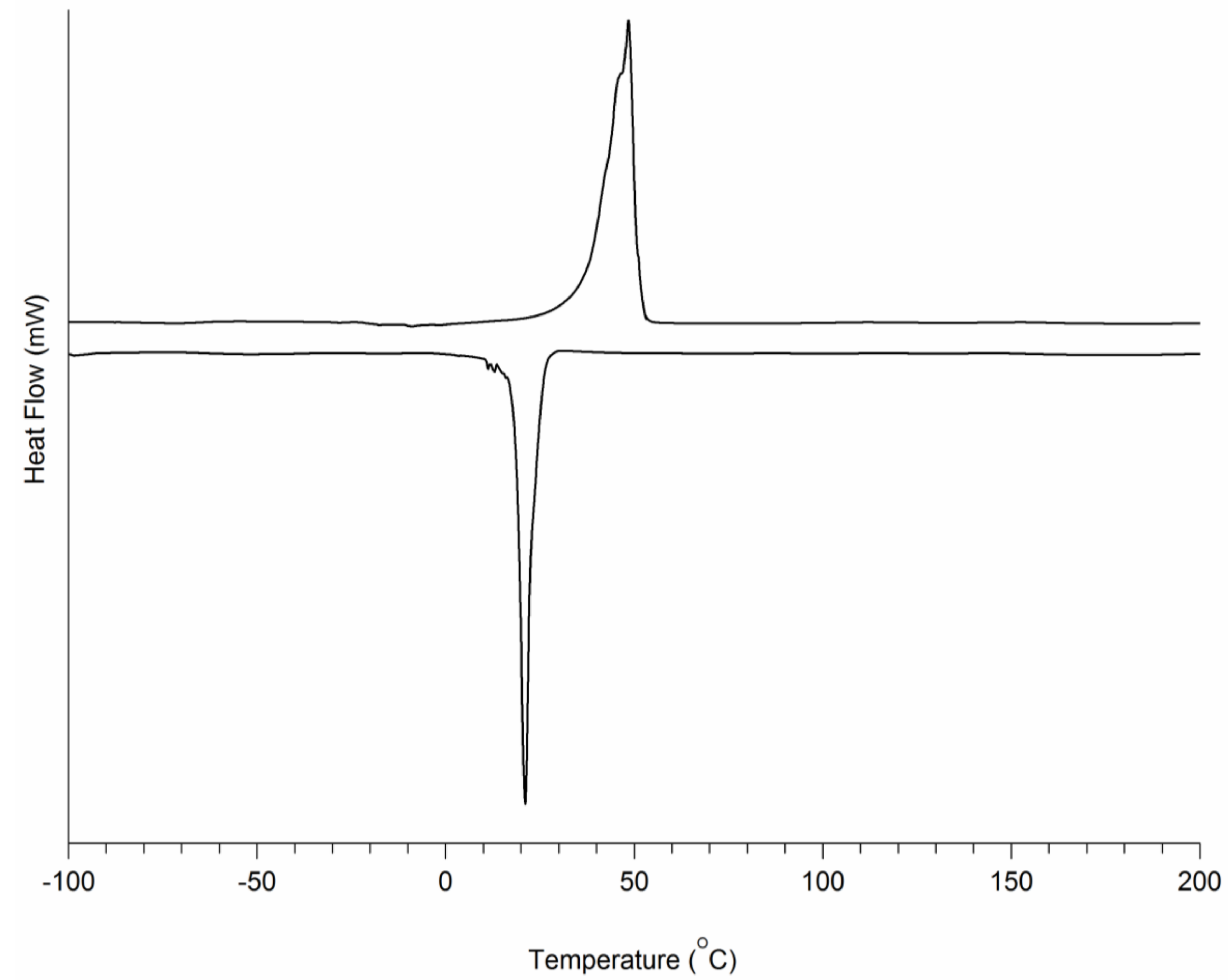
3.1. PCL-b-PEG-b-PCL Triblock Copolymers
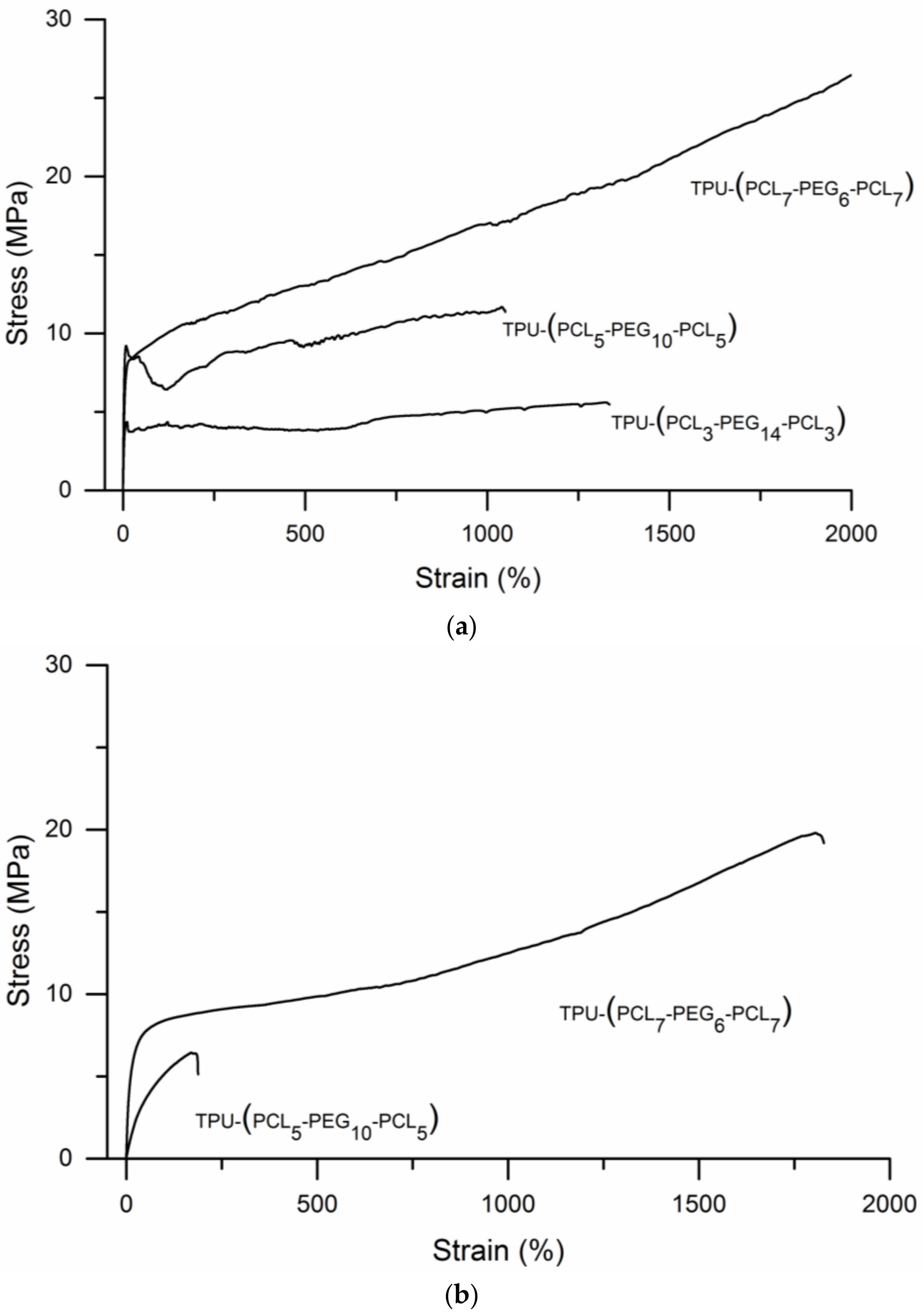
3.2. TPU-(PCL-b-PEG-b-PCL) Multi-Block Copolymers
3.3. 3D Printing of TPU-(PCL-b-PEG-b-PCL) Multi-Block Copolymers
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bae, J.; Bende, N.P.; Evans, A.A.; Na, J.H.; Santangelo, C.D.; Hayward, R.C. Programmable and reversible assembly of soft capillary multipoles. Mater. Horizons. 2017, 4, 228–235. [Google Scholar] [CrossRef]
- Yuk, H.; Zhang, T.; Parada, G.A.; Liu, X.; Zhao, X. Skin-inspired hydrogel-elastomer hybrids with robust interfaces and functional microstructures. Nat. Commun. 2016, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Fung, Y.C. Biomechanics. In Mechanical Properties of Living Tissues, 2nd ed.; Springer: New York, NY, USA, 1993. [Google Scholar]
- Dissanayaka, W.L.; Hargreaves, K.M.; Jin, L.; Samaranayake, L.P.; Zhang, C. The interplay of dental pulp stem cells and endothelial cells in an injectable peptide hydrogel on angiogenesis and pulp regeneration in vivo. Tissue Eng. A 2015, 21, 550–563. [Google Scholar] [CrossRef] [PubMed]
- Koutsopoulos, S. Self-assembling peptide nanofiber hydrogels in tissue engineering and regenerative medicine: Progress, design guidelines, and applications. J. Biomed. Mater. Res. A 2016, 104, 1002–1016. [Google Scholar] [CrossRef] [PubMed]
- Kamata, H.; Akagi, Y.; Kayasuga-kariya, Y.; Chung, U.; Sakai, T. Nonswellable hydrogel without mechanical hysteresis. Science 2014, 343, 873–875. [Google Scholar] [CrossRef] [PubMed]
- Zant, E.; Grijpma, D.W. Tough biodegradable mixed-macromer networks and hydrogels by photo-crosslinking in solution. Acta Biomater. 2016, 31, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Zant, E.; Grijpma, D.W. Synthetic biodegradable hydrogels with excellent mechanical properties and good cell adhesion characteristics obtained by the combinatorial synthesis of photo-cross-linked networks. Biomacromolecules 2016, 17, 1582–1592. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.L.; Kurokawa, T.; Kuroda, S.; Bin Ihsan, A.; Akasaki, T.; Sato, K.; Haque, M.A.; Nakajima, T.; Gong, J.P. Physical hydrogels composed of polyampholytes demonstrate high toughness and viscoelasticity. Nat. Mater. 2013, 12, 932–937. [Google Scholar] [CrossRef] [PubMed]
- Costa, A.M.S.; Mano, J.F. Extremely strong and tough hydrogels as prospective candidates for tissue repair—A review. Eur. Polym. J. 2015, 72, 344–364. [Google Scholar] [CrossRef]
- Ni, Y.; Tang, Z.; Cao, W.; Lin, H.; Fan, Y.; Guo, L; Zhang, X. Tough and elastic hydrogel of hyaluronic acid and chondroitin sulfate as potential cell scaffold materials. Int. J. Biol. Macromol. 2015, 74, 367–375. [Google Scholar] [CrossRef] [PubMed]
- Malda, J.; Visser, J.; Melchels, F.P.; Jüngst, T.; Hennink, W.E.; Dhert, W.J.A.; Groll, J.; Hutmacher, D.W. 25th anniversary article: Engineering hydrogels for biofabrication. Adv. Mater. 2013, 25, 5011–5028. [Google Scholar] [CrossRef] [PubMed]
- Mouser, V.H.M.; Levato, R.; Bonassar, L.J.; D’Lima, D.D.; Grande, D.A.; Klein, T.J.; Saris, D.B.F.; Zenobi-Wong, M.; Gawlitta, D.; Malda, J. Three-dimensional bioprinting and its potential in the field of articular cartilage regeneration. Cartilage 2017, 8, 327–340. [Google Scholar] [CrossRef] [PubMed]
- Kirchmajer, D.M.; Gorkin, R., III. An overview of the suitability of hydrogel-forming polymers for extrusion-based 3D-printing. J. Mater. Chem. B 2015, 3, 4105–4117. [Google Scholar] [CrossRef]
- Shelke, N.B.; Nagarale, R.K.; Kumbar, S.G. Polyurethanes. In Natural and Synthetic Biomedical Polymers, 1st ed.; Kumbar, S.G., Laurencin, C., Deng, M., Eds.; Elsevier: Amsterdam, The Netherlands, 2014; pp. 123–144. [Google Scholar]
- Wu, F.; Chen, L.; Li, Y.; Lee, K.I.; Fei, B. Super-tough hydrogels from shape-memory polyurethane with wide-adjustable mechanical properties. J. Mater. Sci. 2017, 52, 4421–4434. [Google Scholar] [CrossRef]
- Jen, A.C.; Wake, M.C.; Mikos, A.G. Hydrogels for cell immobilization. Biotechnol. Bioeng. 1996, 50, 357–364. [Google Scholar] [CrossRef]
- Lendlein, A.; Langer, R. Biodegradable, elastic shape-memory polymers for potential biomedical applications. Science 2002, 296, 1673–1676. [Google Scholar] [CrossRef] [PubMed]
- Yen, M.S.; Kuo, S.C. PCL-PEG-PCL Triblock Ester-Ether Copolydiol-Based Waterborne Polyurethane. II. Effect of NCO/OH Mole Ratio and DMPA Content on the Physical Properties. J. Appl. Polym. Sci. 1998, 67, 1301–1311. [Google Scholar] [CrossRef]
- Yen, M.S.; Kuo, S.C. Effects of a soft segment component on the physical properties of synthesized waterborne polyurethanes by using triblock ester-ether copolydiol as the soft segment. J. Polym. Res. 1998, 5, 125–131. [Google Scholar] [CrossRef]
- Dušek, K.; Ilavský, M.; Matějka, L. Statistical treatment of allophanate crosslinking in the formation of polyurethane networks. Polym. Bull. 1984, 12, 33–40. [Google Scholar] [CrossRef]
- Hoo Fatt, M.S.; Chen, L.; Al-Quraishi, A.A. Fracture parameters for natural rubber under dynamic loading. Strain 2011, 47, 505–518. [Google Scholar] [CrossRef]
- Tonsomboon, K.; Butcher, A.L.; Oyen, M.L. Strong and tough nanofibrous hydrogel composites based on biomimetic principles. Mater. Sci. Eng. C 2017, 72, 220–227. [Google Scholar] [CrossRef] [PubMed]
- Naficy, S.; Brown, H.R.; Razal, J.M.; Spinks, G.M.; Whitten, P.G. Progress toward robust polymer hydrogels. Aust. J. Chem. 2011, 64, 1007–1025. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Structure | Mn of PEG Block a | CL Conversion b | Mn of PCL Blocks | PEG Content | Mn of Triblock Copolymer |
|---|---|---|---|---|---|
| (kg/mol) | (%) | (kg/mol) | (mol%) | (kg/mol) | |
| PCL3-PEG14-PCL3 | 13.9 | 99.2 | ½ × 5.9 | 70.2 | 19.8 |
| PCL5-PEG10-PCL5 | 9.9 | 99.4 | ½ × 10.0 | 49.8 | 19.9 |
| PCL7-PEG6-PCL7 | 5.9 | 99.8 | ½ × 14.1 | 29.5 | 20.0 |
| Tg (°C) | Tc (°C) | Tm (°C) | ΔH (J/g) | |
|---|---|---|---|---|
| PCL3-PEG14-PCL3 | −61 | 20 | 51 | 115 |
| PCL5-PEG10-PCL5 | −64 | 17 | 52 | 101 |
| PCL7-PEG6-PCL7 | −67 | 15 | 47 | 69 |
| Water Uptake (wt.%) | Tg (°C) | Tc (°C) | Tm (°C) | ΔH (J/g) | ||
|---|---|---|---|---|---|---|
| DRY | TPU-(PCL3-PEG14-PCL3) | - | −58 | 26 | 48 | 100 |
| TPU-(PCL5-PEG10-PCL5) | - | −60 | 21 | 49 | 81 | |
| TPU-(PCL7-PEG6-PCL7) | - | −60 | 23 | 51 | 37 | |
| WET | TPU-(PCL3-PEG14-PCL3) a | soluble | - | - | - | - |
| TPU-(PCL5-PEG10-PCL5) | 534 | −73 | b | 42 | 10 | |
| TPU-(PCL7-PEG6-PCL7) | 122 | −69 | b | 49 | 19 | |
| PEG/PCL | Water Uptake | E | σmax | ɛbreak | σyield | ɛyield | Wtensile | |
|---|---|---|---|---|---|---|---|---|
| (mol/mol) | (wt.%) | (MPa) | (MPa) | (%) | (MPa) | (%) | (N/mm2) | |
| TPU-(PCL3-PEG14-PCL3) | 70.2/29.8 | dry | 100 ± 15 | 6.1 ± 0.4 | 1080 ± 242 | 3.7 ± 0.5 | 5.1 ± 0.2 | 6244 ± 725 |
| TPU-(PCL5-PEG10-PCL5) | 49.8/50.2 | dry | 176 ± 16 | 8 ± 2 | 766 ± 282 | 7.5 ± 1 | 3.2 ± 0.6 | 5865 ± 3224 |
| TPU-(PCL7-PEG6-PCL7) | 29.5/70.5 | dry | 103 ± 16 | 15 ± 7 | 1566 ± 326 | 6.9 ± 0.6 | 6.1 ± 0.8 | 21841 ± 8667 |
| TPU-(PCL3-PEG14-PCL3) a | 70.2/29.8 | soluble | - | - | - | - | - | - |
| TPU-(PCL5-PEG10-PCL5) | 49.8/50.2 | 534 | 7 ± 2 | 4 ± 1 | 147 ± 41 | 3.0 ± 0.6 | 3.0 ± 0.5 | 513 ± 267 |
| TPU-(PCL7-PEG6-PCL7) | 29.5/70.5 | 122 | 52 ± 10 | 17 ± 2 | 1553 ± 155 | 7.3 ± 0.7 | 13 ± 2 | 17976 ± 3011 |
| TPSave (N/mm) | TPSmax (N/mm) | G (kJ/m2) | ||||
|---|---|---|---|---|---|---|
| Dry | Wet | Dry | Wet | Dry | Wet | |
| TPU-(PCL3-PEG14-PCL3) | 44 ± 12 | a | 71 ± 4 | a | 89 ± 12 | a |
| TPU-(PCL5-PEG10-PCL5) | 66 ± 14 | 8 ± 2 | 94 ± 3 | 13 ± 4 | 132 ± 21 | 16 ± 4 |
| TPU-(PCL7-PEG6-PCL7) | 64 ± 18 | 36 ± 20 | 93 ± 13 | 50 ± 24 | 127 ± 31 | 72 ± 41 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Güney, A.; Gardiner, C.; McCormack, A.; Malda, J.; Grijpma, D.W. Thermoplastic PCL-b-PEG-b-PCL and HDI Polyurethanes for Extrusion-Based 3D-Printing of Tough Hydrogels. Bioengineering 2018, 5, 99. https://doi.org/10.3390/bioengineering5040099
Güney A, Gardiner C, McCormack A, Malda J, Grijpma DW. Thermoplastic PCL-b-PEG-b-PCL and HDI Polyurethanes for Extrusion-Based 3D-Printing of Tough Hydrogels. Bioengineering. 2018; 5(4):99. https://doi.org/10.3390/bioengineering5040099
Chicago/Turabian StyleGüney, Aysun, Christina Gardiner, Andrew McCormack, Jos Malda, and Dirk W. Grijpma. 2018. "Thermoplastic PCL-b-PEG-b-PCL and HDI Polyurethanes for Extrusion-Based 3D-Printing of Tough Hydrogels" Bioengineering 5, no. 4: 99. https://doi.org/10.3390/bioengineering5040099
APA StyleGüney, A., Gardiner, C., McCormack, A., Malda, J., & Grijpma, D. W. (2018). Thermoplastic PCL-b-PEG-b-PCL and HDI Polyurethanes for Extrusion-Based 3D-Printing of Tough Hydrogels. Bioengineering, 5(4), 99. https://doi.org/10.3390/bioengineering5040099