
Human Cell Line-Derived Monoclonal IgA Antibodies for Cancer Immunotherapy

Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Consumables
2.2. Recombinant Antibody Production
2.3. Eukaryotic Cell Lines
2.4. Perfusion Process in the 2-L Stirred Tank Bioreactor
2.5. Antibody Purification by Affinity Chromatography
2.6. Sodium Dodecyl Sulfate Polyacrylamide Gel Electrophoresis and Western Blots
2.7. Size Exclusion Chromatography
2.8. Surface Plasmon Resonance
2.9. N-Glycan Profiling
2.10. Flow Cytometry
2.11. Proliferation Inhibition
2.12. Antibody-Dependent Cellular Cytotoxicity Assay
2.13. Antibody-Dependent Cellular Phagocytosis Assay
2.14. B Cell Depletion in Whole Blood
2.15. Software and Statistical Analysis
3. Results
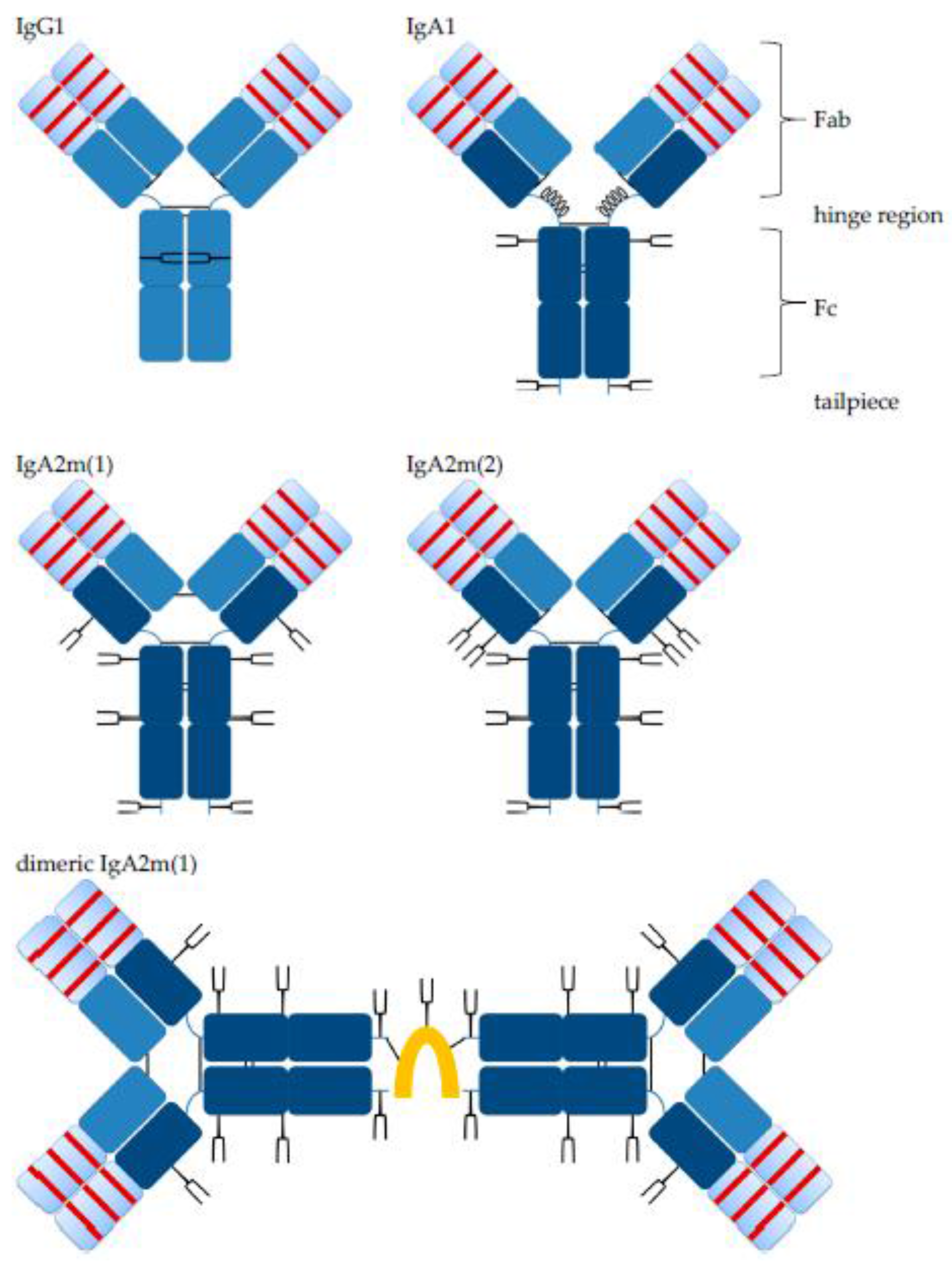
3.1. Generation of Human Cell Line-Derived Monoclonal IgA Antibodies Directed against Five Targets for Cancer Therapy
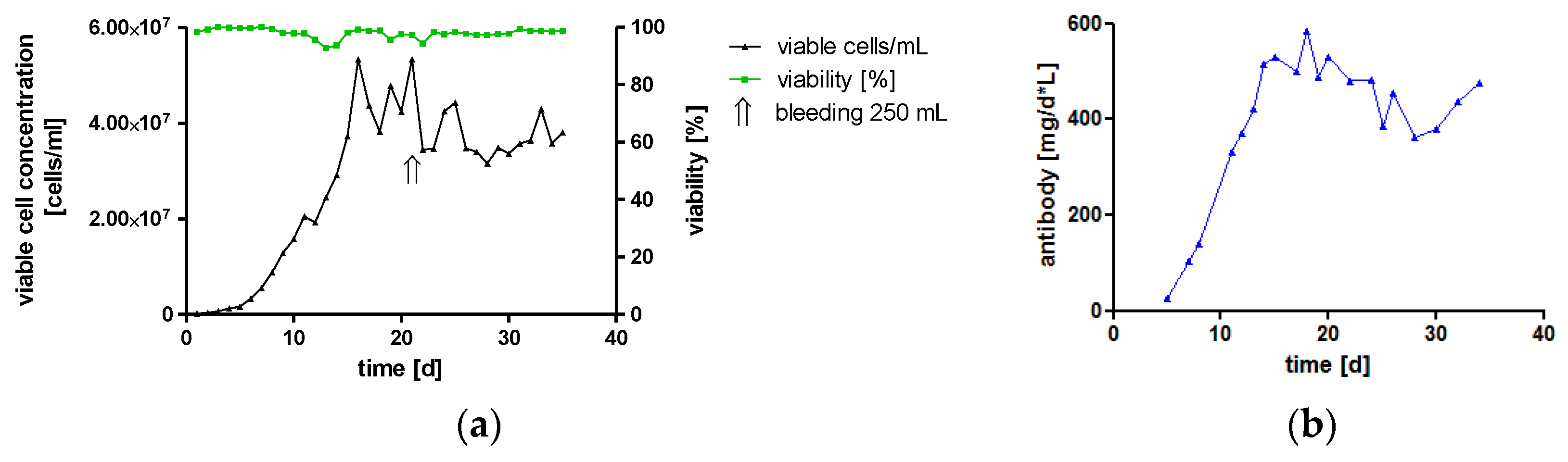
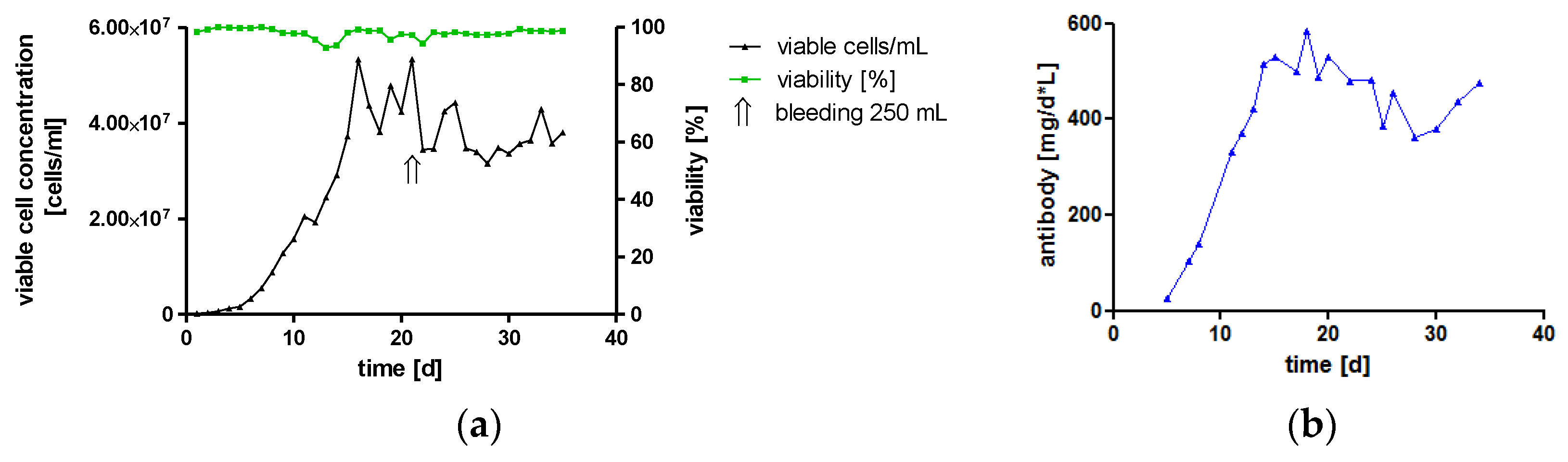
High-Yield Production of hTM IgA2 by Cultivation in a Bioreactor
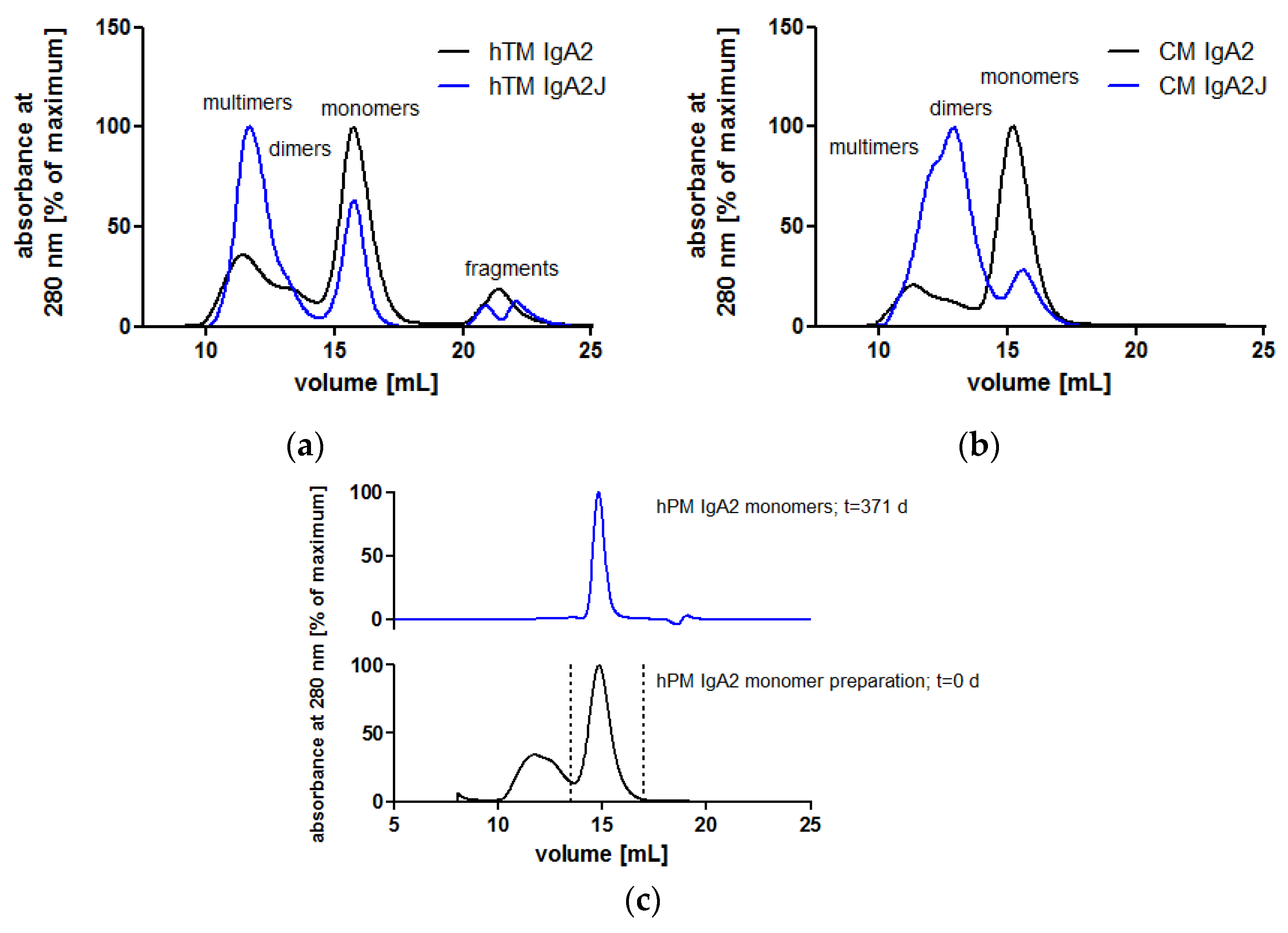
3.2. Biochemical Integrity of Novel Recombinant Monoclonal IgA Antibodies
3.2.1. Human N-Glycosylation Profiles of Recombinant IgA Antibodies
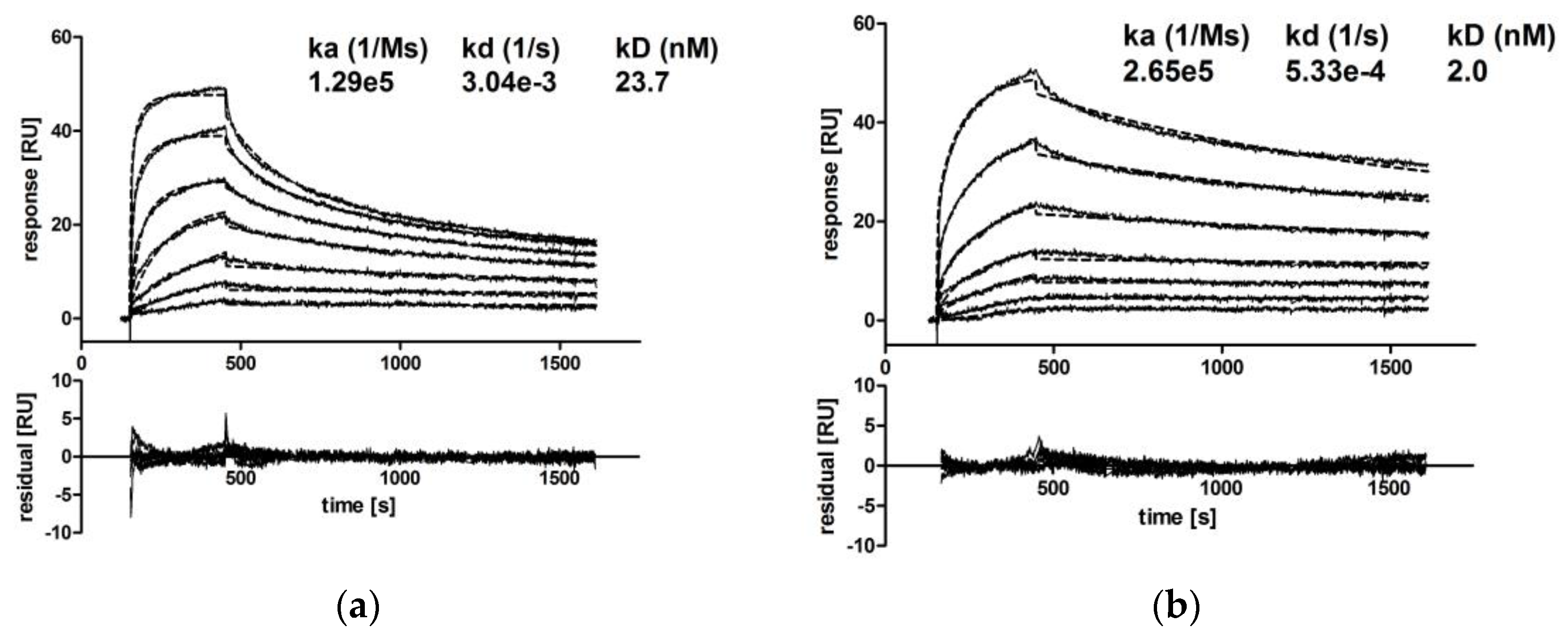
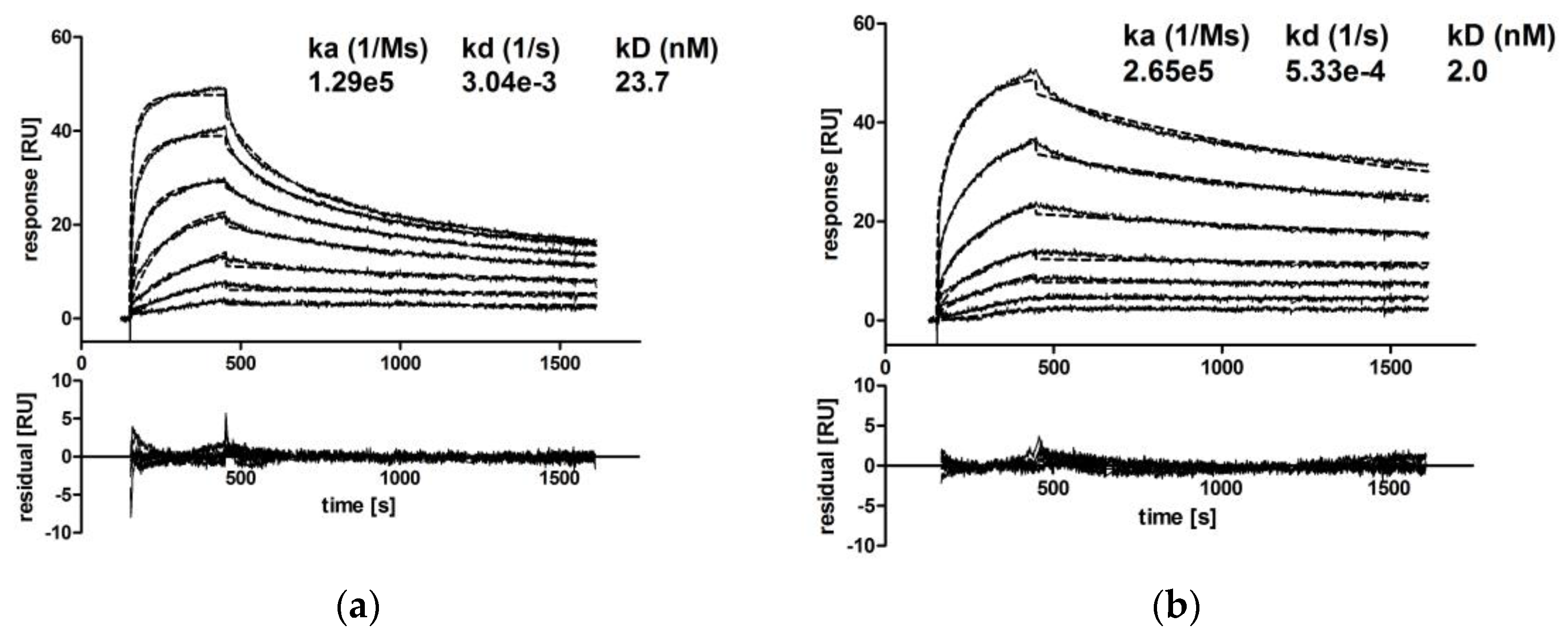
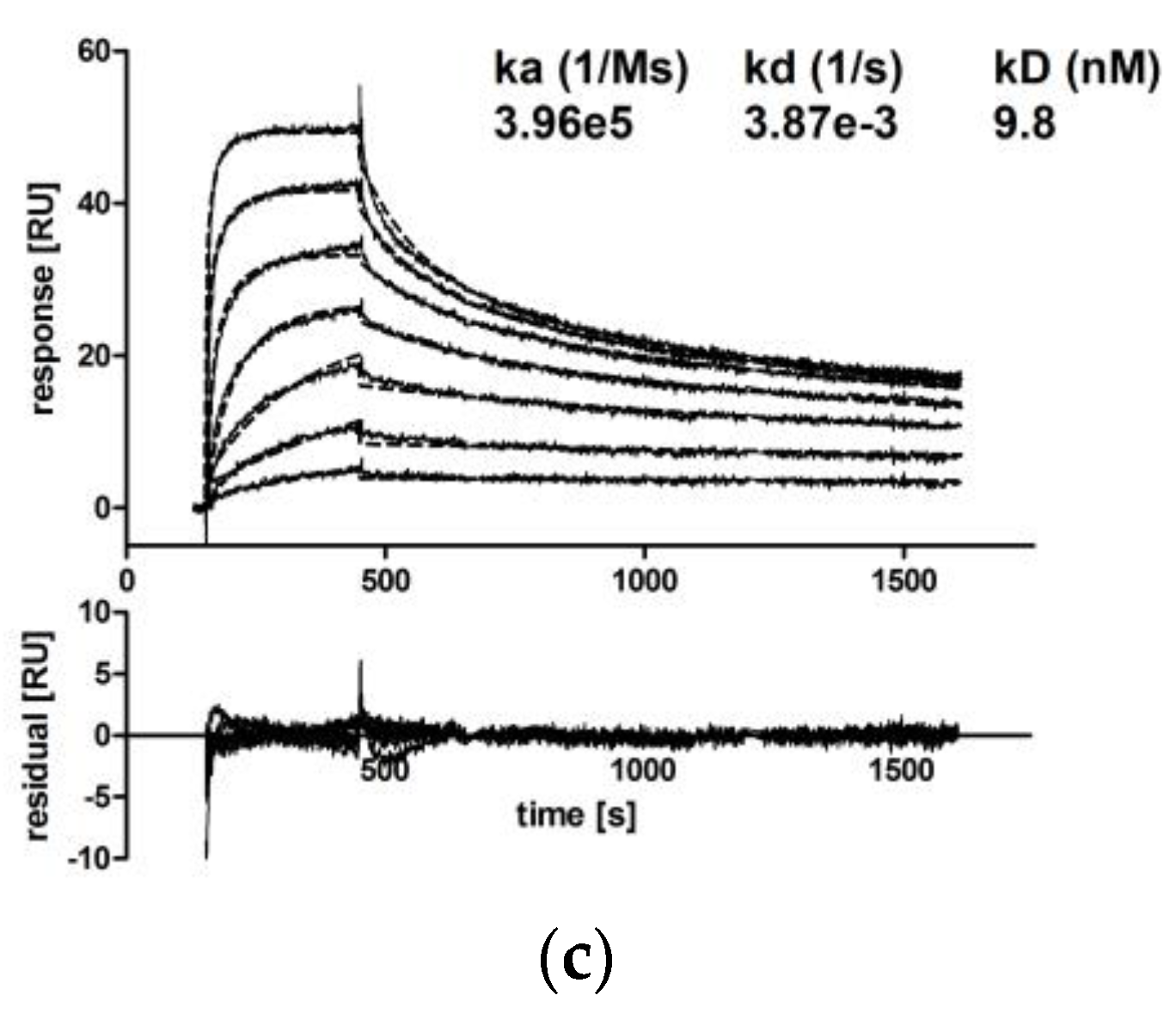
3.2.2. IgA Dimers Show Increased Antigen Binding Avidity Compared to IgG Antibodies
3.3. Recombinant IgA Antibodies Are Biofunctional against Cancer Target Cell Lines
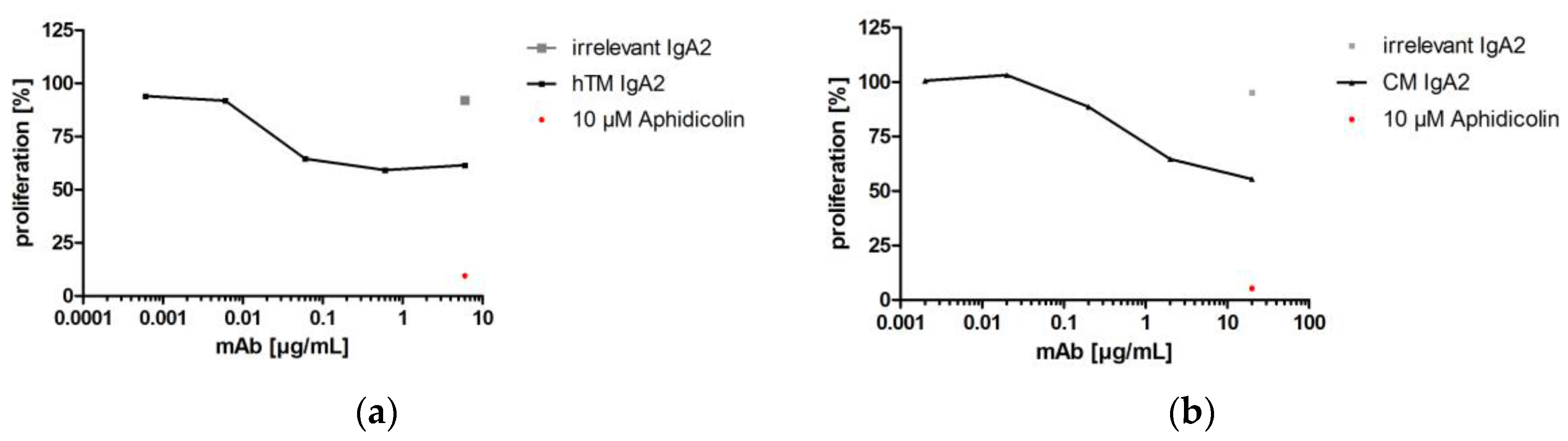
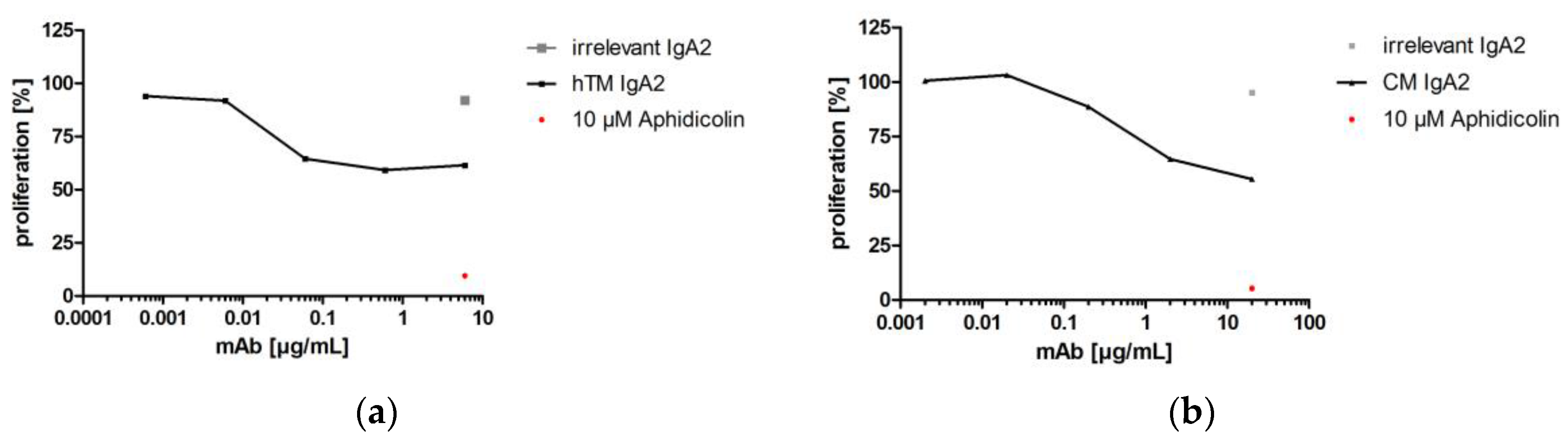
3.3.1. Proliferation Inhibition
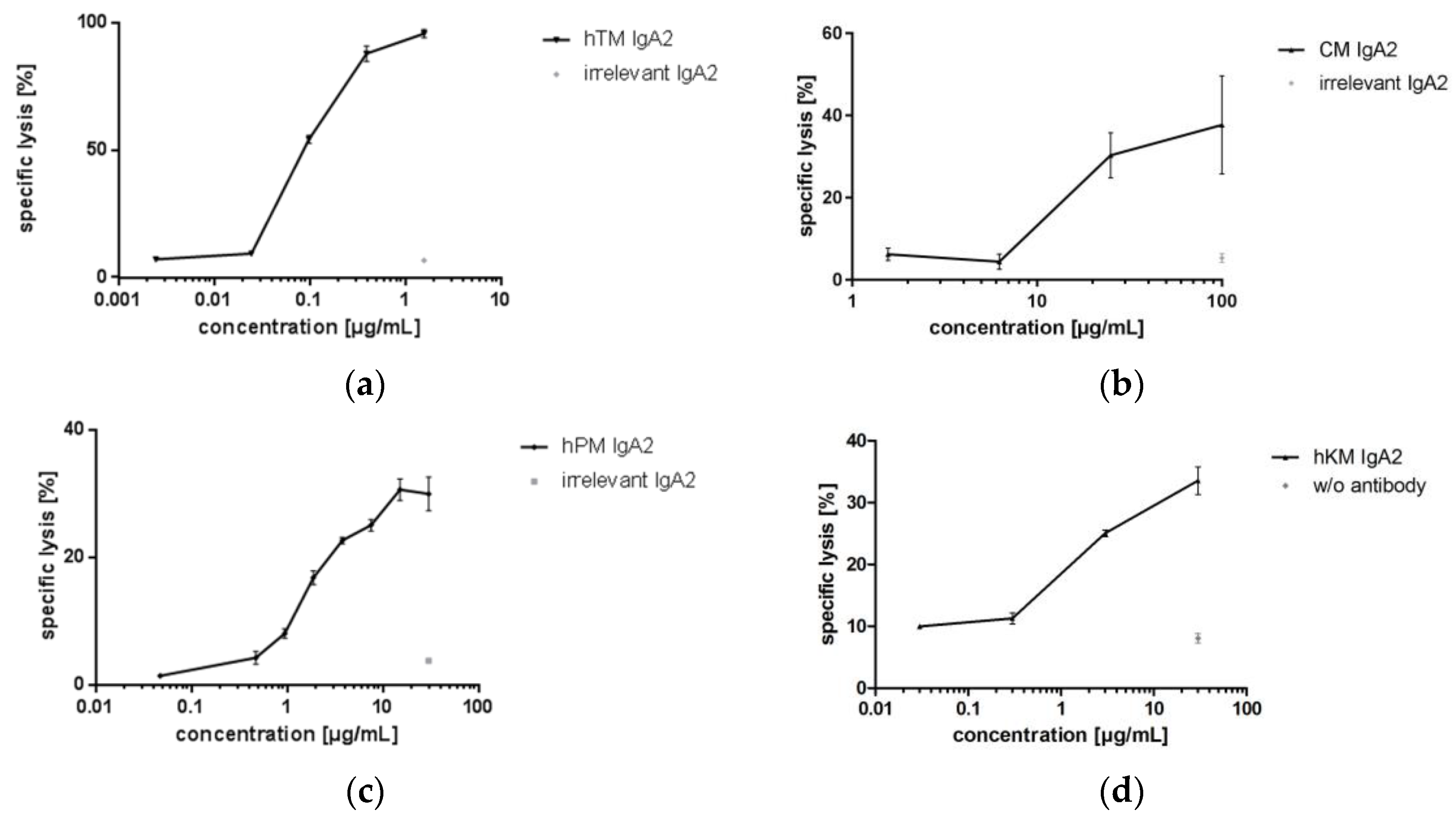
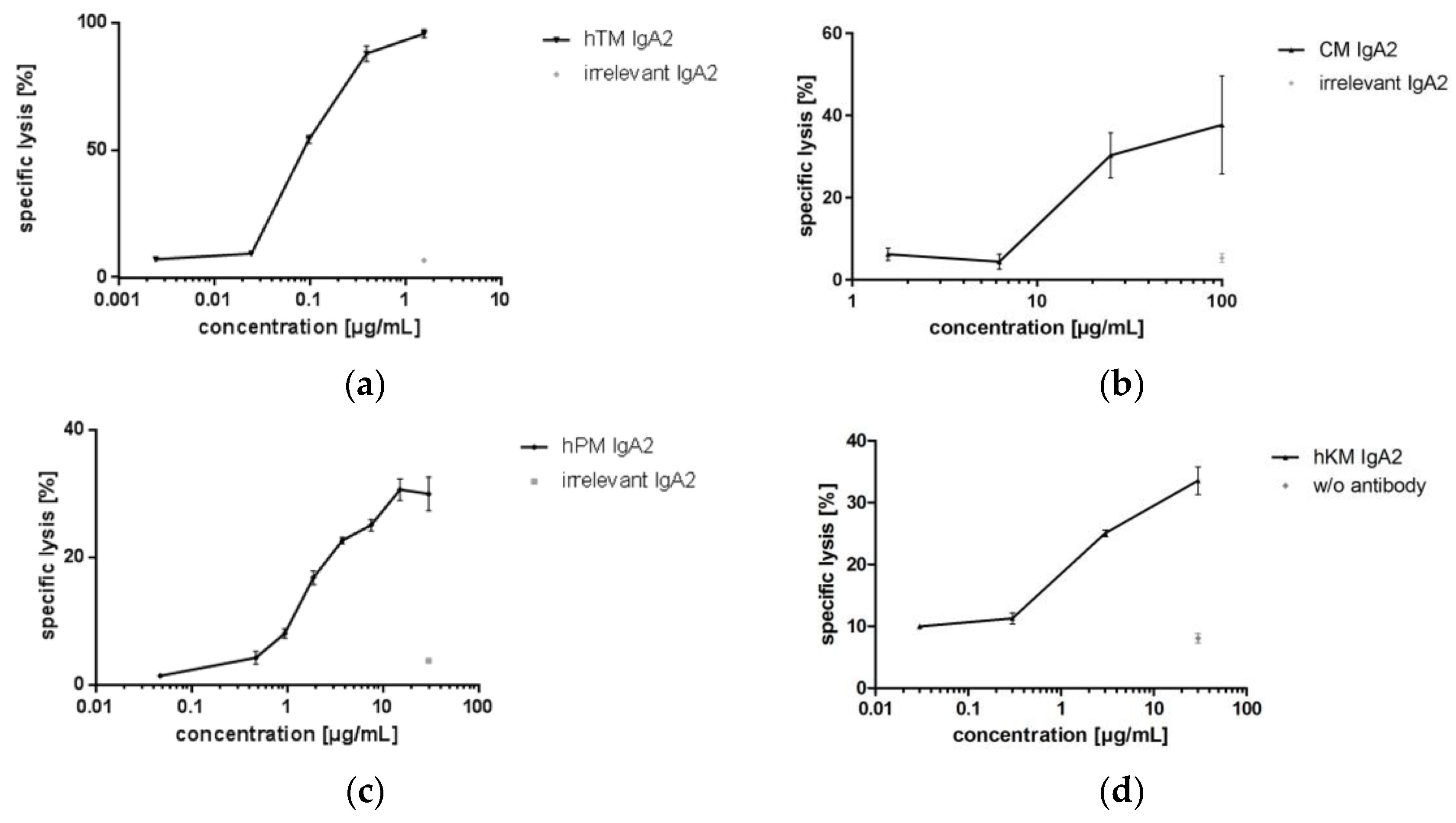
3.3.2. IgA Antibodies Mediate Antibody-Dependent Cellular Cytotoxicity
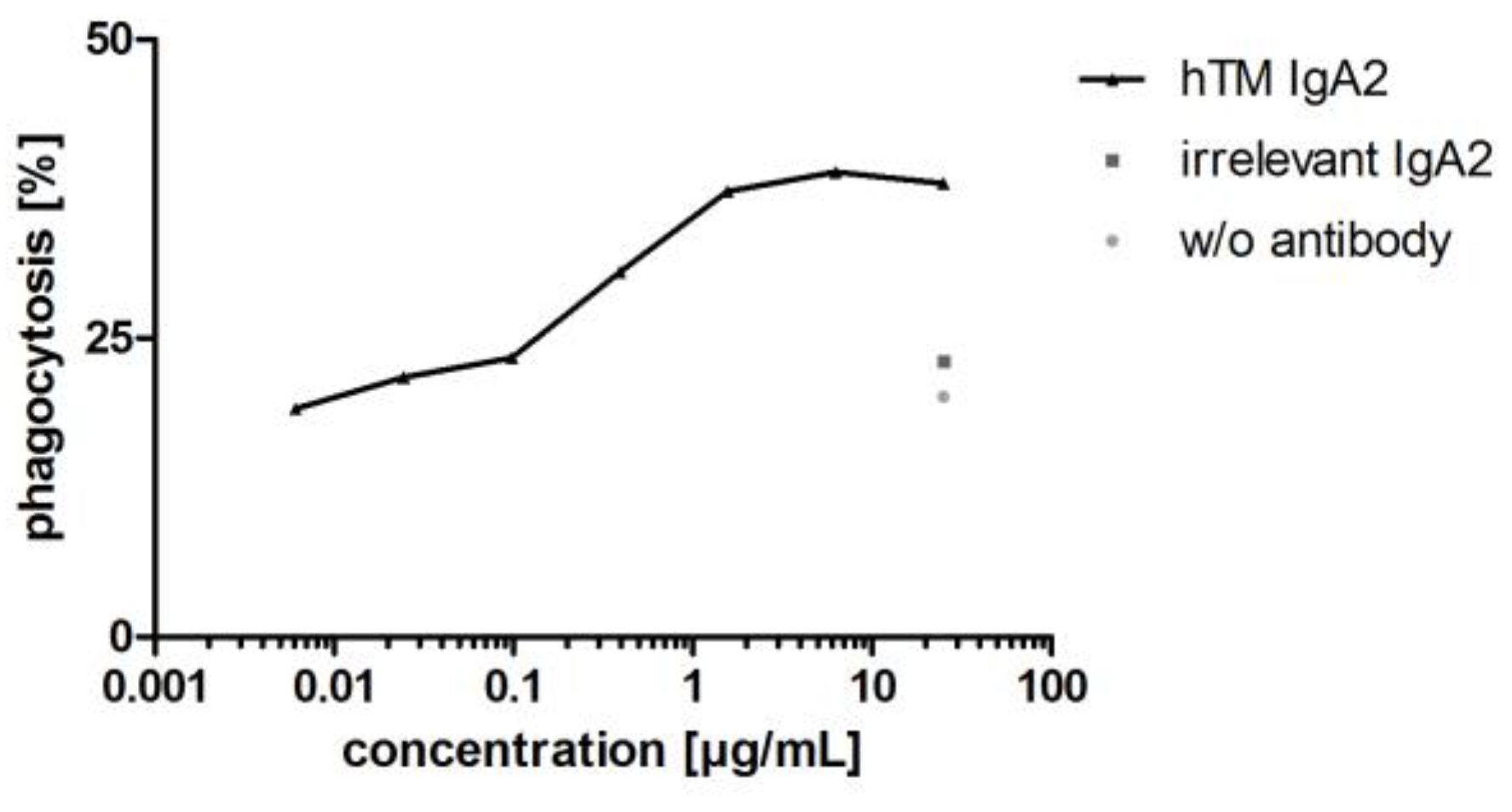
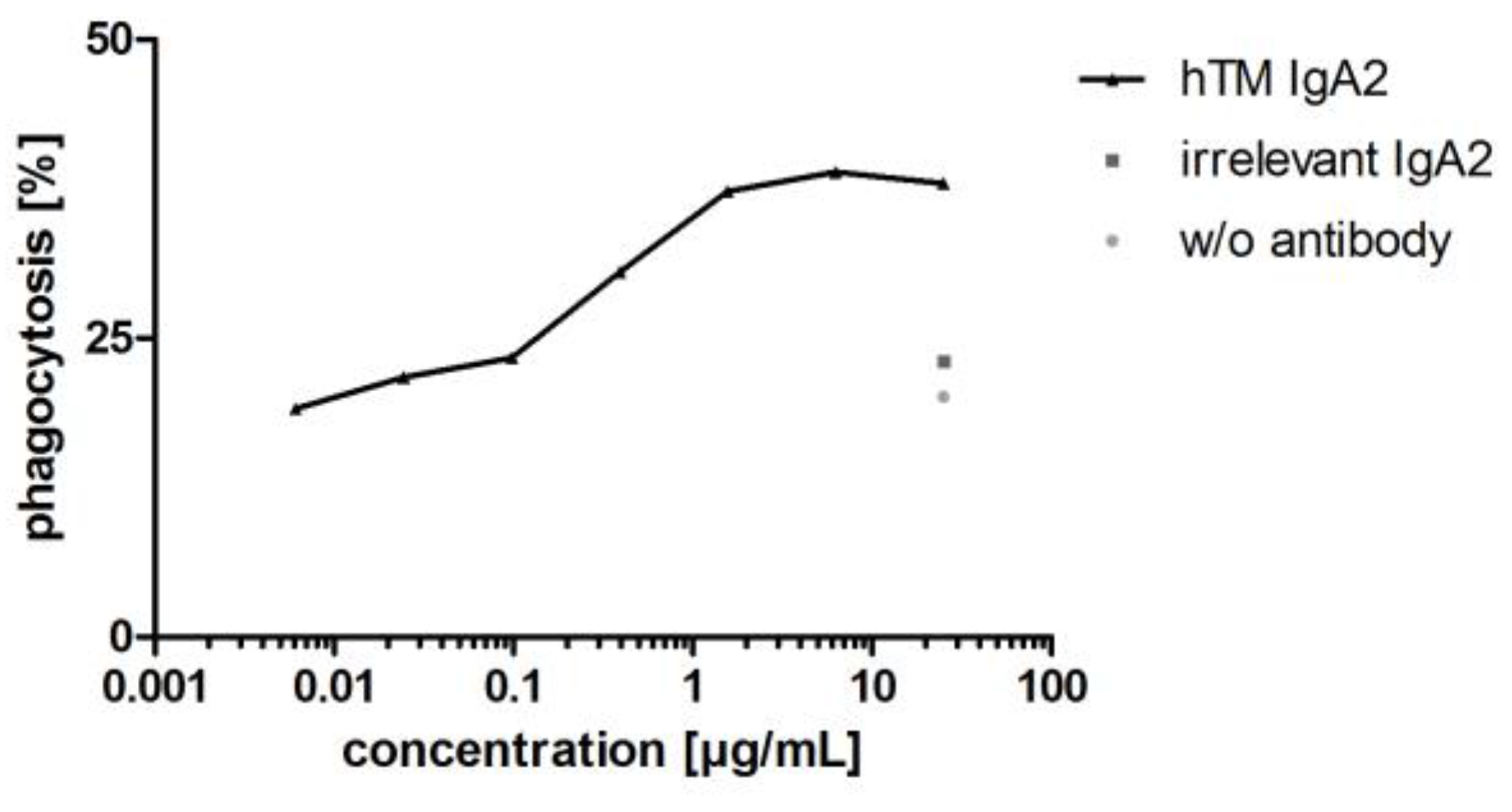
3.3.3. IgA Antibodies Mediate Antibody-Dependent Cellular Phagocytosis
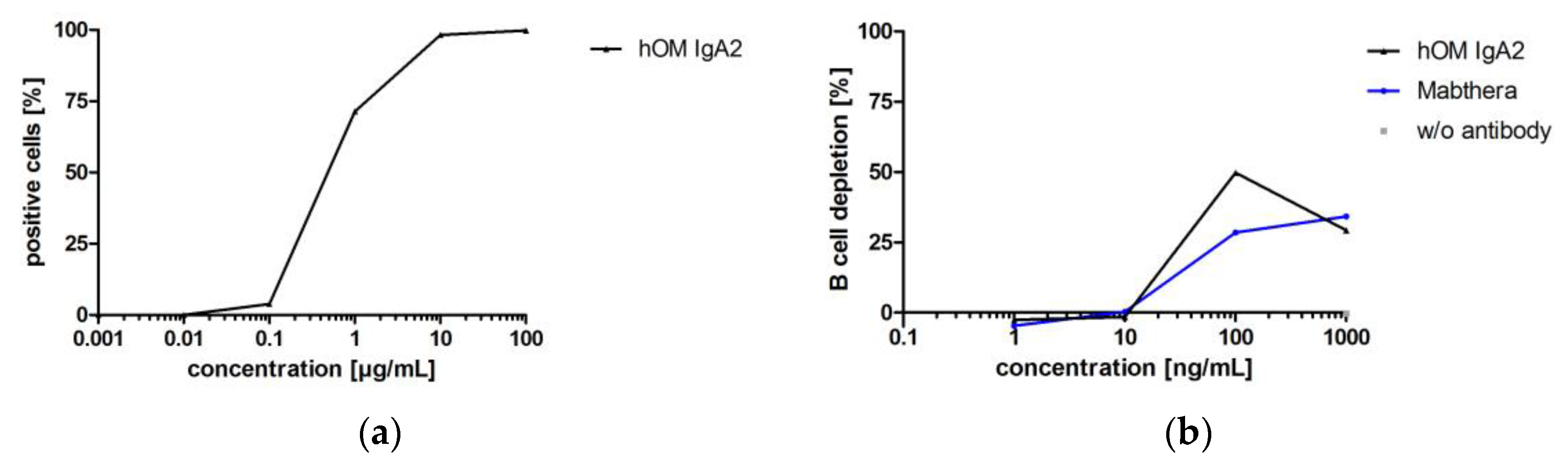
3.3.4. Biofunctionality of Anti-CD20 IgA2 Antibody
4. Discussion
4.1. Novel Tumor-Specific Monoclonal IgA Antibodies with Human Glycosylation
4.2. Biochemical Integrity and Antigen Binding Characteristics
4.3. IgA Antibodies as a Novel Class for Cancer Immunotherapy: Modes of Action
5. Conclusions
Supplementary Materials
Author Contributions
Conflicts of Interest
References
- Scott, A.M.; Wolchok, J.D.; Old, L.J. Antibody therapy of cancer. Nat. Rev. Cancer 2012, 12, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Beck, A.; Reichert, J.M. Antibody-drug conjugates. MAbs 2014, 6, 15–17. [Google Scholar] [CrossRef] [PubMed]
- Valerius, T.; Stockmeyer, B.; van Spriel, A.B.; Graziano, R.F.; van den Herik-Oudijk, I.E.; Repp, R.; Deo, Y.M.; Lund, J.; Kalden, J.R.; Gramatzki, M.; et al. FcalphaRI (CD89) as a novel trigger molecule for bispecific antibody therapy. Blood 1997, 90, 4485–4492. [Google Scholar] [PubMed]
- Huls, G.; Heijnen, I.A.F.M.; Cuomo, E.; van der Linden, J.; Boel, E.; van de Winkel, J.G.J.; Logtenberg, T. Antitumor immune effector mechanisms recruited by phage display-derived fully human IgG1 and IgA1 monoclonal antibodies. Cancer Res. 1999, 59, 5778–5784. [Google Scholar] [PubMed]
- Dechant, M.; Vidarsson, G.; Stockmeyer, B.; Repp, R.; Glennie, M.J.; Gramatzki, M.; van de Winkel, J.G.J.; Valerius, T. Chimeric IgA antibodies against HLA class II effectively trigger lymphoma cell killing. Blood 2002, 100, 4574–4580. [Google Scholar] [CrossRef] [PubMed]
- Van Egmond, M.; van Spriel, A.B.; Vermeulen, H.; Huls, G.; van Garderen, E.; van de Winkel, J.G. Enhancement of polymorphonuclear cell-mediated tumor cell killing on simultaneous engagement of fcgammaRI (CD64) and fcalphaRI (CD89). Cancer Res. 2001, 61, 4055–4060. [Google Scholar] [PubMed]
- Otten, M.A.; Rudolph, E.; Dechant, M.; Tuk, C.W.; Reijmers, R.M.; Beelen, R.H.J.; van de Winkel, J.G.J.; van Egmond, M. Immature neutrophils mediate tumor cell killing via IgA but not IgG Fc receptors. J. Immunol. 2005, 174, 5472–5480. [Google Scholar] [CrossRef] [PubMed]
- Dechant, M.; Beyer, T.; Schneider-Merck, T.; Weisner, W.; Peipp, M.; van de Winkel, J.G.J.; Valerius, T. Effector Mechanisms of Recombinant IgA Antibodies against Epidermal Growth Factor Receptor. J. Immunol. 2007, 179, 2936–2943. [Google Scholar] [CrossRef] [PubMed]
- Beyer, T.; Lohse, S.; Berger, S.; Peipp, M.; Valerius, T.; Dechant, M. Serum-free production and purification of chimeric IgA antibodies. J. Immunol. Methods 2009, 346, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Lohse, S.; Derer, S.; Beyer, T.; Klausz, K.; Peipp, M.; Leusen, J.H.W.; van de Winkel, J.G.J.; Dechant, M.; Valerius, T. Recombinant Dimeric IgA Antibodies against the Epidermal Growth Factor Receptor Mediate Effective Tumor Cell Killing. J. Immunol. 2011, 186, 3770–3778. [Google Scholar] [CrossRef] [PubMed]
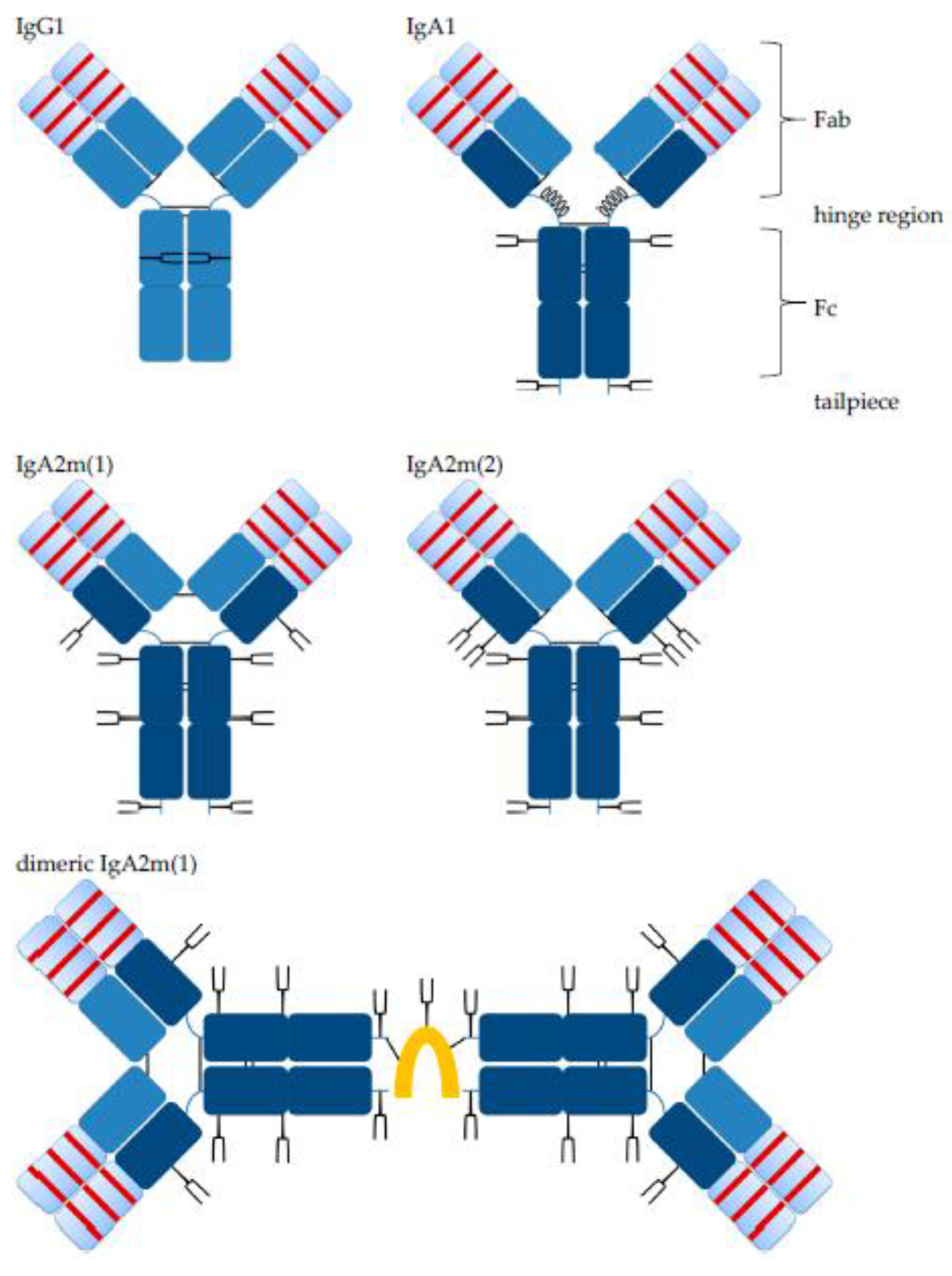
- Woof, J.M.; Kerr, M.A. IgA function—Variations on a theme. Immunology 2004, 113, 175–177. [Google Scholar] [CrossRef] [PubMed]
- Kerr, M.A. The structure and function of human IgA. Hum. Cell Off. J. Hum. Cell Res. Soc. 1990, 271, 285–296. [Google Scholar] [CrossRef]
- Murphy, K.M. Janeway’s Immunobiology; Garland Science: New York, NY, USA, 2011. [Google Scholar]
- Woof, J.M.; Kerr, M.A. The function of immunoglobulin A in immunity. J. Pathol. 2006, 208, 270–282. [Google Scholar] [CrossRef] [PubMed]
- Brandtzaeg, P.; Prydz, H. Direct evidence for an integrated function of J chain and secretory component in epithelial transport of immunoglobulins. Nature 1984, 311, 71–73. [Google Scholar] [CrossRef] [PubMed]
- Tomasi, T.B.; Tan, E.M.; Solomon, A.; Prendergast, R.A. Characteristics of an Immune System Common To Certain External Secretions. J. Exp. Med. 1965, 121, 101–124. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, R.C.; van de Winkel, J.G.J. IgA Fc receptors. Annu. Rev. Immunol. 2003, 21, 177–204. [Google Scholar] [CrossRef] [PubMed]
- Woof, J.M.; Burton, D.R. Human antibody-Fc receptor interactions illuminated by crystal structures. Nat. Rev. Immunol. 2004, 4, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Woof, J.M.; Russell, M.W. Structure and function relationships in IgA. Mucosal Immunol. 2011, 4, 590–597. [Google Scholar] [CrossRef] [PubMed]
- Rifai, A.; Fadden, K.; Morrison, S.L.; Chintalacharuvu, K.R. The N-glycans determine the differential blood clearance and hepatic uptake of human immunoglobulin (Ig)A1 and IgA2 isotypes. J. Exp. Med. 2000, 191, 2171–2182. [Google Scholar] [CrossRef] [PubMed]
- Chintalacharuvu, K.R.; Raines, M.; Morrison, S.L. Divergence of human alpha-chain constant region gene sequences. A novel recombinant alpha 2 gene. J. Immunol. 1994, 152, 5299–5304. [Google Scholar] [PubMed]
- Novak, J.; Tomana, M.; Kilian, M.; Coward, L.; Kulhavy, R.; Barnes, S.; Mestecky, J. Heterogeneity of O-glycosylation in the hinge region of human IgA1. Mol. Immunol. 2001, 37, 1047–1056. [Google Scholar] [CrossRef]
- Pouria, S.; Corran, P.H.; Smith, A.C.; Smith, H.W.; Hendry, B.M.; Challacombe, S.J.; Tarelli, E. Glycoform composition profiling of O-glycopeptides derived from human serum IgA1 by matrix-assisted laser desorption ionization-time of flight-mass spectrometry. Anal. Biochem. 2004, 330, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Mattu, T.S.; Pleass, R.J.; Willis, A.C.; Kilian, M.; Wormald, M.R.; Lellouch, A.C.; Rudd, P.M.; Woof, J.M.; Dwek, R.A. The glycosylation and structure of human serum IgA1, Fab, and Fc regions and the role of N-glycosylation on Fcα receptor interactions. J. Biol. Chem. 1998, 273, 2260–2272. [Google Scholar] [CrossRef] [PubMed]
- Allen, A.C.; Bailey, E.M.; Barratt, J.; Buck, K.S.; Feehally, J. Analysis of IgA1 O-glycans in IgA nephropathy by fluorophore-assisted carbohydrate electrophoresis. J. Am. Soc. Nephrol. 1999, 10, 1763–1771. [Google Scholar] [PubMed]
- Arnold, J.N.; Wormald, M.R.; Sim, R.B.; Rudd, P.M.; Dwek, R.A. The impact of glycosylation on the biological function and structure of human immunoglobulins. Annu. Rev. Immunol. 2007, 25, 21–50. [Google Scholar] [CrossRef] [PubMed]
- Hiki, Y.; Kokubo, T.; Iwase, H.; Masaki, Y.; Sano, T.; Tanaka, A.; Toma, K.; Hotta, K.; Kobayashi, Y. Underglycosylation of IgA1 hinge plays a certain role for its glomerular deposition in IgA nephropathy. J. Am. Soc. Nephrol. 1999, 10, 760–769. [Google Scholar] [PubMed]
- Umaña, P.; Jean-Mairet, J.; Moudry, R.; Amstutz, H.; Bailey, J.E. Engineered glycoforms of an antineuroblastoma IgG1 with optimized antibody-dependent cellular cytotoxic activity. Nat. Biotechnol. 1999, 17, 176–180. [Google Scholar] [PubMed]
- Jefferis, R. Glycosylation as a strategy to improve antibody-based therapeutics. Nat. Rev. Drug Discov. 2009, 8, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Anthony, R.M.; Ravetch, J.V. A Novel Role for the IgG Fc Glycan: The Anti-inflammatory Activity of Sialylated IgG Fcs. J. Clin. Immunol. 2010, 30, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, C.; Grau, S.; Jäger, C.; Sondermann, P.; Brünker, P.; Waldhauer, I.; Hennig, M.; Ruf, A.; Rufer, A.C.; Stihle, M.; et al. Unique carbohydrate-carbohydrate interactions are required for high affinity binding between FcgammaRIII and antibodies lacking core fucose. Proc. Natl. Acad. Sci. USA 2011, 108, 12669–12674. [Google Scholar] [CrossRef] [PubMed]
- Herter, S.; Birk, M.C.; Klein, C.; Gerdes, C.; Umana, P.; Bacac, M. Glycoengineering of Therapeutic Antibodies Enhances Monocyte/Macrophage-Mediated Phagocytosis and Cytotoxicity. J. Immunol. 2014, 192, 2252–2260. [Google Scholar] [CrossRef] [PubMed]
- Van Egmond, M.; Damen, C.A.; van Spriel, A.B.; Vidarsson, G.; van Garderen, E.; van de Winkel, J.G.J. IgA and the IgA Fc receptor. Trends Immunol. 2001, 22, 205–211. [Google Scholar] [CrossRef]
- Herr, A.B.; White, C.L.; Milburn, C.; Wu, C.; Bjorkman, P.J. Bivalent binding of IgA1 to FcαRI suggests a mechanism for cytokine activation of IgA phagocytosis. J. Mol. Biol. 2003, 327, 645–657. [Google Scholar] [CrossRef]
- Herr, A.B.; Ballister, E.R.; Bjorkman, P.J. Insights into IgA-mediated immune responses from the crystal structures of human FcalphaRI and its complex with IgA1-Fc. Nature 2003, 423, 614–620. [Google Scholar] [CrossRef] [PubMed]
- Bakema, J.E.; van Egmond, M. The human immunoglobulin A Fc receptor FcαRI: A multifaceted regulator of mucosal immunity. Mucosal Immunol. 2011, 4, 612–624. [Google Scholar] [CrossRef] [PubMed]
- Mkaddem, S.B.; Rossato, E.; Heming, N.; Monteiro, R.C. Anti-inflammatory role of the IgA Fc receptor (CD89): From autoimmunity to therapeutic perspectives. Autoimmun. Rev. 2013, 12, 666–669. [Google Scholar] [CrossRef] [PubMed]
- Pfirsch-Maisonnas, S.; Aloulou, M.; Xu, T.; Claver, J.; Kanamaru, Y.; Tiwari, M.; Launay, P.; Monteiro, R.C.; Blank, U. Inhibitory ITAM signaling traps activating receptors with the phosphatase SHP-1 to form polarized ‘inhibisome’ clusters. Sci. Signal. 2011, 4, ra24. [Google Scholar] [CrossRef] [PubMed]
- Ivashkiv, L.B. How ITAMs inhibit signaling. Sci. Signal. 2011, 4, pe20. [Google Scholar] [CrossRef] [PubMed]
- Balu, S.; Reljic, R.; Lewis, M.J.; Pleass, R.J.; McIntosh, R.; van Kooten, C.; van Egmond, M.; Challacombe, S.; Woof, J.M.; Ivanyi, J. A novel human IgA monoclonal antibody protects against tuberculosis. J. Immunol. 2011, 186, 3113–3119. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; McIntosh, R.S.; Pleass, R.J. Antibody- and Fc-receptor-based therapeutics for malaria. Clin. Sci. 2006, 110, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Muramatsu, M.; Yoshida, R.; Yokoyama, A.; Miyamoto, H.; Kajihara, M.; Maruyama, J.; Nao, N.; Manzoor, R.; Takada, A. Comparison of antiviral activity between IgA and IgG specific to Influenza Virus Hemagglutinin: Increased potential if IgA for heterosubtypic immunity. PLoS ONE 2014, 9, e85582. [Google Scholar] [CrossRef] [PubMed]
- Boross, P.; Lohse, S.; Nederend, M.; Jansen, J.H.M.; van Tetering, G.; Dechant, M.; Peipp, M.; Royle, L.; Liew, L.P.; Boon, L.; et al. IgA EGFR antibodies mediate tumour killing in vivo. EMBO Mol. Med. 2013, 5, 1213–1226. [Google Scholar] [CrossRef] [PubMed]
- Tanikawa, T.; Ishikawa, T.; Maekawa, T.; Kuronane, K.; Imai, Y. Characterization of Monoclonal Immunoglobulin A and G Against Shiga Toxin Binding Subunits Produced by Intranasal Immunization. Scand. J. Immunol. 2008, 68, 414–422. [Google Scholar] [CrossRef] [PubMed]
- Chintalacharuvu, K.R.; Gurbaxani, B.; Morrison, S.L. Incomplete assembly of IgA2m(2) in Chinese hamster ovary cells. Mol. Immunol. 2007, 44, 3445–3452. [Google Scholar] [CrossRef] [PubMed]
- Beyer, T.; Lohse, S.; Dechant, M.; Valerius, T. Expression of IgA Molecules in Mammalian Cells. In Antibody Engineering; Kontermann, R., Dübel, S., Eds.; Springer: Berlin/Heidelberg, Germany, 2010; Volume 1, pp. 471–486. [Google Scholar]
- Reinhart, D.; Weik, R.; Kunert, R. Recombinant IgA production: Single step affinity purification using camelid ligands and product characterization. J. Immunol. Methods 2012, 378, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Field, M.C.; Amatayakul-Chantler, S.; Rademacher, T.W.; Rudd, P.M.; Dwek, R.A. Structural analysis of the N-glycans from human immunoglobulin A1: Comparison of normal human serum immunoglobulin A1 with that isolated from patients with rheumatoid arthritis. Biochem. J. 1994, 299, 261–275. [Google Scholar] [CrossRef] [PubMed]
- Kokubo, T.; Hiki, Y.; Iwase, H.; Horii, A.; Tanaka, A.; Nishikido, J.; Hotta, K.; Kobayashi, Y. Evidence for involvement of IgA1 hinge glycopeptide in the IgA1-IgA1 interaction in IgA nephropathy. J. Am. Soc. Nephrol. 1997, 8, 915–919. [Google Scholar] [PubMed]
- Royle, L.; Roos, A.; Harvey, D.J.; Wormald, M.R.; van Gijlswijk-Janssen, D.; Redwan, E.R.M.; Wilson, I.A.; Daha, M.R.; Dwek, R.A.; Rudd, P.M. Secretory IgA N- and O-glycans provide a link between the innate and adaptive immune systems. J. Biol. Chem. 2003, 278, 20140–20153. [Google Scholar] [CrossRef] [PubMed]
- Walsh, G. Biopharmaceutical benchmarks 2014. Nat. Biotechnol. 2014, 32, 992–1000. [Google Scholar] [CrossRef] [PubMed]
- Gerdes, C.A.; Nicolini, V.G.; Herter, S.; van Puijenbroek, E.; Lang, S.; Roemmele, M.; Moessner, E.; Freytag, O.; Friess, T.; Ries, C.H.; et al. GA201 (RG7160): A novel, humanized, glycoengineered anti—EGFR antibody with enhanced ADCC and superior in vivo efficacy compared with cetuximab. Clin. Cancer Res. 2013, 19, 1126–1138. [Google Scholar] [CrossRef] [PubMed]
- Sambrook, J.; Russell, D.W. Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2001; Volume 1. [Google Scholar]
- Mossner, E.; Brunker, P.; Moser, S.; Puntener, U.; Schmidt, C.; Herter, S.; Grau, R.; Gerdes, C.; Nopora, A.; van Puijenbroek, E.; et al. Increasing the efficacy of CD20 antibody therapy through the engineering of a new type II anti-CD20 antibody with enhanced direct and immune effector cell-mediated B-cell cytotoxicity. Blood 2010, 115, 4393–4402. [Google Scholar] [CrossRef] [PubMed]
- Varki, A.; Cummings, R.D.; Esko, J.D.; Freeze, H.H.; Stanley, P.; Bertozzi, C.R.; Hart, G.W.; Etzler, M.E. Essentials of Glycobiology; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2009. [Google Scholar]
- Krawczyk, P.A.; Kowalski, D.M. Review Genetic and immune factors underlying the efficacy of cetuximab and panitumumab in the treatment of patients with metastatic colorectal cancer. Współczesna Onkol. 2014, 1, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Baselga, J.; Swain, S.M. Novel anticancer targets: Revisiting ERBB2 and discovering ERBB3. Nat. Rev. Cancer 2009, 9, 463–475. [Google Scholar] [CrossRef] [PubMed]
- Weiner, L.M.; Surana, R.; Wang, S. Monoclonal antibodies: Versatile platforms for cancer immunotherapy. Nat. Rev. Immunol. 2010, 10, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Lohse, S.; Brunke, C.; Derer, S.; Peipp, M.; Boross, P.; Kellner, C.; Beyer, T.; Dechant, M.; van der Winkel, J.G.J.; Leusen, J.H.W.; et al. Characterization of a mutated IgA2 antibody of the m(1) allotype against the epidermal growth factor receptor for the recruitment of monocytes and macrophages. J. Biol. Chem. 2012, 287, 25139–25150. [Google Scholar] [CrossRef] [PubMed]
- Akewanlop, C.; Watanabe, M.; Singh, B.; Walker, M.; Kufe, D.W.; Hayes, D.F. Phagocytosis of breast cancer cells mediated by anti-MUC-1 monoclonal antibody, DF3, and its bispecific antibody. Cancer Res. 2001, 61, 4061–4065. [Google Scholar] [PubMed]
- Dhodapkar, K.M.; Krasovsky, J.; Williamson, B.; Dhodapkar, M.V. Antitumor monoclonal antibodies enhance cross-presentation of cellular antigens and the generation of myeloma-specific killer T cells by dendritic cells. J. Exp. Med. 2002, 195, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Golay, J.; da Roit, F.; Bologna, L.; Ferrara, C.; Leusen, J.H.; Rambaldi, A.; Klein, C.; Introna, M. Glycoengineered CD20 antibody obinutuzumab activates neutrophils and mediates phagocytosis through CD16B more efficiently than rituximab. Blood 2013, 122, 3482–3491. [Google Scholar] [CrossRef] [PubMed]
- Lewis, C.E.; Leek, R.; Harris, A.; McGee, J.O. Cytokine regulation of angiogenesis in breast cancer: The role of tumor-associated macrophages. J. Leukoc. Biol. 1995, 57, 747–751. [Google Scholar] [PubMed]
- Herter, S.; Herting, F.; Mundigl, O.; Waldhauer, I.; Weinzierl, T.; Fauti, T.; Muth, G.; Ziegler-Landesberger, D.; van Puijenbroek, E.; Lang, S.; et al. Preclinical activity of the type II CD20 antibody GA101 (obinutuzumab) compared with rituximab and ofatumumab in vitro and in xenograft models. Mol. Cancer Ther. 2013, 12, 2031–2042. [Google Scholar] [CrossRef] [PubMed]
- Brunke, C.; Lohse, S.; Derer, S.; Peipp, M.; Boross, P.; Kellner, C.; Beyer, T.; Dechant, M.; Royle, L.; Liew, L.P.; et al. Effect of a tail piece cysteine deletion on biochemical and functional properties of an epidermal growth factor receptor-directed IgA2m(1) antibody. MAbs 2013, 5, 936–945. [Google Scholar] [CrossRef] [PubMed]
- Pascal, V.; Laffleur, B.; Debin, A.; Cuvillier, A.; van Egmond, M.; Drocourt, D.; Imbertie, L.; Pangault, C.; Tarte, K.; Tiraby, G.; et al. Anti-CD20 IgA can protect mice against lymphoma development: Evaluation of the direct impact of IgA and cytotoxic effector recruitment on CD20 target cells. Haematologica 2012, 97, 1686–1694. [Google Scholar] [CrossRef] [PubMed]
- Deo, Y.M.; Sundarapandiyan, K.; Keler, T.; Wallace, P.K.; Graziano, R.F. Bispecific molecules directed to the Fc receptor for IgA (Fc alpha RI, CD89) and tumor antigens efficiently promote cell-mediated cytotoxicity of tumor targets in whole blood. J. Immunol. 1998, 160, 1677–1686. [Google Scholar] [PubMed]
- Stockmeyer, B.; Dechant, M.; van Egmond, M.; Tutt, A.L.; Sundarapandiyan, K.; Graziano, R.F.; Repp, R.; Kalden, J.R.; Gramatzki, M.; et al. Triggering Fc alpha-receptor I (CD89) recruits neutrophils as effector cells for CD20-directed antibody therapy. J. Immunol. 2000, 165, 5954–5961. [Google Scholar] [CrossRef] [PubMed]
- Bakema, J.E.; Ganzevles, S.H.; Fluitsma, D.M.; Schilham, M.W.; Beelen, R.H.J.; Valerius, T.; Lohse, S.; Glennie, M.J.; Medema, J.P.; van Egmond, M. Targeting FcαRI on Polymorphonuclear Cells Induces Tumor Cell Killing through Autophagy. J. Immunol. 2011, 187, 726–732. [Google Scholar] [CrossRef] [PubMed]
- Atkin, J.D.; Pleass, R.J.; Owens, R.J.; Woof, J.M. Mutagenesis of the Human IgA1 Heavy Chain Tailpiece That Prevents Dimer Assembly. J. Immunol. 1996, 157, 156–159. [Google Scholar] [PubMed]
- Yoo, E.M.; Yu, L.J.; Wims, L.A.; Goldberg, D.; Morrison, S.L. Differences in N-glycan structures found on recombinant IgA1 and IgA2 produced in murine myeloma and CHO cell lines. MAbs 2010, 2, 320–334. [Google Scholar] [CrossRef] [PubMed]
- Morton, H.C.; Atkin, J.D.; Owens, R.J.; Woof, J.M. Purification and characterization of chimeric human IgA1 and IgA2 expressed in COS and Chinese hamster ovary cells. J. Immunol. 1993, 151, 4743–4752. [Google Scholar] [PubMed]
- Carayannopoulos, L.; Max, E.E.; Capra, J.D. Recombinant human IgA expressed in insect cells. Proc. Natl. Acad. Sci. USA 1994, 91, 8348–8352. [Google Scholar] [CrossRef] [PubMed]
- Juarez, P.; Huet-Trujillo, E.; Sarrion-Perdigones, A.; Falconi, E.; Granell, A.; Orzaez, D. Combinatorial Analysis of Secretory Immunoglobulin A (sIgA) Expression in Plants. Int. J. Mol. Sci. 2013, 14, 6205–6222. [Google Scholar] [CrossRef] [PubMed]
- Larrick, J.W.; Yu, L.; Naftzger, C.; Jaiswal, S.; Wycoff, K. Production of secretory IgA antibodies in plants. Biomol. Eng. 2001, 18, 87–94. [Google Scholar] [CrossRef]
- Pleass, R.J.; Dehal, P.K.; Lewis, M.J.; Woof, J.M. Limited role of charge matching in the interaction of human immunoglobulin A with the immunoglobulin A Fc receptor (Fc alpha RI) CD89. Immunology 2003, 109, 331–335. [Google Scholar] [CrossRef] [PubMed]
- Moldt, B.; Saye-Francisco, K.; Schultz, N.; Burton, D.R.; Hessell, A.J. Simplifying the synthesis of SIgA: Combination of dIgA and rhSC using affinity chromatography. Methods 2014, 65, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.H.; Mirakhur, B.; Chan, E.; Le, Q.-T.; Berlin, J.; Morse, M.; Murphy, B.A.; Satinover, S.M.; Hosen, J.; Mauro, D.; et al. Cetuximab-Induced Anaphylaxis and IgE Specific for Galactose-α-1,3-Galactose. N. Engl. J. Med. 2008, 358, 1109–1117. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, T.; Nakanishi, F.; Ogura, Y.; Oi, D.; Omasa, T.; Katakura, Y.; Kishimoto, M.; Suga, K. Amplified Gene Location in Chromosomal DNA Affected Recombinant Protein Production and Stability of Amplified Genes. Biotechnol. Prog. 2000, 16, 710–715. [Google Scholar] [CrossRef] [PubMed]
- Kawabe, Y.; Makitsubo, H.; Kameyama, Y.; Huang, S.; Ito, A.; Kamihira, M. Repeated integration of antibody genes into a pre-selected chromosomal locus of CHO cells using an accumulative site-specific gene integration system. Cytotechnology 2012, 64, 267–279. [Google Scholar] [CrossRef] [PubMed]
- Lipscomb, M.L.; Palomares, L.A.; Hernández, V.; Ramírez, O.T.; Kompala, D.S. Effect of Production Method and Gene Amplification on the Glycosylation Pattern of a Secreted Reporter Protein in CHO Cells. Biotechnol. Prog. 2008, 21, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Goldman, M.H.; James, D.C.; Rendall, M.; Ison, A.P.; Hoare, M.; Bull, A.T. Monitoring recombinant human interferon-gamma N-glycosylation during perfused fluidized-bed and stirred-tank batch culture of CHO cells. Biotechnol. Bioeng. 1998, 60, 596–607. [Google Scholar] [CrossRef]
- Oortwijn, B.D.; Roos, A.; Royle, L.; van Gijlswijk-Janssen, D.J.; Faber-Krol, M.C.; Eijgenraam, J.-W.; Dwek, R.A.; Daha, M.R.; Rudd, P.M.; van Kooten, C. Differential glycosylation of polymeric and monomeric IgA: A possible role in glomerular inflammation in IgA nephropathy. J. Am. Soc. Nephrol. 2006, 17, 3529–3539. [Google Scholar] [CrossRef] [PubMed]
- Hexham, J.M.; White, K.D.; Carayannopoulos, L.N.; Mandecki, W.; Brisette, R.; Yang, Y.S.; Capra, J.D. A human immunoglobulin (Ig)A Cα3 domain motif directs polymeric Ig receptor-mediated secretion. J. Exp. Med. 1999, 189, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; McIntosh, R.S.; Adame-Gallegos, J.; Dehal, P.K.; van Egmond, M.; van de Winkel, J.; Draper, S.J.; Forbes, E.K.; Corran, P.H.; Holder, A.A.; et al. The generation and evaluation of recombinant human IgA specific for Plasmodium falciparum merozoite surface protein 1-19 (PfMSP119). BMC Biotechnol. 2011, 11, 77. [Google Scholar] [CrossRef] [PubMed]
- Basset, C.; Devauchelle, V.; Durand, V.; Jamin, C.; Pennec, Y.L.; Youinou, P.; Dueymes, M. Glycosylation of immunoglobulin a influences its receptor binding. Scand. J. Immunol. 1999, 50, 572–579. [Google Scholar] [CrossRef] [PubMed]
- Gomes, M.M.; Wall, S.B.; Takahashi, K.; Novak, J.; Renfrow, M.B.; Herr, A.B. Analysis of IgA1 N -Glycosylation and Its Contribution to FcαRI Binding. Biochemistry 2008, 47, 11285–11299. [Google Scholar] [CrossRef] [PubMed]
- Kumpel, B.M.; Rademacher, T.W.; Rook, G.A.; Williams, P.J.; Wilson, I.B. Galactosylation of human IgG monoclonal anti-D produced by EBV- transformed B-lymphoblastoid cell lines is dependent on culture method and affects Fc receptor-mediated functional activity. Hum. Antib. Hybrid. 1994, 5, 143–151. [Google Scholar]
- Derer, S.; Bauer, P.; Lohse, S.; Scheel, A.H.; Berger, S.; Kellner, C.; Peipp, M.; Valerius, T. Impact of epidermal growth factor receptor (EGFR) cell surface expression levels on effector mechanisms of EGFR antibodies. J. Immunol. 2012, 189, 5230–5239. [Google Scholar] [CrossRef] [PubMed]
- Keler, T.; Wallace, P.K.; Vitale, L.A.; Russoniello, C.; Sundarapandiyan, K.; Graziano, R.F.; Deo, Y.M. Differential effect of cytokine treatment on Fc alpha receptor I- and Fc gamma receptor I-mediated tumor cytotoxicity by monocyte-derived macrophages. J. Immunol. 2000, 164, 5746–5752. [Google Scholar] [CrossRef] [PubMed]
- Nishie, A.; Ono, M.; Shono, T.; Fukushi, J.; Otsubo, M.; Onoue, H.; Ito, Y.; Inamura, T.; Ikezaki, K.; Fukui, M.; et al. Macrophage Infiltration and Heme Oxygenase-1 Expression Correlate with Angiogenesis in Human Gliomas. Clin. Cancer Res. 1999, 5, 1107–1113. [Google Scholar] [PubMed]
- Mantovani, A.; Schioppa, T.; Porta, C.; Allavena, P.; Sica, A. Role of tumor-associated macrophages in tumor progression and invasion. Cancer Metastasis Rev. 2006, 25, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Alduaij, W.; Ivanov, A.; Honeychurch, J.; Cheadle, E.J.; Potluri, S.; Lim, S.H.; Shimada, K.; Chan, C.H.T.; Tutt, A.; Beers, S.A.; et al. Novel type II anti-CD20 monoclonal antibody (GA101) evokes homotypic adhesion and actin-dependent, lysosome-mediated cell death in B-cell malignancies. Blood 2011, 117, 4519–4529. [Google Scholar] [CrossRef] [PubMed]
- Van Egmond, M.; van Vuuren, A.J.; Morton, H.C.; van Spriel, A.B.; Shen, L.; Hofhuis, F.M.; Saito, T.; Mayadas, T.N.; Verbeek, J.S.; van de Winkel, J.G. Human immunoglobulin A receptor (FcalphaRI, CD89) function in transgenic mice requires both FcR gamma chain and CR3 (CD11b/CD18). Blood 1999, 93, 4387–4394. [Google Scholar] [PubMed]
- Meyer, S.; Nederend, M.; Jansen, J.H.M.; Reiding, K.R.; Jacobino, S.R.; Meeldijk, J.; Bovenschen, N.; Wuhrer, M.; Valerius, T.; Ubink, R.; et al. Improved in vivo anti-tumor effects of IgA-Her2 antibodies through half-life extension and serum exposure enhancement by FcRn targeting. MAbs 2015, 0862, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Lohse, S.; Meyer, S.; Meulenbroek, L.A.P.M.; Jansen, J.H.M.; Nederend, M.; Kretschmer, A.; Klausz, K.; Moginger, U.; Derer, S.; Rosner, T.; et al. An Anti-EGFR IgA That Displays Improved Pharmacokinetics and Myeloid Effector Cell Engagement In Vivo. Cancer Res. 2016, 76, 403–417. [Google Scholar] [CrossRef] [PubMed]
- Rouwendal, G.J.; van der Lee, M.M.; Meyer, S.; Reiding, K.R.; Schouten, J.; de Roo, G.; Egging, D.F.; Leusen, J.H.; Boross, P.; Wuhrer, M.; et al. A comparison of anti-HER2 IgA and IgG1 in vivo efficacy is facilitated by high N-glycan sialylation of the IgA. MAbs 2016, 8, 74–86. [Google Scholar] [CrossRef] [PubMed]
- Staff, C.; Magnusson, C.G.M.; Hojjat-Farsangi, M.; Mosolits, S.; Liljefors, M.; Frödin, J.-E.; Wahrén, B.; Mellstedt, H.; Ullenhag, G.J. Induction of IgM, IgA and IgE Antibodies in Colorectal Cancer Patients Vaccinated with a Recombinant CEA Protein. J. Clin. Immunol. 2012, 32, 855–865. [Google Scholar] [CrossRef] [PubMed]
- Welinder, C.; Baldetorp, B.; Blixt, O.; Grabau, D.; Jansson, B. Primary Breast Cancer Tumours Contain High Amounts of IgA1 Immunoglobulin: An Immunohistochemical Analysis of a Possible Carrier of the Tumour-Associated Tn Antigen. PLoS ONE 2013, 8, e61749. [Google Scholar] [CrossRef] [PubMed]
- Kalia, M. Biomarkers for personalized oncology: Recent advances and future challenges. Metabolism 2015, 64, S16–S21. [Google Scholar] [CrossRef] [PubMed]
- Gravitz, L. Therapy: This time it’s personal. Nature 2014, 509, S52–S54. [Google Scholar] [CrossRef] [PubMed]
- Henderson, D.; Ogilvie, L.A.; Hoyle, N.; Keilholz, U.; Lange, B.; Lehrach, H.; OncoTrack Consortium. Personalized medicine approaches for colon cancer driven by genomics and systems biology: OncoTrack. Biotechnol. J. 2014, 9, 1104–1114. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name (Abbreviation) | Target | Variants Generated | Titer (mg/L) |
|---|---|---|---|
| PankoMab (hPM) | Tumor-associated mucin 1 (TA-MUC1) | IgA1 | 41.8 |
| IgA2 | 4.0 * | ||
| TrasGEX (hTM) | Epidermal growth factor receptor 2 (Her2) | IgA2 | 55.8 |
| IgA2J | 41.5 | ||
| CetuGEX (CM) | Epidermal growth factor receptor 1 (EGFR) | IgA2 | 6.5 |
| IgA2J | 9.8 | ||
| KaroMab (hKM) | Thomsen-Friedenreich (TF) antigen, core 1 | IgA2 | 24.7 |
| ObiGEX (hOM) | B-lymphocyte antigen CD20 | IgA2 | 15.3 |
| IgA2 | Relative Molar Amount of Glycan Parameters (%) | |||||||
|---|---|---|---|---|---|---|---|---|
| Antibody | F | B | S0 | S > 0 | G0 | G > 0 | αGal | NeuGc |
| hTM | 64 | 15 | 32 | 62 | 3 | 91 | 0 | 0 |
| hOM | 79 | 9 | 29 | 64 | 11 | 81 | 0 | 0 |
| hPM | 82 | 12 | 29 | 68 | 4 | 92 | 0 | 0 |
| CM | 70 | 12 | 27 | 63 | 10 | 81 | 0 | 0 |
| hKM | 84 | 7 | 34 | 62 | 12 | 83 | 0 | 0 |
| IgG Erbitux | 85 | 0 | 81 | 16 | 30 | 66 | 28 | 15 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hart, F.; Danielczyk, A.; Goletz, S. Human Cell Line-Derived Monoclonal IgA Antibodies for Cancer Immunotherapy. Bioengineering 2017, 4, 42. https://doi.org/10.3390/bioengineering4020042
Hart F, Danielczyk A, Goletz S. Human Cell Line-Derived Monoclonal IgA Antibodies for Cancer Immunotherapy. Bioengineering. 2017; 4(2):42. https://doi.org/10.3390/bioengineering4020042
Chicago/Turabian StyleHart, Felix, Antje Danielczyk, and Steffen Goletz. 2017. "Human Cell Line-Derived Monoclonal IgA Antibodies for Cancer Immunotherapy" Bioengineering 4, no. 2: 42. https://doi.org/10.3390/bioengineering4020042
APA StyleHart, F., Danielczyk, A., & Goletz, S. (2017). Human Cell Line-Derived Monoclonal IgA Antibodies for Cancer Immunotherapy. Bioengineering, 4(2), 42. https://doi.org/10.3390/bioengineering4020042