
Engineering of CHO Cells for the Production of Recombinant Glycoprotein Vaccines with Xylosylated N-glycans


 ,
, 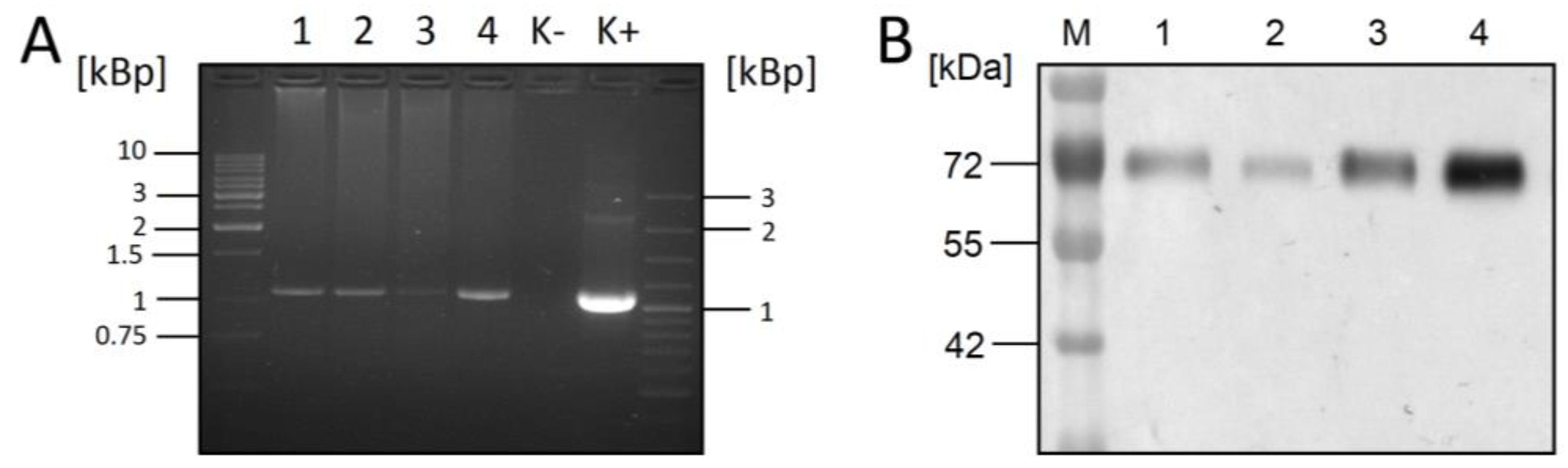{kind=link}
{kind=link}
{kind=link}
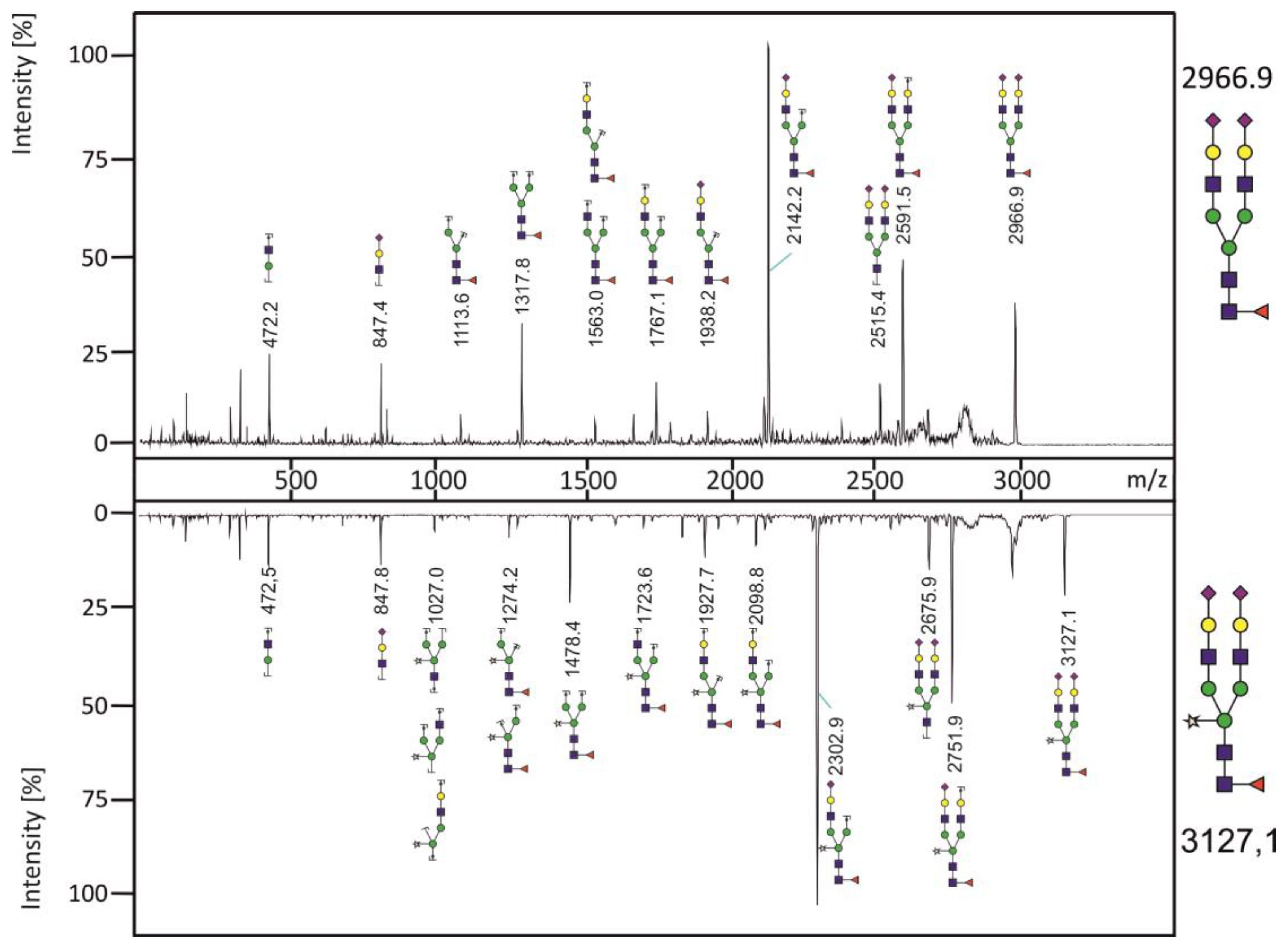{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture
2.3. Construction of Expression Vectors
2.4. Transfection of CHO Cells
2.5. Protein Production
2.6. Purification of Proteins
2.7. High-pH Anion-Exchange Chromatography with Pulsed Amperometric Detection (HPAEC-PAD) Monosaccharide Analysis
2.8. Release and Separation of N-Linked Glycans
2.9. Permethylation and Matrix-Assisted Laser Desorption/Ionization Time-of-Flight (MALDI-TOF) Mass Spectrometry
2.10. Cytokine and Gene Expression Analysis
3. Results and Discussion
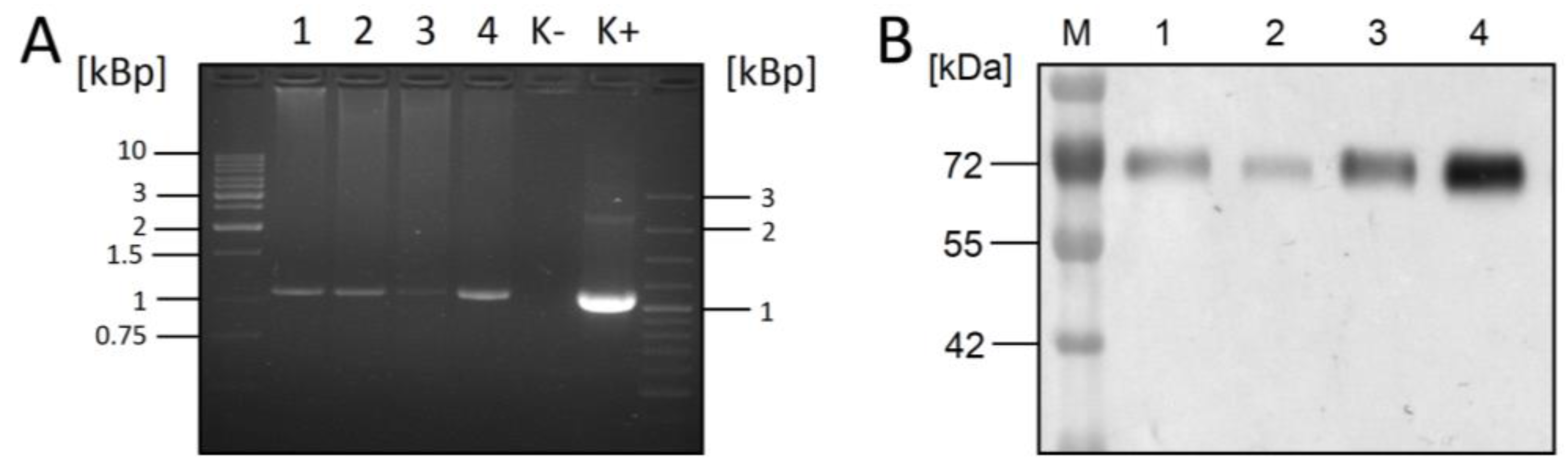
3.1. Co-Expression of RSV-F and XylT in CHO DG44 Cells
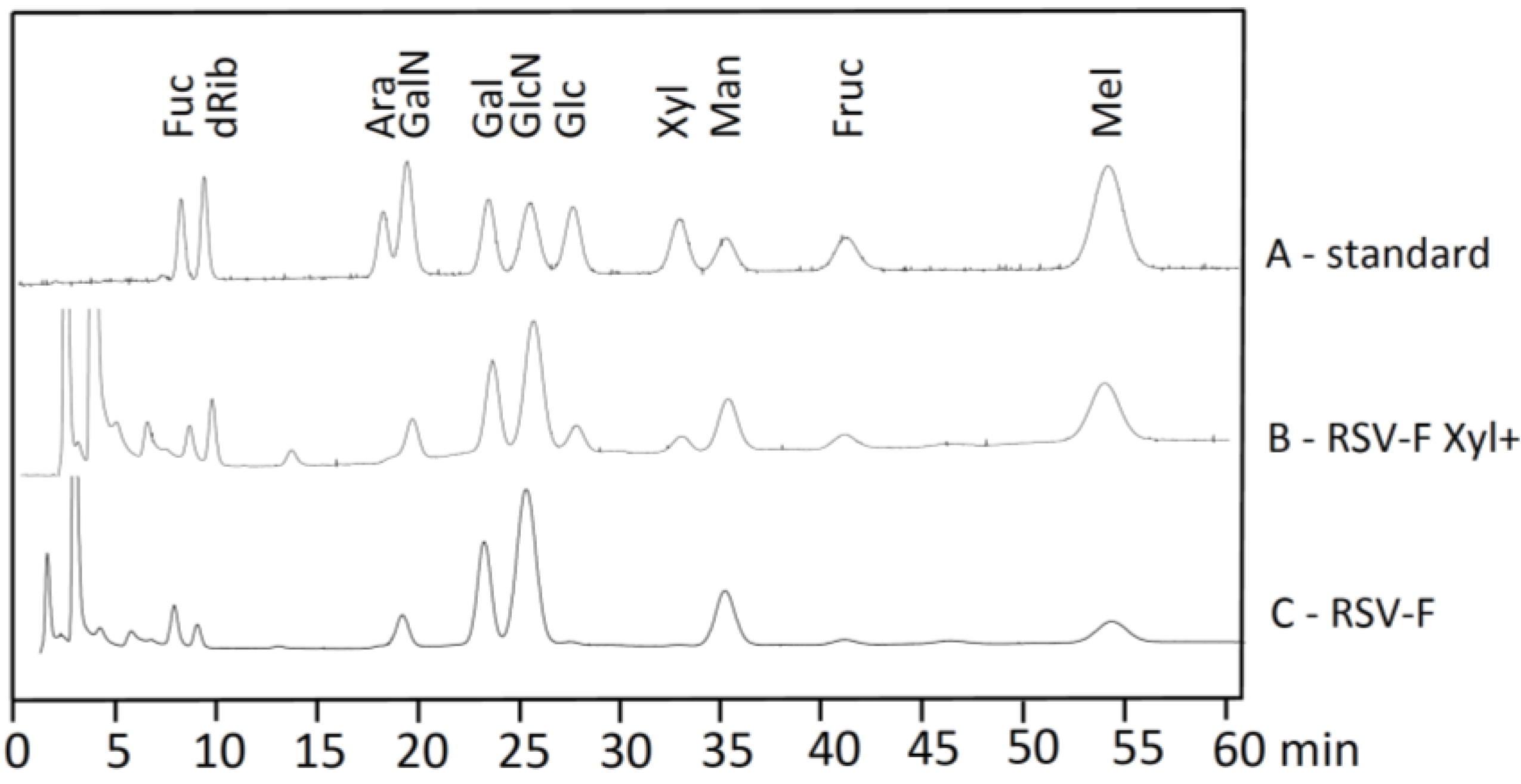
3.2. Monosaccharide Analysis
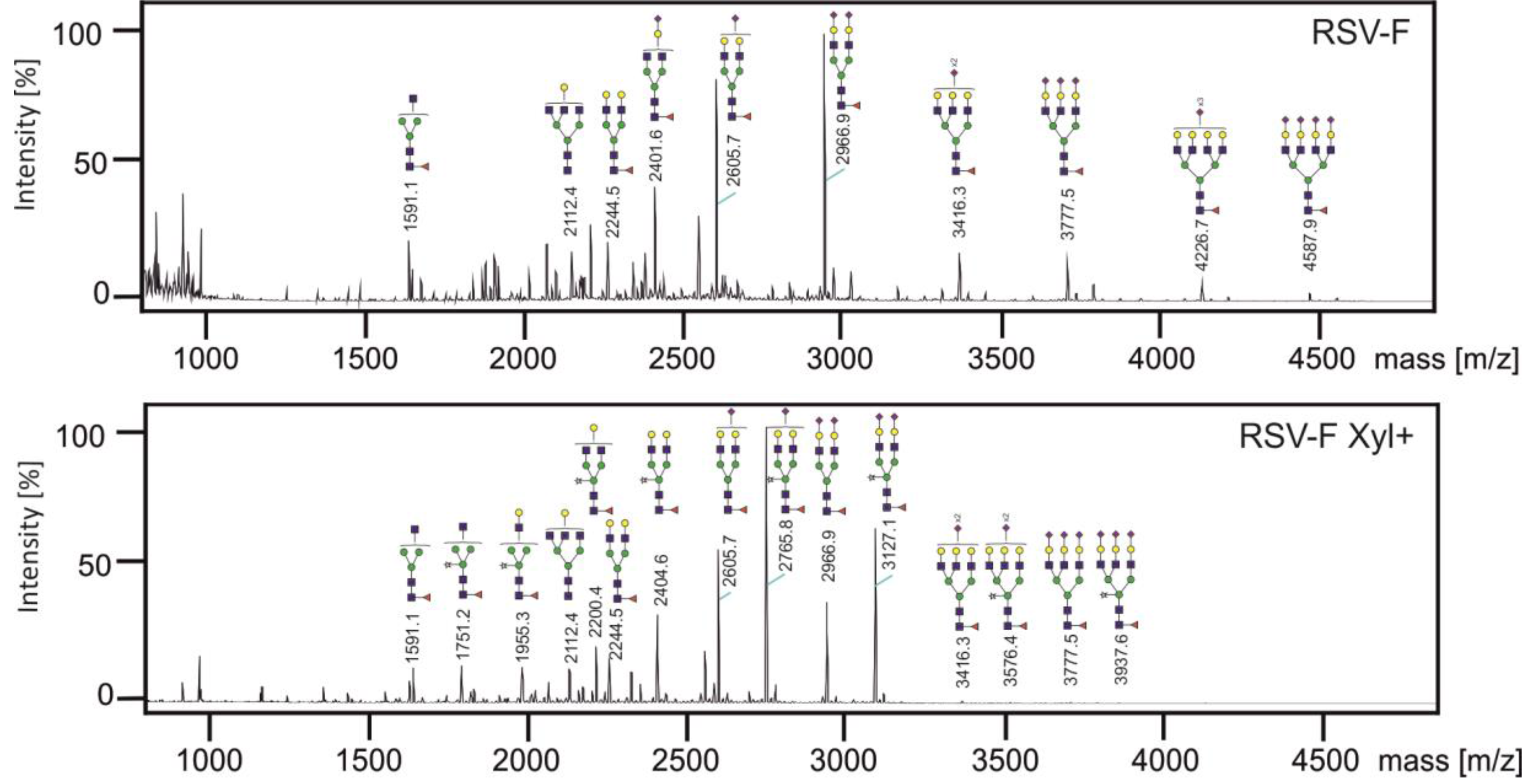
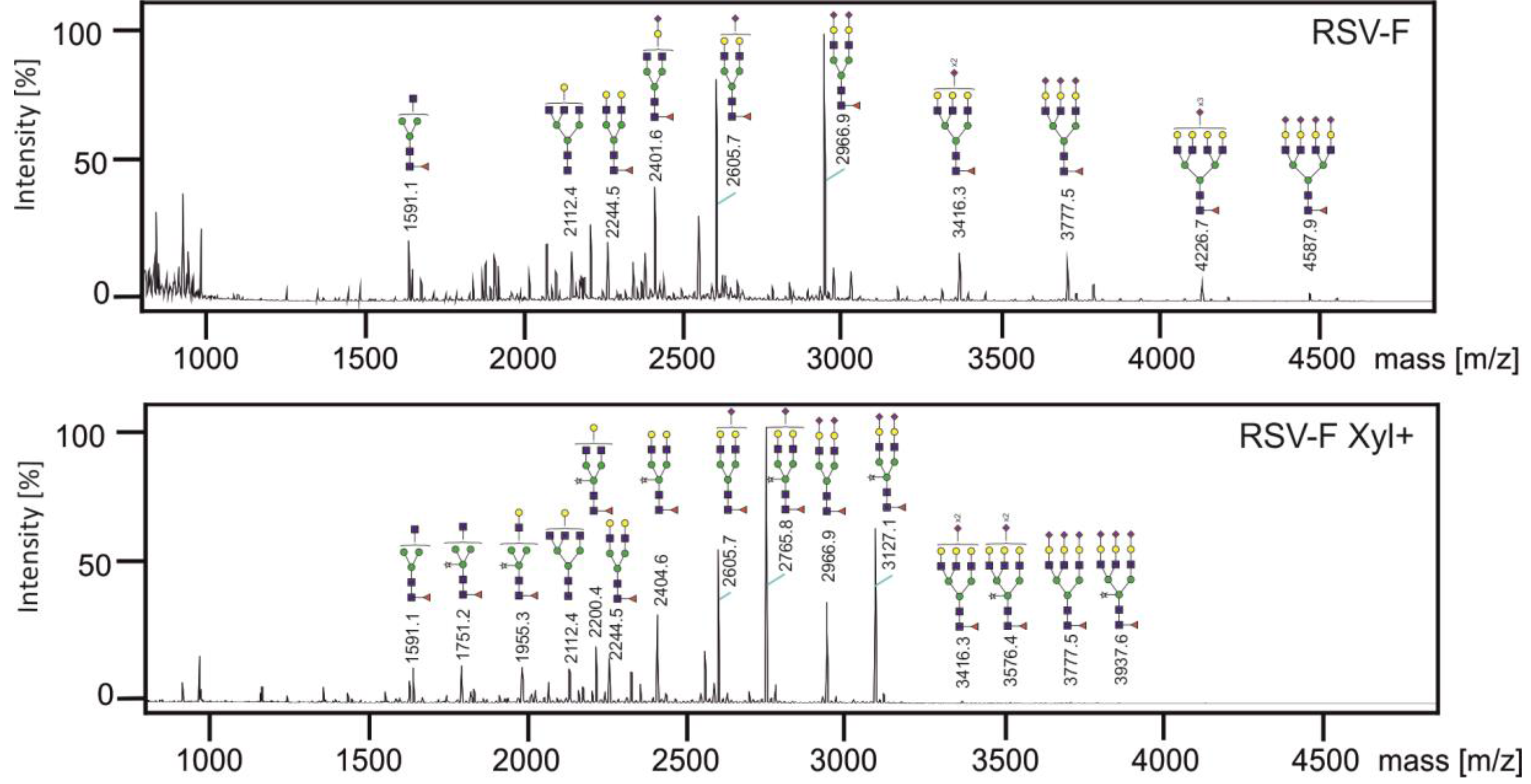
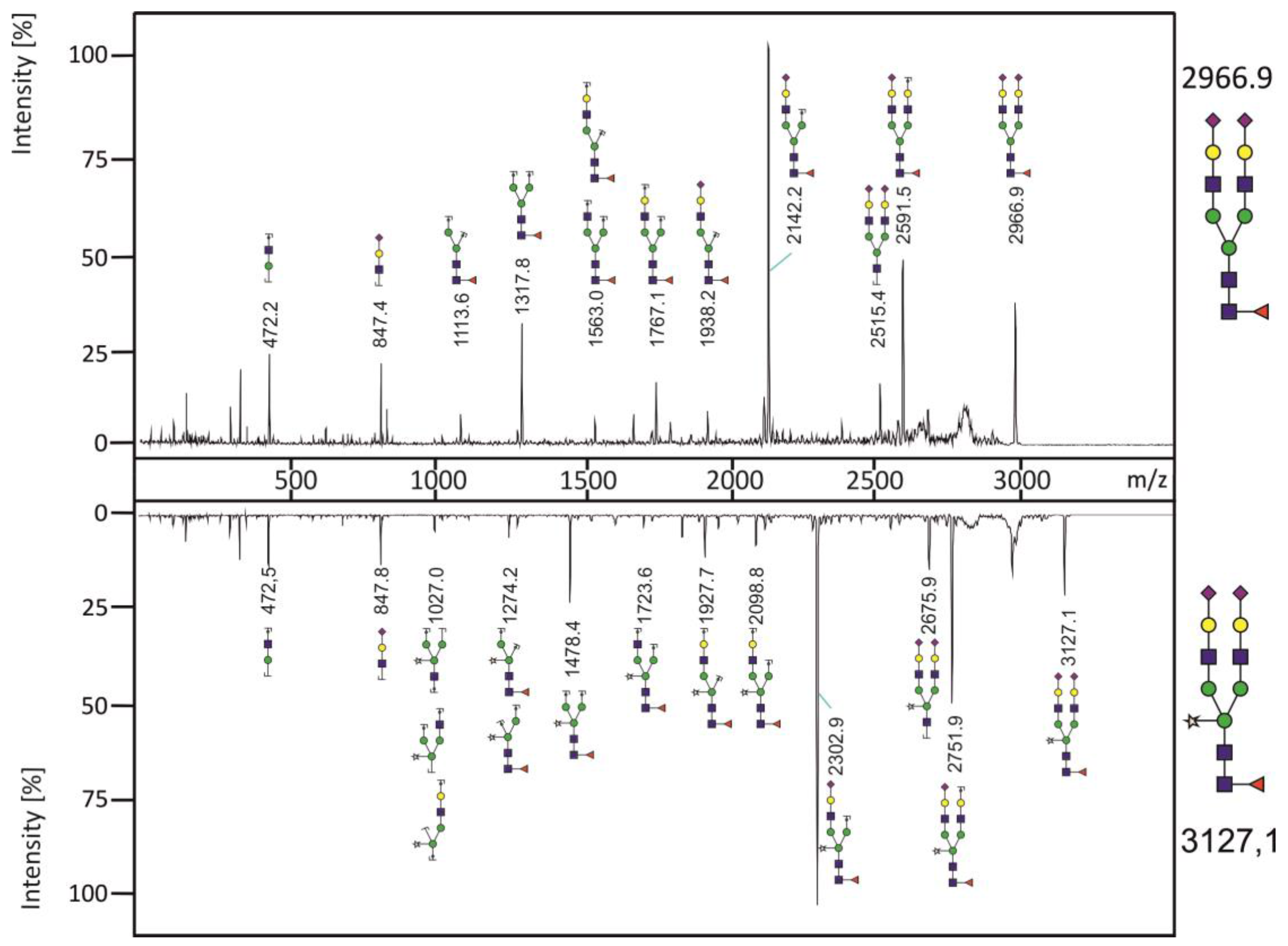
3.3. Analysis of N-glycans of RSV-F Proteins
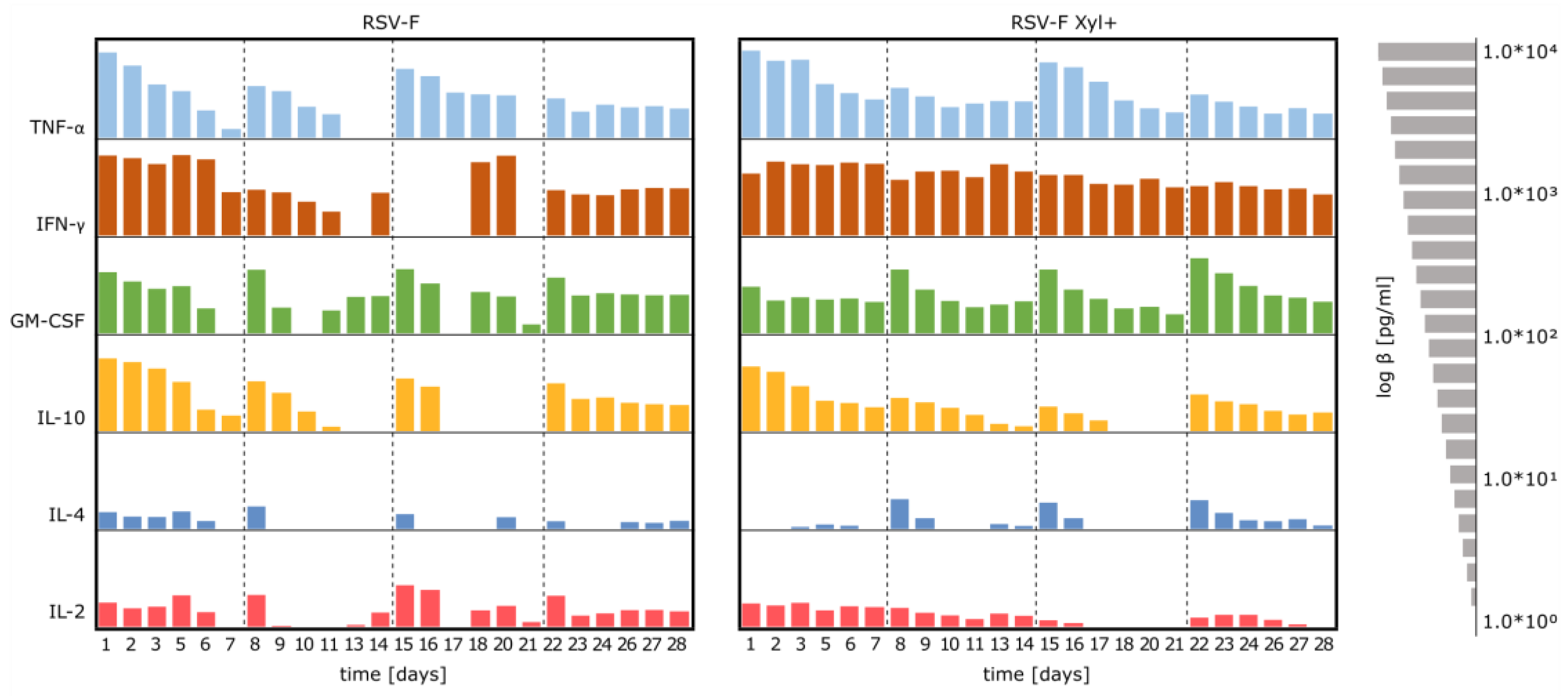
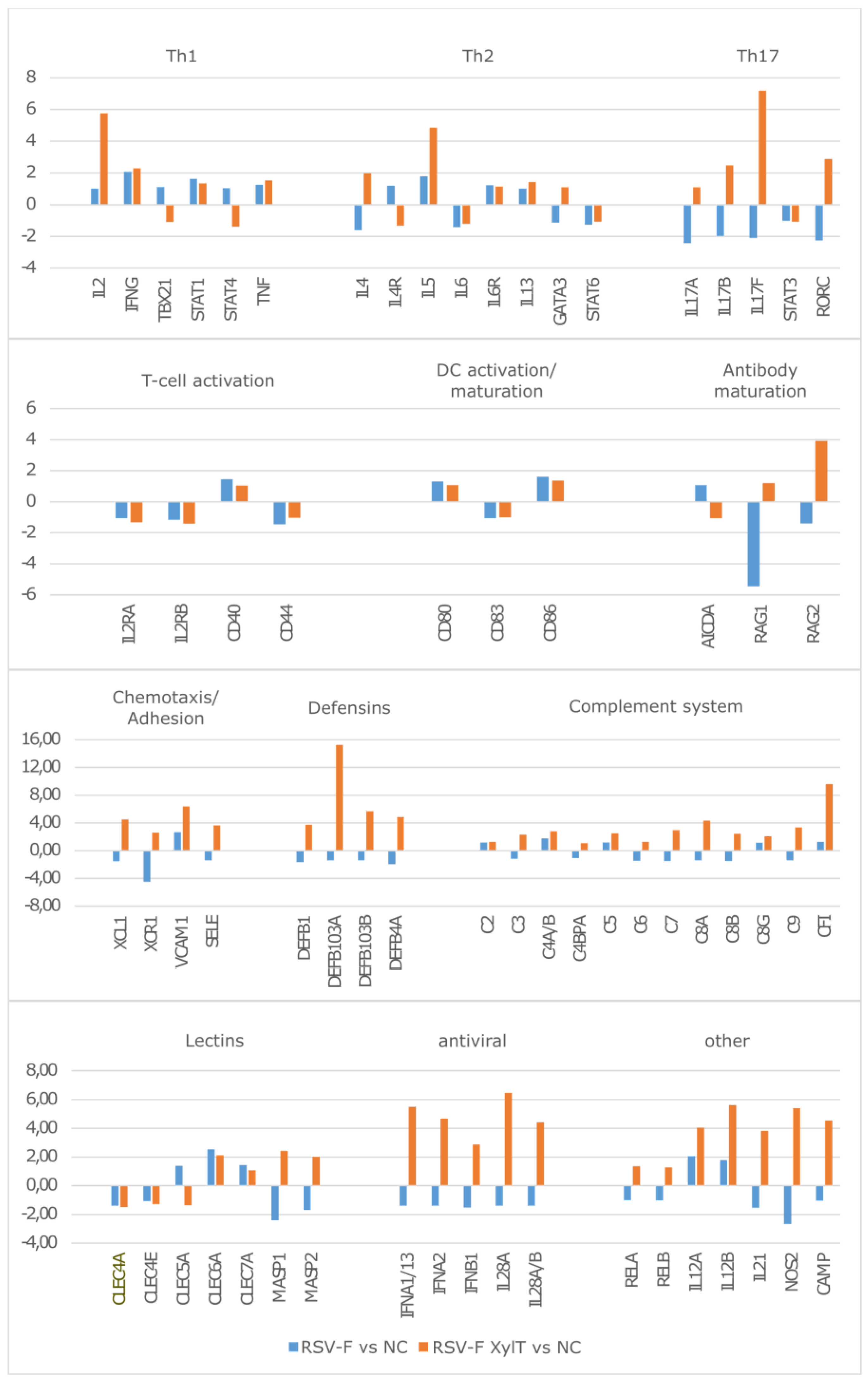
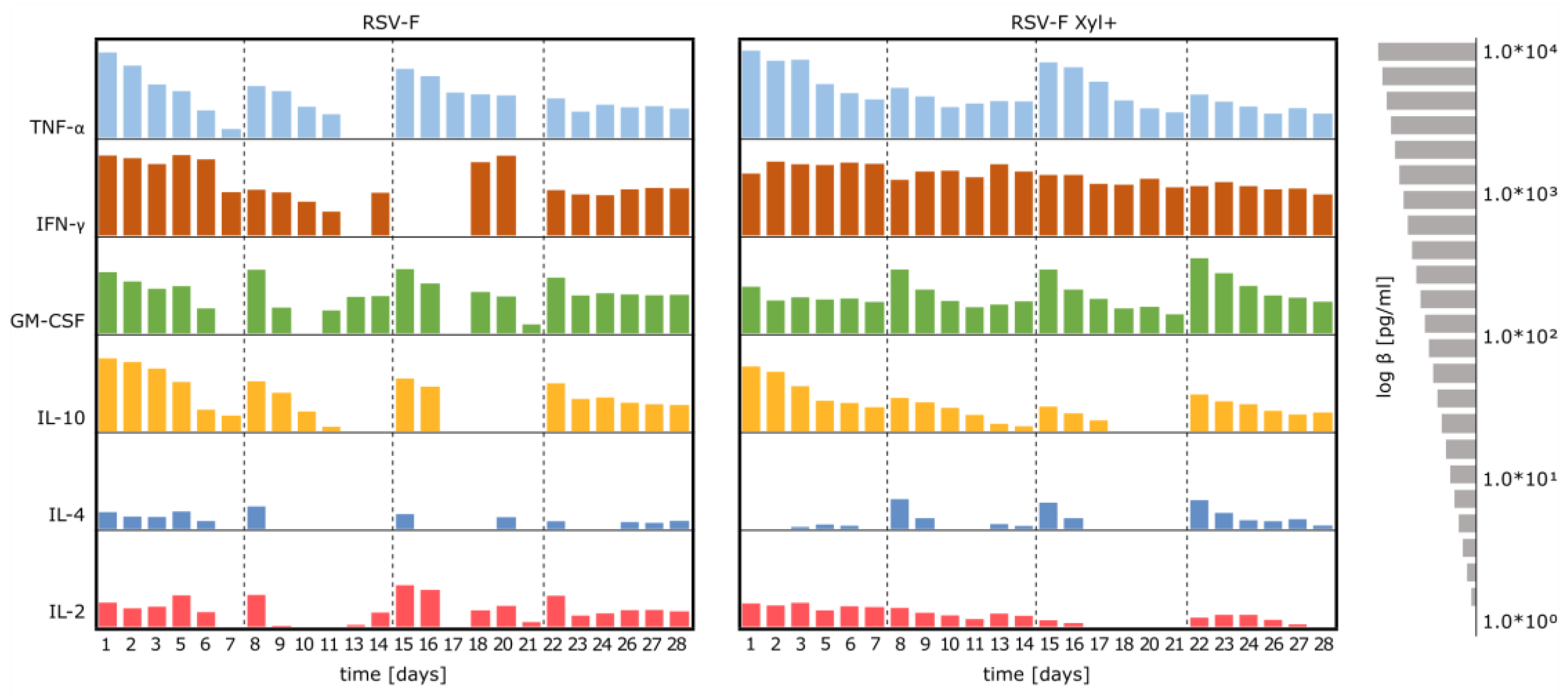
3.4. Stimulation of Immune Cells with RSV-F Proteins
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Varki, A. Biological roles of oligosaccharides: All of the theories are correct. Glycobiology 1993, 3, 97–130. [Google Scholar] [CrossRef] [PubMed]
- Brockhausen, I.; Schachter, H.; Stanley, P. O-GalNAc Glycans. In Essentials of Glycobiology, 2nd ed.; Varki, A., Ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2009; Chapter 9. [Google Scholar]
- Götting, C.; Kuhn, J.; Kleesiek, K. Human xylosyltransferases in health and disease. Cell. Mol. Life Sci. 2007, 64, 1498–1517. [Google Scholar] [CrossRef] [PubMed]
- Inamori, K.; Yoshida-Moriguchi, T.; Hara, Y.; Anderson, M.E.; Yu, L.; Campbell, K.P. Dystroglycan function requires xylosyl- and glucuronyltransferase activities of LARGE. Science 2012, 335, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Altmann, F. The role of protein glycosylation in allergy. Int. Arch. Allergy Immunol. 2007, 142, 99–115. [Google Scholar] [CrossRef] [PubMed]
- Strasser, R. Plant protein glycosylation. Glycobiology 2016, 26, 926–939. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Weng, Y.; Dickey, A.; Wang, K.Y. Plants as Factories for Human Pharmaceuticals: Applications and Challenges. Int. J. Mol. Sci. 2015, 16, 28549–28565. [Google Scholar] [CrossRef] [PubMed]
- Strasser, R.; Altmann, F.; Mach, L.; Glössl, J.; Steinkellner, H. Generation of Arabidopsis thaliana plants with complex N-glycans lacking beta1,2-linked xylose and core alpha1,3-linked fucose. FEBS Lett. 2004, 561, 132–136. [Google Scholar] [CrossRef]
- Castilho, A.; Steinkellner, H. Glyco-engineering in plants to produce human-like N-glycan structures. Biotechnol. J. 2012, 7, 1088–1098. [Google Scholar] [CrossRef] [PubMed]
- Bakker, H.; Oka, T.; Ashikov, A.; Yadav, A.; Berger, M.; Rana, N.A.; Bai, X.; Jigami, Y.; Haltiwanger, R.S.; Esko, J.D.; et al. Functional UDP-xylose transport across the endoplasmic reticulum/Golgi membrane in a Chinese hamster ovary cell mutant defective in UDP-xylose Synthase. J. Biol. Chem. 2009, 284, 2576–2583. [Google Scholar] [CrossRef] [PubMed]
- Gornik, O.; Pavić, T.; Lauc, G. Alternative glycosylation modulates function of IgG and other proteins—Implications on evolution and disease. Biochim. Biophys. Acta 2012, 1820, 1318–1326. [Google Scholar] [CrossRef] [PubMed]
- Nishat, S.; Andreana, P.R. Entirely Carbohydrate-Based Vaccines: An Emerging Field for Specific and Selective Immune Responses. Vaccines (Basel) 2016, 4, E19. [Google Scholar] [CrossRef]
- Brzezicka, K.; Echeverria, B.; Serna, S.; van Diepen, A.; Hokke, C.H.; Reichardt, N.C. Synthesis and microarray-assisted binding studies of core xylose and fucose containing N-glycans. ACS Chem. Biol. 2015, 10, 1290–1302. [Google Scholar] [CrossRef] [PubMed]
- Glezen, W.P.; Taber, L.H.; Frank, A.L.; Kasel, J.A. Risk of primary infection and reinfection with respiratory syncytial virus. Am. J. Dis. Child. 1986, 140, 543–546. [Google Scholar] [CrossRef] [PubMed]
- Thorburn, K. Pre-existing disease is associated with a significantly higher risk of death in severe respiratory syncytial virus infection. Arch. Dis. Child. 2009, 94, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Anderson, L.J.; Dormitzer, P.R.; Nokes, D.J.; Rappuoli, R.; Roca, A.; Graham, B.S. Strategic priorities for respiratory syncytial virus (RSV) vaccine development. Vaccine 2013, 31 (Suppl. 2), B209–B215. [Google Scholar] [CrossRef] [PubMed]
- Gruber, C.; Levine, S. Respiratory syncytial virus polypeptides. IV. The oligosaccharides of the glycoproteins. J. Gen. Virol. 1985, 66, 417–432. [Google Scholar] [CrossRef] [PubMed]
- Scheid, A.; Choppin, P.W. Two disulfide-linked polypeptide chains constitute the active F protein of paramyxoviruses. Virology 1977, 80, 54–66. [Google Scholar] [CrossRef]
- Arumugham, R.G.; Hildreth, S.W.; Paradiso, P.R. Interprotein disulfide bonding between F and G glycoproteins of human respiratory syncytial virus. Arch. Virol. 1989, 105, 65–79. [Google Scholar] [CrossRef]
- Ternette, N.; Tippler, B.; Uberla, K.; Grunwald, T. Immunogenicity and efficacy of codon optimized DNA vaccines encoding the F-protein of respiratory syncytial virus. Vaccine 2007, 25, 7271–7279. [Google Scholar] [CrossRef] [PubMed]
- Reinke, S.O.; Bayer, M.; Berger, M.; Blanchard, V.; Hinderlich, S. Analysis of cell surface N-glycosylation of the human embryonic kidney 293t cell line. J. Carbohydr. Chem. 2011, 30, 218–232. [Google Scholar] [CrossRef]
- Wada, Y.; Azadi, P.; Costello, C.E.; Dell, A.; Dwek, R.A.; Geyer, H.; Geyer, R.; Kakehi, K.; Karlsson, N.G.; Kato, K.; et al. Comparison of the methods for profiling glycoprotein glycans—HUPO Human Disease Glycomics/Proteome Initiative multi-institutional study. Glycobiology 2007, 17, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Dell, A.; Khoo, K.H.; Panico, M.; McDowell, R.A.; Etienne, A.T.; Reason, A.J.; Morris, H.R. FAB-MS and ES-MS of glycoproteins. In Glycobiology: A Practical Approach; Fukuda, M., Kobata, A., Eds.; IRL Press at Oxford University Press: Oxford, UK, 1993; pp. 187–222. [Google Scholar]
- Frisch, E.; Kaup, M.; Egerer, K.; Weimann, A.; Tauber, R.; Berger, M.; Blanchard, V. Profiling of endo H-released serum N-glycans using CE-LIF and MALDI-TOF-MS--application to rheumatoid arthritis. Electrophoresis 2011, 32, 3510–3515. [Google Scholar] [CrossRef] [PubMed]
- Varki, A.; Cummings, R.D.; Esko, J.D.; Freeze, H.H.; Stanley, P.; Marth, J.D.; Bertozzi, C.R.; Hart, G.W.; Etzler, M.E. Symbol nomenclature for glycan representation. Proteomics 2009, 9, 5398–5399. [Google Scholar] [CrossRef] [PubMed]
- Giese, C.; Lubitz, A.; Demmler, C.D.; Reuschel, J.; Bergner, K.; Marx, U. Immunological substance testing on human lymphatic micro-organoids in vitro. J. Biotechnol. 2010, 148, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Seifert, M.; Lubitz, A.; Trommer, J.; Könnig, D.; Korus, G.; Marx, U.; Volk, H.D.; Duda, G.; Kasper, G.; Lehmann, K.; et al. Crosstalk between immune cells and mesenchymal stromal cells in a 3D bioreactor system. Int. J. Artif. Organs 2012, 35, 986–995. [Google Scholar] [CrossRef]
- Geiss, G.K.; Bumgarner, R.E.; Birditt, B.; Dahl, T.; Dowidar, N.; Fell, H.P.; Dunaway, D.L.; Ferree, S.; George, R.D.; Grogan, T.; et al. Direct multiplexed measurement of gene expression with color-coded probe pairs. Nat. Biotechnol. 2008, 26, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Radke, L.; Giese, C.; Lubitz, A.; Hinderlich, S.; Sandig, G.; Hummel, M.; Frohme, M. Reference gene stability in peripheral blood mononuclear cells determined by qPCR and NanoString. Microchim. Acta 2014, 181, 1733–1742. [Google Scholar] [CrossRef]
- Esko, J.D.; Stanley, P. Glycosylation Mutants of Cultured Cells. In Essentials of Glycobiology, 2nd ed.; Varki, A., Ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2009; Chapter 46. [Google Scholar]
- Stanley, P.; Schachter, H.; Taniguchi, N. N-Glycans. In Essentials of Glycobiology, 2nd ed.; Varki, A., Ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2009; Chapter 8. [Google Scholar]
- Radke, L.; Lopez-Hemmerling, D.A.; Lubitz, A.; Giese, C.; Frohme, M. Induced cytokine response of human PMBC-cultures: Correlation of gene expression and secretion profiling and the effect of cryopreservation. Cell Immunol. 2012, 272, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Gabr, M.A.; Jung, L.; Helbling, A.R.; Sinclair, S.M.; Allen, K.D.; Shamji, M.F.; Richardson, W.J.; Fitch, R.D.; Setton, L.A.; Chen, L. Interleukin-17 synergizes with IFNγ or TNFα to promote inflammatory mediator release and intercellular adhesion molecule-1 (ICAM-1) expression in human intervertebral disc cells. J. Orthop. Res. 2011, 29, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Brzezicka, K.; Vogel, V.; Serna, S.; Johannssen, T.; Lepenies, B.; Reichardt, N.C. Influence of Core β-1,2-Xylosylation on Glycoprotein Recognition by Murine C-type Lectin Receptors and Its Impact on Dendritic Cell Targeting. ACS Chem. Biol. 2016, 11, 2347–2356. [Google Scholar] [CrossRef] [PubMed]
- Ghaderi, D.; Taylor, R.E.; Padler-Karavani, V.; Diaz, S.; Varki, A. Implications of the presence of N-glycolylneuraminic acid in recombinant therapeutic glycoproteins. Nat. Biotechnol. 2010, 28, 863–867. [Google Scholar] [CrossRef] [PubMed]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sandig, G.; Von Horsten, H.H.; Radke, L.; Blanchard, V.; Frohme, M.; Giese, C.; Sandig, V.; Hinderlich, S. Engineering of CHO Cells for the Production of Recombinant Glycoprotein Vaccines with Xylosylated N-glycans. Bioengineering 2017, 4, 38. https://doi.org/10.3390/bioengineering4020038
Sandig G, Von Horsten HH, Radke L, Blanchard V, Frohme M, Giese C, Sandig V, Hinderlich S. Engineering of CHO Cells for the Production of Recombinant Glycoprotein Vaccines with Xylosylated N-glycans. Bioengineering. 2017; 4(2):38. https://doi.org/10.3390/bioengineering4020038
Chicago/Turabian StyleSandig, Grit, Hans Henning Von Horsten, Lars Radke, Véronique Blanchard, Marcus Frohme, Christoph Giese, Volker Sandig, and Stephan Hinderlich. 2017. "Engineering of CHO Cells for the Production of Recombinant Glycoprotein Vaccines with Xylosylated N-glycans" Bioengineering 4, no. 2: 38. https://doi.org/10.3390/bioengineering4020038
APA StyleSandig, G., Von Horsten, H. H., Radke, L., Blanchard, V., Frohme, M., Giese, C., Sandig, V., & Hinderlich, S. (2017). Engineering of CHO Cells for the Production of Recombinant Glycoprotein Vaccines with Xylosylated N-glycans. Bioengineering, 4(2), 38. https://doi.org/10.3390/bioengineering4020038