

Exploration of Cytokines That Impact the Therapeutic Efficacy of Mesenchymal Stem Cells in Alzheimer’s Disease

 ,
,
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Mouse Grouping and Drug Administration Protocol
2.2. Novel Object Recognition Test
2.3. Morris Water Maze Test
2.4. Detection of Brain Oxidative Stress and Inflammatory Cytokine Levels
2.5. MSCs Preparation
2.6. Statistical Analysis and Graphs
3. Results
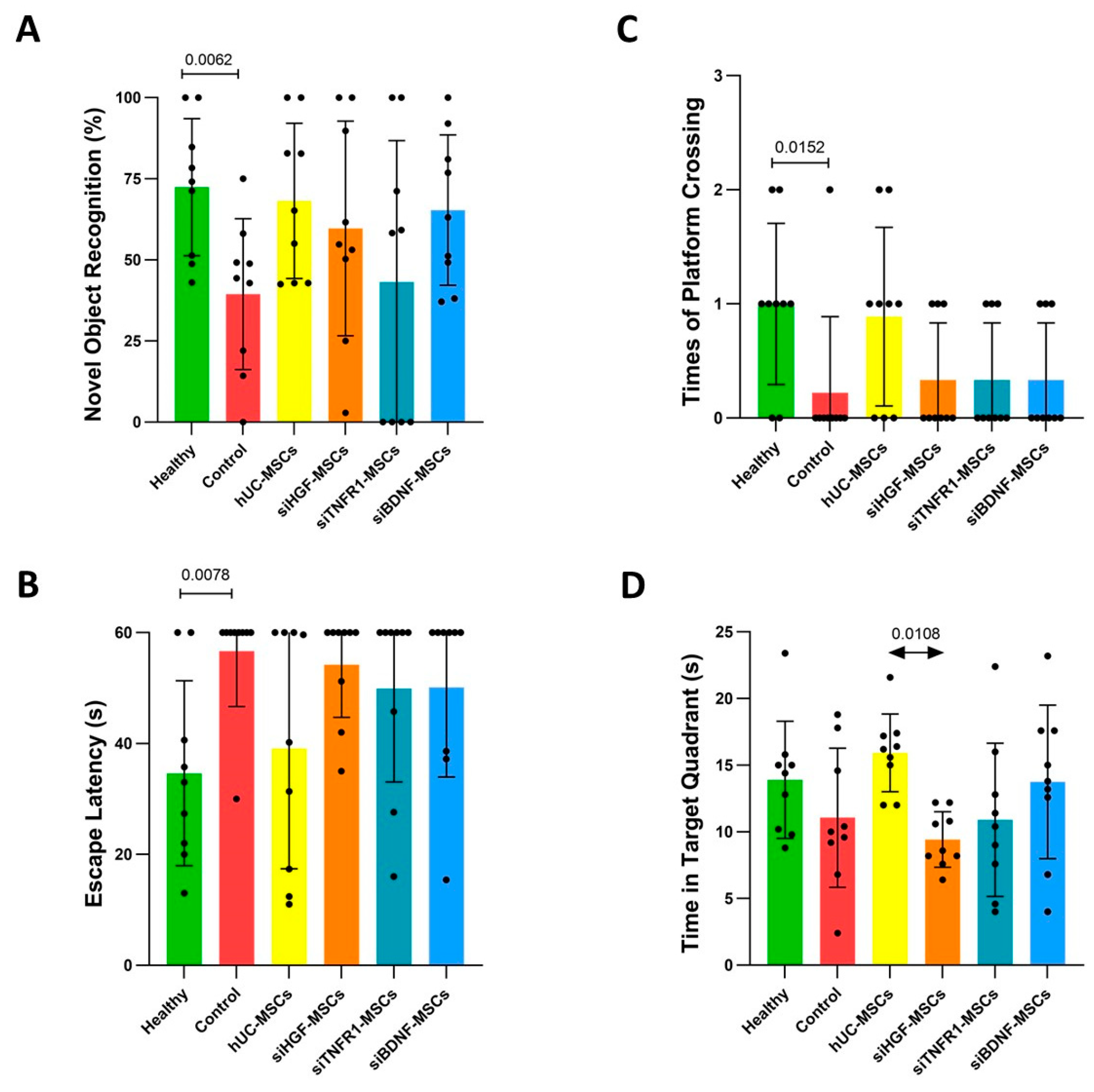
3.1. Effects of MSCs on Behavioral Performance in AD Mice
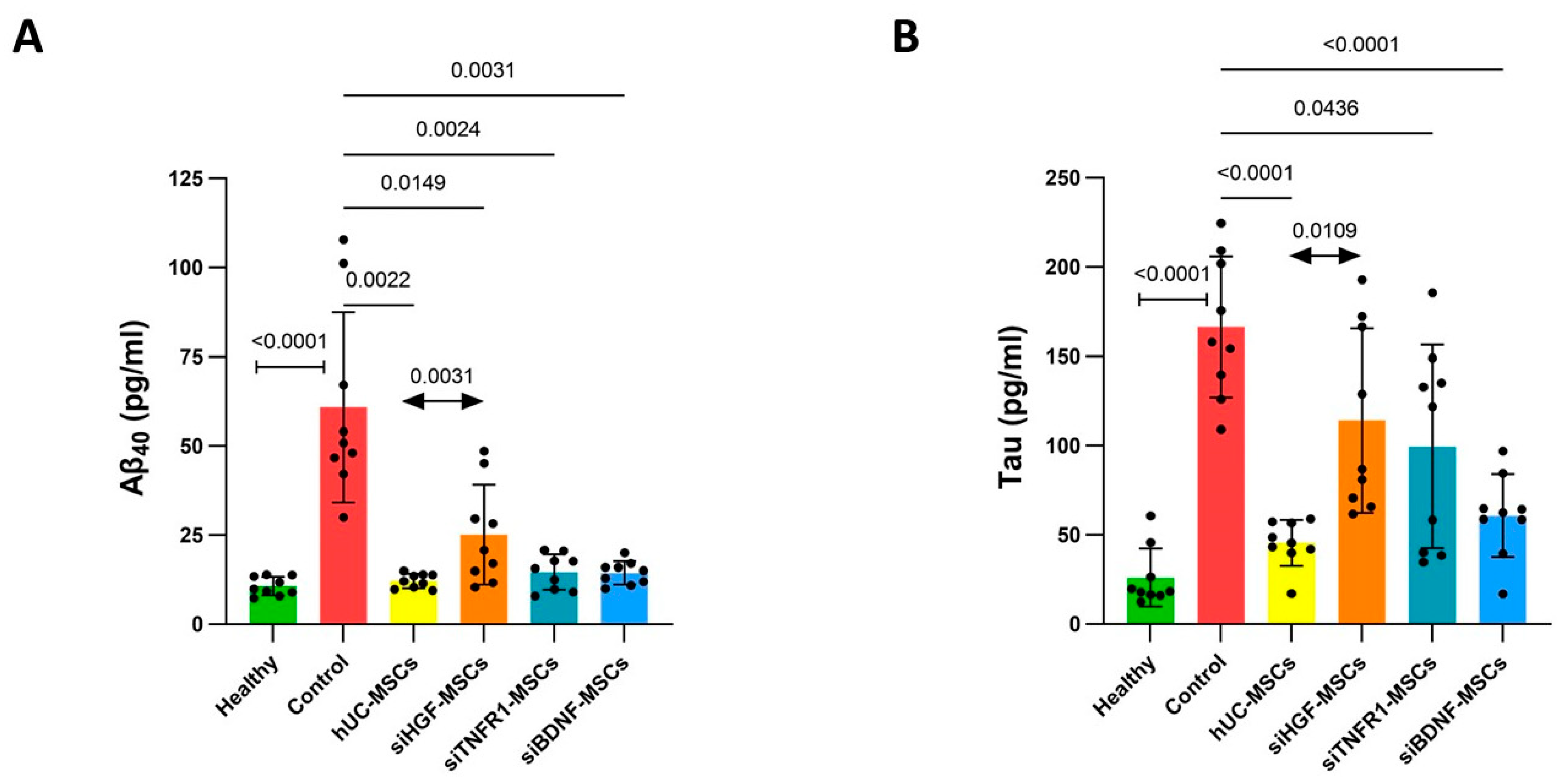
3.2. Effects of MSCs on Aβ40 and Tau Levels in AD Mouse Brain Tissue
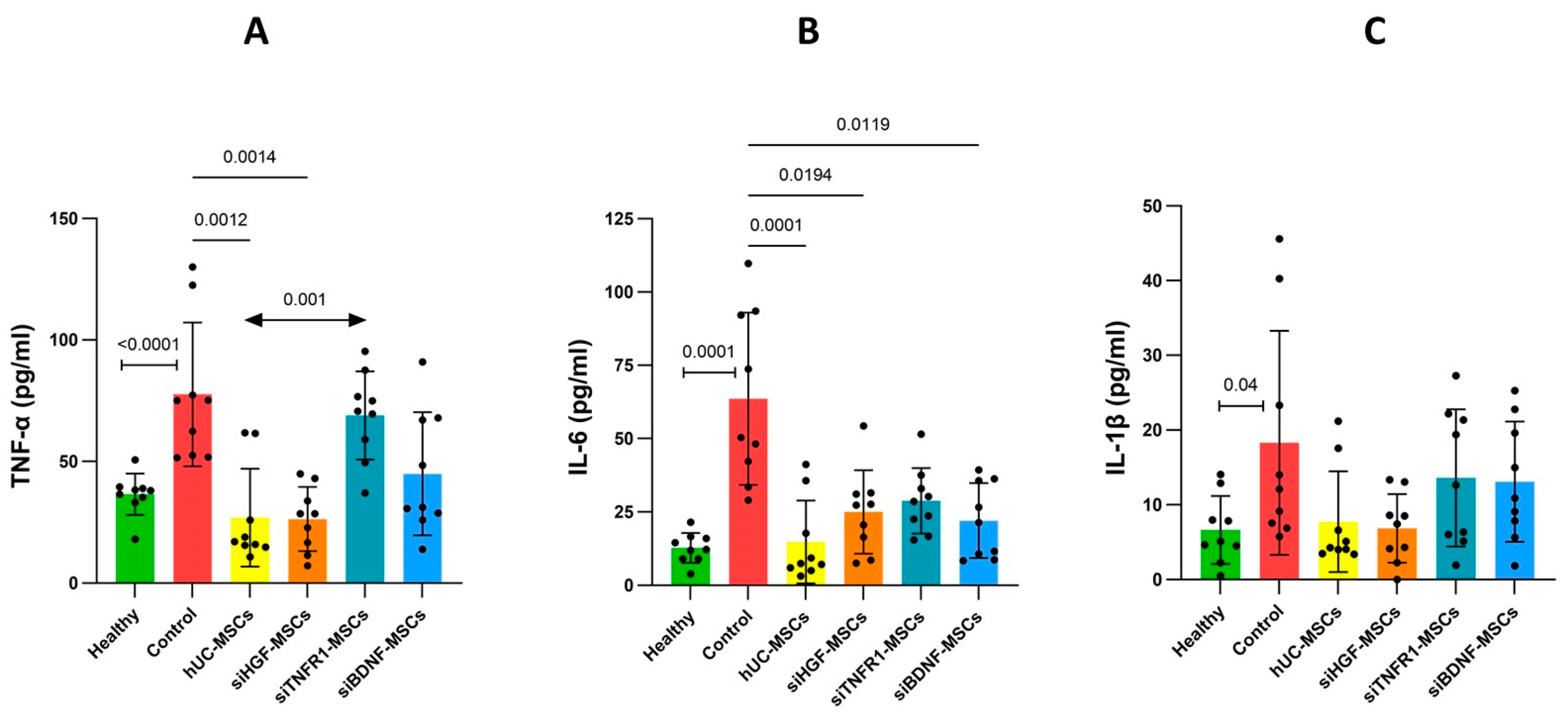
3.3. Effects of MSCs on Neuroinflammation in AD Mice
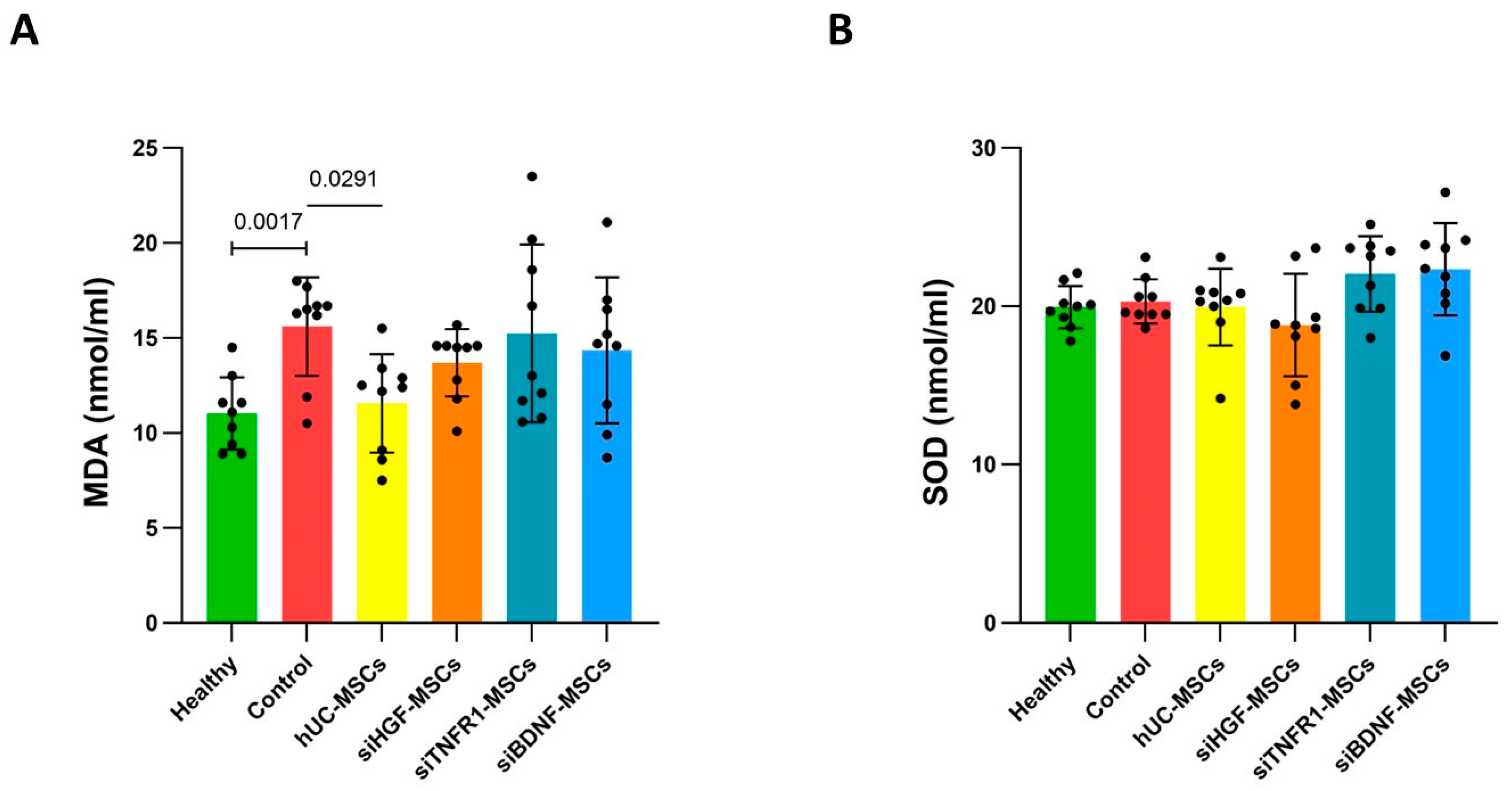
3.4. Effects of MSCs on Brain Oxidative Stress in AD Mice
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jucker, M.; Walker, L.C. Alzheimer’s disease: From immunotherapy to immunoprevention. Cell 2023, 186, 4260–4270. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.; Zhang, D.; Zeng, Y.; Huang, T.Y.; Xu, H.; Zhao, Y. Molecular and cellular mechanisms underlying the pathogenesis of Alzheimer’s disease. Mol. Neurodegener. 2020, 15, 40. [Google Scholar] [CrossRef] [PubMed]
- Self, W.K.; Holtzman, D.M. Emerging diagnostics and therapeutics for Alzheimer disease. Nat. Med. 2023, 29, 2187–2199. [Google Scholar] [CrossRef]
- Yiannopoulou, K.G.; Papageorgiou, S.G. Current and Future Treatments in Alzheimer Disease: An Update. J. Central Nerv. Syst. Dis. 2020, 12, 1179573520907397. [Google Scholar] [CrossRef]
- Boxer, A.L.; Sperling, R. Accelerating Alzheimer’s therapeutic development: The past and future of clinical trials. Cell 2023, 186, 4757–4772. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- Sun, M.-K.; Alkon, D.L. Alkon, Alzheimer’s therapeutic development: Shifting neurodegeneration to neuroregeneration. Trends Pharmacol. Sci. 2024, 45, 197–209. [Google Scholar] [CrossRef]
- Qin, C.; Li, Y.; Wang, K. Functional Mechanism of Bone Marrow-Derived Mesenchymal Stem Cells in the Treatment of Animal Models with Alzheimer’s Disease: Inhibition of Neuroinflammation. J. Inflamm. Res. 2021, 14, 4761–4775. [Google Scholar] [CrossRef]
- Hernández, A.E.; García, E. Mesenchymal Stem Cell Therapy for Alzheimer’s Disease. Stem Cells Int. 2021, 2021, 7834421. [Google Scholar] [CrossRef]
- Han, Y.; Yang, J.; Fang, J.; Zhou, Y.; Candi, E.; Wang, J.; Hua, D.; Shao, C.; Shi, Y. The secretion profile of mesenchymal stem cells and potential applications in treating human diseases. Signal Transduct. Target. Ther. 2022, 7, 92. [Google Scholar] [CrossRef]
- Ebrahim, N.; Al Saihati, H.A.; Alali, Z.; Aleniz, F.Q.; Mahmoud, S.Y.M.; Badr, O.A.; Dessouky, A.A.; Mostafa, O.; Hussien, N.I.; Farid, A.S.; et al. Exploring the molecular mechanisms of MSC-derived exosomes in Alzheimer’s disease: Autophagy, insulin and the PI3K/Akt/mTOR signaling pathway. Biomed. Pharmacother. 2024, 176, 116836. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Castillo, A.I.; Sepúlveda, M.R.; Marín-Teva, J.L.; Cuadros, M.A.; Martín-Oliva, D.; González-Rey, E.; Delgado, M.; Neubrand, V.E. Switching Roles: Beneficial Effects of Adipose Tissue-Derived Mesenchymal Stem Cells on Microglia and Their Implication in Neurodegenerative Diseases. Biomolecules 2022, 12, 219. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Cho, K.R.; Jang, H.; Lee, N.K.; Jung, Y.H.; Kim, J.P.; Lee, J.I.; Chang, J.W.; Park, S.; Kim, S.T.; et al. Intracerebroventricular injection of human umbilical cord blood mesenchymal stem cells in patients with Alzheimer’s disease dementia: A phase I clinical trial. Alzheimer′s Res. Ther. 2021, 13, 154. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Cao, N.; Zhai, J.; Zeng, Q.; Zheng, P.; Su, R.; Liao, T.; Liu, J.; Pei, H.; Fan, Z.; et al. HGF Mediates Clinical-Grade Human Umbilical Cord-Derived Mesenchymal Stem Cells Improved Functional Recovery in a Senescence-Accelerated Mouse Model of Alzheimer’s Disease. Adv. Sci. 2020, 7, 1903809. [Google Scholar] [CrossRef]
- Li, Y.; Ye, R.; Dai, H.; Lin, J.; Cheng, Y.; Zhou, Y.; Lu, Y. Exploring TNFR1: From discovery to targeted therapy development. J. Transl. Med. 2025, 23, 71. [Google Scholar] [CrossRef]
- Liu, G.; Wang, H.; Zhang, C.; Li, X.; Mi, Y.; Chen, Y.; Xu, L.; Miao, L.; Long, H.; Liu, Y. Tumor Necrosis Factor Receptor 1 Is Required for Human Umbilical Cord-Derived Mesenchymal Stem Cell-Mediated Rheumatoid Arthritis Therapy. Cell Transplant. 2025, 34, 9636897241301703. [Google Scholar] [CrossRef]
- Song, M.-S.; Learman, C.; Ahn, K.-C.; Baker, G.; Kippe, J.; Field, E.; Dunbar, G. In vitro validation of effects of BDNF-expressing mesenchymal stem cells on neurodegeneration in primary cultured neurons of APP/PS1 mice. Neuroscience 2015, 307, 37–50. [Google Scholar] [CrossRef]
- Liang, J.; Deng, G.; Huang, H. The activation of BDNF reduced inflammation in a spinal cord injury model by TrkB/p38 MAPK signaling. Exp. Ther. Med. 2019, 17, 1688–1696. [Google Scholar] [CrossRef]
- Liu, G.; Wang, H.; Li, X.; Mi, Y.; Zhang, C.; Chen, Y.; Miao, L.; Long, H.; He, J.; Ge, Q.; et al. Biodistribution and persistence of human umbilical cord-derived mesenchymal stem cells in NCG mice: A comparative study. Future Sci. OA 2025, 11, 2471723. [Google Scholar] [CrossRef]
- Gallego-Rudolf, J.; Wiesman, A.I.; Binette, A.P.; Villeneuve, S.; Baillet, S.; PREVENT-AD Research Group. Synergistic association of Aβ and tau pathology with cortical neurophysiology and cognitive decline in asymptomatic older adults. Nat. Neurosci. 2024, 27, 2130–2137. [Google Scholar] [CrossRef]
- Thal, D.R.; Ruüb, U.; Orantes, M.; Braak, H. Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology 2002, 58, 1791–1800. [Google Scholar] [CrossRef] [PubMed]
- Roemer-Cassiano, S.N.; Wagner, F.; Evangelista, L.; Rauchmann, B.-S.; Dehsarvi, A.; Steward, A.; Dewenter, A.; Biel, D.; Zhu, Z.; Pescoller, J.; et al. Amyloid-associated hyperconnectivity drives tau spread across connected brain regions in Alzheimer’s disease. Sci. Transl. Med. 2025, 17, eadp2564. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yu, Y. Tau and neuroinflammation in Alzheimer’s disease: Interplay mechanisms and clinical translation. J. Neuroinflamm. 2023, 20, 165. [Google Scholar] [CrossRef]
- Madsen, L.S.; Ismail, R.; Parbo, P.; Kjeldsen, P.L.; Schaldemose, J.L.; Hansen, K.V.; Gottrup, H.; Aanerud, J.; Eskildsen, S.F.; Brooks, D.J. Microglial responses partially mediate the effect of Aβ on cognition in Alzheimer’s disease. Alzheimer′s Dement. 2024, 20, 8028–8037. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Li, H.; Li, Z.; Muñoz-Castro, C.; Jaisa-Aad, M.; Healey, M.A.; Welikovitch, L.A.; Jayakumar, R.; Bryant, A.G.; Noori, A.; et al. Astrocyte transcriptomic changes along the spatiotemporal progression of Alzheimer’s disease. Nat. Neurosci. 2024, 27, 2384–2400. [Google Scholar] [CrossRef]
- Xu, D.; Lian, D.; Wu, J.; Liu, Y.; Zhu, M.; Sun, J.; He, D.; Li, L. Brain-derived neurotrophic factor reduces inflammation and hippocampal apoptosis in experimental Streptococcus pneumoniae meningitis. J. Neuroinflamm. 2017, 14, 156. [Google Scholar] [CrossRef]
- Chandimali, N.; Bak, S.G.; Park, E.H.; Lim, H.-J.; Won, Y.-S.; Kim, E.-K.; Park, S.-I.; Lee, S.J. Free radicals and their impact on health and antioxidant defenses: A review. Cell Death Discov. 2025, 11, 19. [Google Scholar] [CrossRef]
- Habib, N.; McCabe, C.; Medina, S.; Varshavsky, M.; Kitsberg, D.; Dvir-Szternfeld, R.; Green, G.; Dionne, D.; Nguyen, L.; Marshall, J.L.; et al. Disease-associated astrocytes in Alzheimer’s disease and aging. Nat. Neurosci. 2020, 23, 701–706. [Google Scholar] [CrossRef]
- Wareham, L.K.; Liddelow, S.A.; Temple, S.; Benowitz, L.I.; Di Polo, A.; Wellington, C.; Goldberg, J.L.; He, Z.; Duan, X.; Bu, G.; et al. Solving neurodegeneration: Common mechanisms and strategies for new treatments. Mol. Neurodegener. 2022, 17, 23. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Ruganzu, J.B.; Zheng, Q.; Wu, X.; He, Y.; Peng, X.; Jin, H.; Zhou, J.; Ma, R.; Ji, S.; Ma, Y.; et al. TREM2 overexpression rescues cognitive deficits in APP/PS1 transgenic mice by reducing neuroinflammation via the JAK/STAT/SOCS signaling pathway. Exp. Neurol. 2021, 336, 113506. [Google Scholar] [CrossRef] [PubMed]
- Ding, M.; Shen, Y.; Wang, P.; Xie, Z.; Xu, S.; Zhu, Z.; Wang, Y.; Lyu, Y.; Wang, D.; Xu, L.; et al. Exosomes Isolated From Human Umbilical Cord Mesenchymal Stem Cells Alleviate Neuroinflammation and Reduce Amyloid-Beta Deposition by Modulating Microglial Activation in Alzheimer’s Disease. Neurochem. Res. 2018, 43, 2165–2177. [Google Scholar] [CrossRef]
- Losurdo, M.; Pedrazzoli, M.; D′Agostino, C.; Elia, C.A.; Massenzio, F.; Lonati, E.; Mauri, M.; Rizzi, L.; Molteni, L.; Bresciani, E.; et al. Intranasal delivery of mesenchymal stem cell-derived extracellular vesicles exerts immunomodulatory and neuroprotective effects in a 3xTg model of Alzheimer’s disease. Stem Cells Transl. Med. 2020, 9, 1068–1084. [Google Scholar] [CrossRef]
- Lin, L.; Huang, L.; Huang, S.; Chen, W.; Huang, H.; Chi, L.; Su, F.; Liu, X.; Yuan, K.; Jiang, Q.; et al. MSC-Derived Extracellular Vesicles Alleviate NLRP3/GSDMD-Mediated Neuroinflammation in Mouse Model of Sporadic Alzheimer’s Disease. Mol. Neurobiol. 2024, 61, 5494–5509. [Google Scholar] [CrossRef]
- Kefaloyianni, E. Soluble forms of cytokine and growth factor receptors: Mechanisms of generation and modes of action in the regulation of local and systemic inflammation. FEBS Lett. 2022, 596, 589–606. [Google Scholar] [CrossRef]
- Gómez, M.I.; Seaghdha, M.O.; Prince, A.S. Staphylococcus aureus protein A activates TACE through EGFR-dependent signaling. EMBO J. 2007, 26, 701–709. [Google Scholar] [CrossRef]
- Kölliker-Frers, R.; Udovin, L.; Otero-Losada, M.; Kobiec, T.; Herrera, M.I.; Palacios, J.; Razzitte, G.; Capani, F. Neuroinflammation: An Integrating Overview of Reactive-Neuroimmune Cell Interactions in Health and Disease. Mediat. Inflamm. 2021, 2021, 9999146. [Google Scholar] [CrossRef]
- Newton, K.; Strasser, A.; Kayagaki, N.; Dixit, V.M. Cell death. Cell 2024, 187, 235–256. [Google Scholar] [CrossRef]
- Dorronsoro, A.; Ferrin, I.; Salcedo, J.M.; Jakobsson, E.; Fernández-Rueda, J.; Lang, V.; Sepulveda, P.; Fechter, K.; Pennington, D.; Trigueros, C. Human mesenchymal stromal cells modulate T-cell responses through TNF-α-mediated activation of NF-κB. Eur. J. Immunol. 2014, 44, 480–488. [Google Scholar] [CrossRef]
- Numakawa, T.; Odaka, H.; Adachi, N. Actions of Brain-Derived Neurotrophin Factor in the Neurogenesis and Neuronal Function, and Its Involvement in the Pathophysiology of Brain Diseases. Int. J. Mol. Sci. 2018, 19, 3650. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.H.; Bylykbashi, E.; Chatila, Z.K.; Lee, S.W.; Pulli, B.; Clemenson, G.D.; Kim, E.; Rompala, A.; Oram, M.K.; Asselin, C.; et al. Combined adult neurogenesis and BDNF mimic exercise effects on cognition in an Alzheimer’s mouse model. Science 2018, 361, eaan8821. [Google Scholar] [CrossRef] [PubMed]
- Wei, P.; Jia, M.; Kong, X.; Lyu, W.; Feng, H.; Sun, X.; Li, J.; Yang, J.-J. Human umbilical cord-derived mesenchymal stem cells ameliorate perioperative neurocognitive disorder by inhibiting inflammatory responses and activating BDNF/TrkB/CREB signaling pathway in aged mice. Stem Cell Res. Ther. 2023, 14, 263. [Google Scholar] [CrossRef]
- Gliwińska, A.; Czubilińska-Łada, J.; Więckiewicz, G.; Świętochowska, E.; Badeński, A.; Dworak, M.; Szczepańska, M. The Role of Brain-Derived Neurotrophic Factor (BDNF) in Diagnosis and Treatment of Epilepsy, Depression, Schizophrenia, Anorexia Nervosa and Alzheimer’s Disease as Highly Drug-Resistant Diseases: A Narrative Review. Brain Sci. 2023, 13, 163. [Google Scholar] [CrossRef]
- AlRuwaili, R.; Al-Kuraishy, H.M.; Al-Gareeb, A.I.; Ali, N.H.; Alexiou, A.; Papadakis, M.; Saad, H.M.; Batiha, G.E.-S. The Possible Role of Brain-derived Neurotrophic Factor in Epilepsy. Neurochem. Res. 2024, 49, 533–547. [Google Scholar] [CrossRef]
- Cano, A.; Fonseca, E.; Ettcheto, M.; Sánchez-López, E.; de Rojas, I.; Alonso-Lana, S.; Morató, X.; Souto, E.B.; Toledo, M.; Boada, M.; et al. Epilepsy in Neurodegenerative Diseases: Related Drugs and Molecular Pathways. Pharmaceuticals 2021, 14, 1057. [Google Scholar] [CrossRef]
- Yang, X.; Huang, Y.A.; Marshall, J. Targeting TrkB-PSD-95 coupling to mitigate neurological disorders. Neural Regen. Res. 2025, 20, 715–724. [Google Scholar] [CrossRef]
- Fenton, H.; Finch, P.; Rubin, J.; Rosenberg, J.; Taylor, W.; Kuo-Leblanc, V.; Rodriguez-Wolf, M.; Baird, A.; Schipper, H.; Stopa, E. Hepatocyte growth factor (HGF/SF) in Alzheimer’s disease. Brain Res. 1998, 779, 262–270. [Google Scholar] [CrossRef]
- Zhao, L.-J.; Wang, Z.-T.; Ma, Y.-H.; Zhang, W.; Dong, Q.; Yu, J.-T.; Tan, L.; Initiative, A.D.N. Associations of the cerebrospinal fluid hepatocyte growth factor with Alzheimer’s disease pathology and cognitive function. BMC Neurol. 2021, 21, 387. [Google Scholar] [CrossRef]
- Hamasaki, H.; Honda, H.; Suzuki, S.O.; Hokama, M.; Kiyohara, Y.; Nakabeppu, Y.; Iwaki, T. Down-regulation of MET in hippocampal neurons of Alzheimer’s disease brains. Neuropathology 2014, 34, 284–290. [Google Scholar] [CrossRef]
- Takeuchi, D.; Sato, N.; Shimamura, M.; Kurinami, H.; Takeda, S.; Shinohara, M.; Suzuki, S.; Kojima, M.; Ogihara, T.; Morishita, R. Alleviation of Abeta-induced cognitive impairment by ultrasound-mediated gene transfer of HGF in a mouse model. Gene Ther. 2008, 15, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Desole, C.; Gallo, S.; Vitacolonna, A.; Montarolo, F.; Bertolotto, A.; Vivien, D.; Comoglio, P.; Crepaldi, T. HGF and MET: From Brain Development to Neurological Disorders. Front. Cell Dev. Biol. 2021, 9, 683609. [Google Scholar] [CrossRef] [PubMed]
- Hecht, M.; Schulte, J.H.; Eggert, A.; Wilting, J.; Schweigerer, L. The neurotrophin receptor TrkB cooperates with c-Met in enhancing neuroblastoma invasiveness. Carcinogenesis 2005, 26, 2105–2115. [Google Scholar] [CrossRef]
- Nishikoba, N.; Kumagai, K.; Kanmura, S.; Nakamura, Y.; Ono, M.; Eguchi, H.; Kamibayashiyama, T.; Oda, K.; Mawatari, S.; Tanoue, S.; et al. HGF-MET Signaling Shifts M1 Macrophages Toward an M2-Like Phenotype Through PI3K-Mediated Induction of Arginase-1 Expression. Front. Immunol. 2020, 11, 2135. [Google Scholar] [CrossRef]
- Chakraborty, S.; Balan, M.; Flynn, E.; Zurakowski, D.; Choueiri, T.K.; Pal, S. Activation of c-Met in cancer cells mediates growth-promoting signals against oxidative stress through Nrf2-HO-1. Oncogenesis 2019, 8, 7. [Google Scholar] [CrossRef]
- Yao, W.; Lin, S.; Su, J.; Cao, Q.; Chen, Y.; Chen, J.; Zhang, Z.; Hashimoto, K.; Qi, Q.; Zhang, J.-C. Activation of BDNF by transcription factor Nrf2 contributes to antidepressant-like actions in rodents. Transl. Psychiatry 2021, 11, 140. [Google Scholar] [CrossRef]
- Ren, H.; Han, R.; Liu, X.; Wang, L.; Koehler, R.C.; Wang, J. Nrf2-BDNF-TrkB pathway contributes to cortical hemorrhage-induced depression, but not sex differences. J. Cereb. Blood Flow Metab. 2021, 41, 3288–3301. [Google Scholar] [CrossRef]
- Sanchez-Diaz, M.; Quiñones-Vico, M.I.; de la Torre, R.S.; Montero-Vílchez, T.; Sierra-Sánchez, A.; Molina-Leyva, A.; Arias-Santiago, S. Biodistribution of Mesenchymal Stromal Cells after Administration in Animal Models and Humans: A Systematic Review. J. Clin. Med. 2021, 10, 2925. [Google Scholar] [CrossRef]
- Khayambashi, P.; Iyer, J.; Pillai, S.; Upadhyay, A.; Zhang, Y.; Tran, S.D. Hydrogel Encapsulation of Mesenchymal Stem Cells and Their Derived Exosomes for Tissue Engineering. Int. J. Mol. Sci. 2021, 22, 684. [Google Scholar] [CrossRef]
- Li, B.; Zhang, L.; Yin, Y.; Chen, A.; Seo, B.R.; Lou, J.; Mooney, D.J.; Weitz, D.A. Stiff Hydrogel Encapsulation Retains Mesenchymal Stem Cell Stemness for Regenerative Medicine. Matter 2024, 7, 3447–3468. [Google Scholar] [CrossRef]
- Sultan, M.T.; Choi, B.Y.; Ajiteru, O.; Hong, D.K.; Lee, S.M.; Kim, H.-J.; Ryu, J.S.; Lee, J.S.; Hong, H.; Lee, Y.J.; et al. Reinforced-hydrogel encapsulated hMSCs towards brain injury treatment by trans-septal approach. Biomaterials 2021, 266, 120413. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, H.; Zhong, C.; Mi, Y.; Li, G.; Zhang, C.; Chen, Y.; Li, X.; Liu, Y.; Liu, G. Exploration of Cytokines That Impact the Therapeutic Efficacy of Mesenchymal Stem Cells in Alzheimer’s Disease. Bioengineering 2025, 12, 646. https://doi.org/10.3390/bioengineering12060646
Wang H, Zhong C, Mi Y, Li G, Zhang C, Chen Y, Li X, Liu Y, Liu G. Exploration of Cytokines That Impact the Therapeutic Efficacy of Mesenchymal Stem Cells in Alzheimer’s Disease. Bioengineering. 2025; 12(6):646. https://doi.org/10.3390/bioengineering12060646
Chicago/Turabian StyleWang, Herui, Chonglin Zhong, Yi Mi, Guo Li, Chenliang Zhang, Yaoyao Chen, Xin Li, Yongjun Liu, and Guangyang Liu. 2025. "Exploration of Cytokines That Impact the Therapeutic Efficacy of Mesenchymal Stem Cells in Alzheimer’s Disease" Bioengineering 12, no. 6: 646. https://doi.org/10.3390/bioengineering12060646
APA StyleWang, H., Zhong, C., Mi, Y., Li, G., Zhang, C., Chen, Y., Li, X., Liu, Y., & Liu, G. (2025). Exploration of Cytokines That Impact the Therapeutic Efficacy of Mesenchymal Stem Cells in Alzheimer’s Disease. Bioengineering, 12(6), 646. https://doi.org/10.3390/bioengineering12060646