
Application of a Rational Crystal Contact Engineering Strategy on a Poly(ethylene terephthalate)-Degrading Cutinase

 , , , ,
, , , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Site-Directed Mutagenesis
2.2. Heterologous Protein Production
2.3. Protein Processing: Purification, Desalting, and Buffer Exchange
2.4. Static Protein Crystallization
2.5. Protein Analytics: Electrophoresis and Esterase Assay
2.6. X-Ray Diffraction, Data Collection, Processing, and Refinement
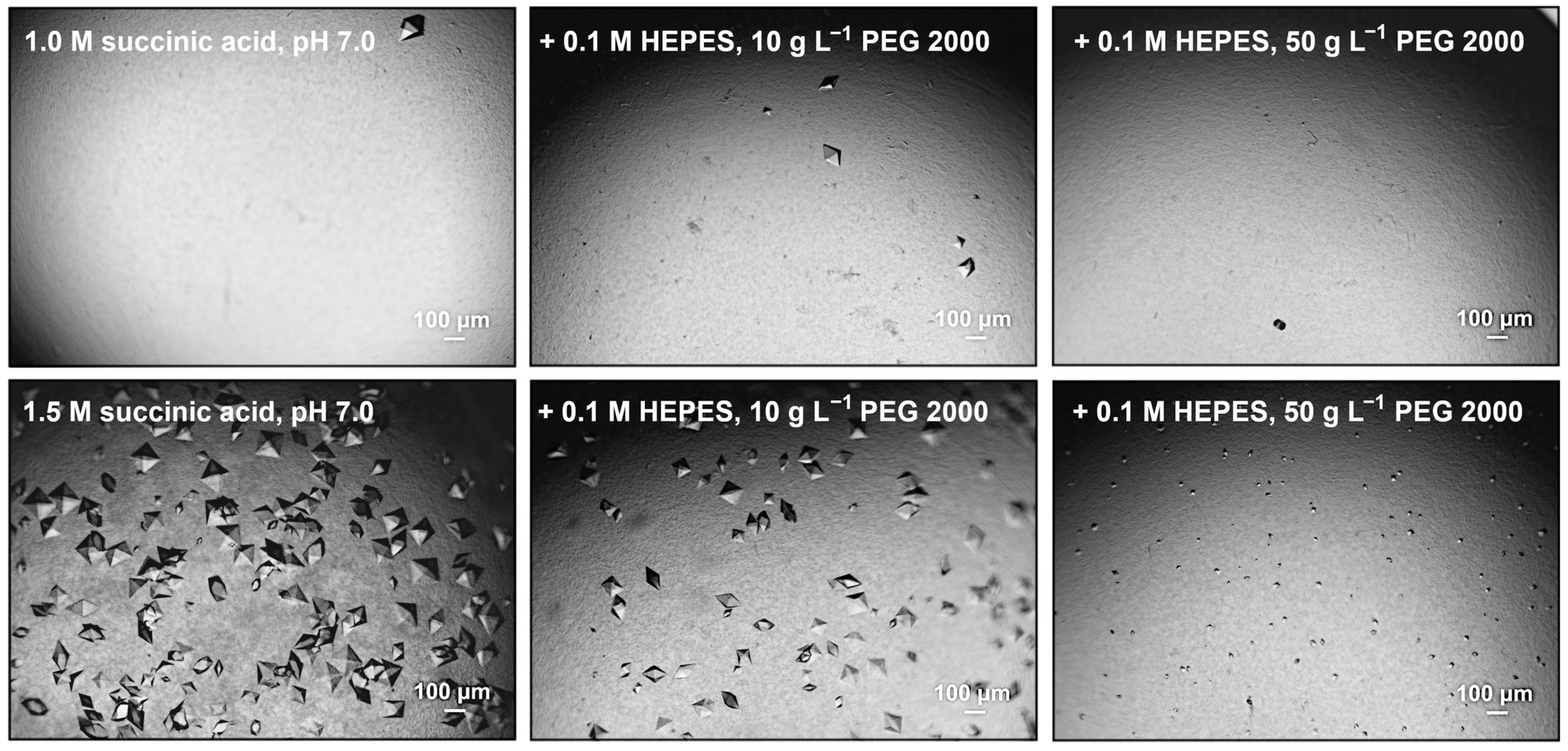
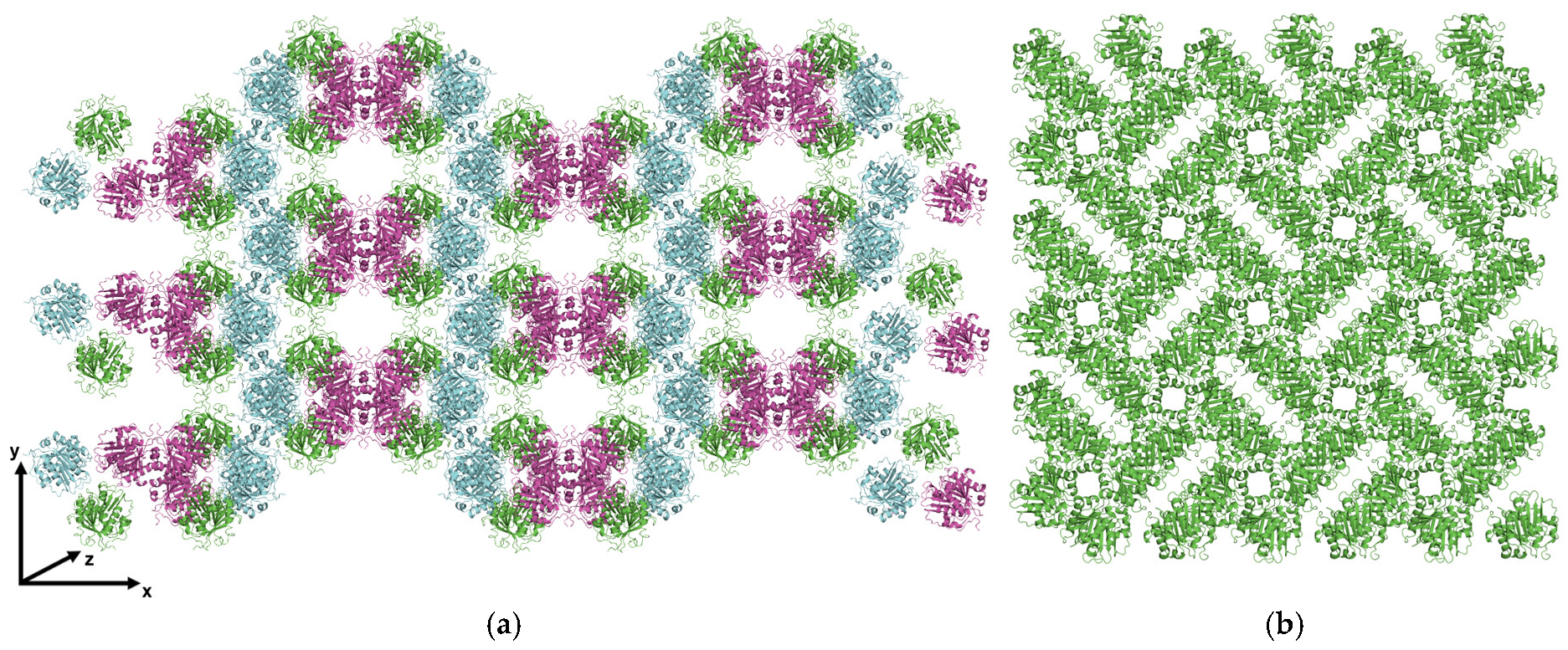
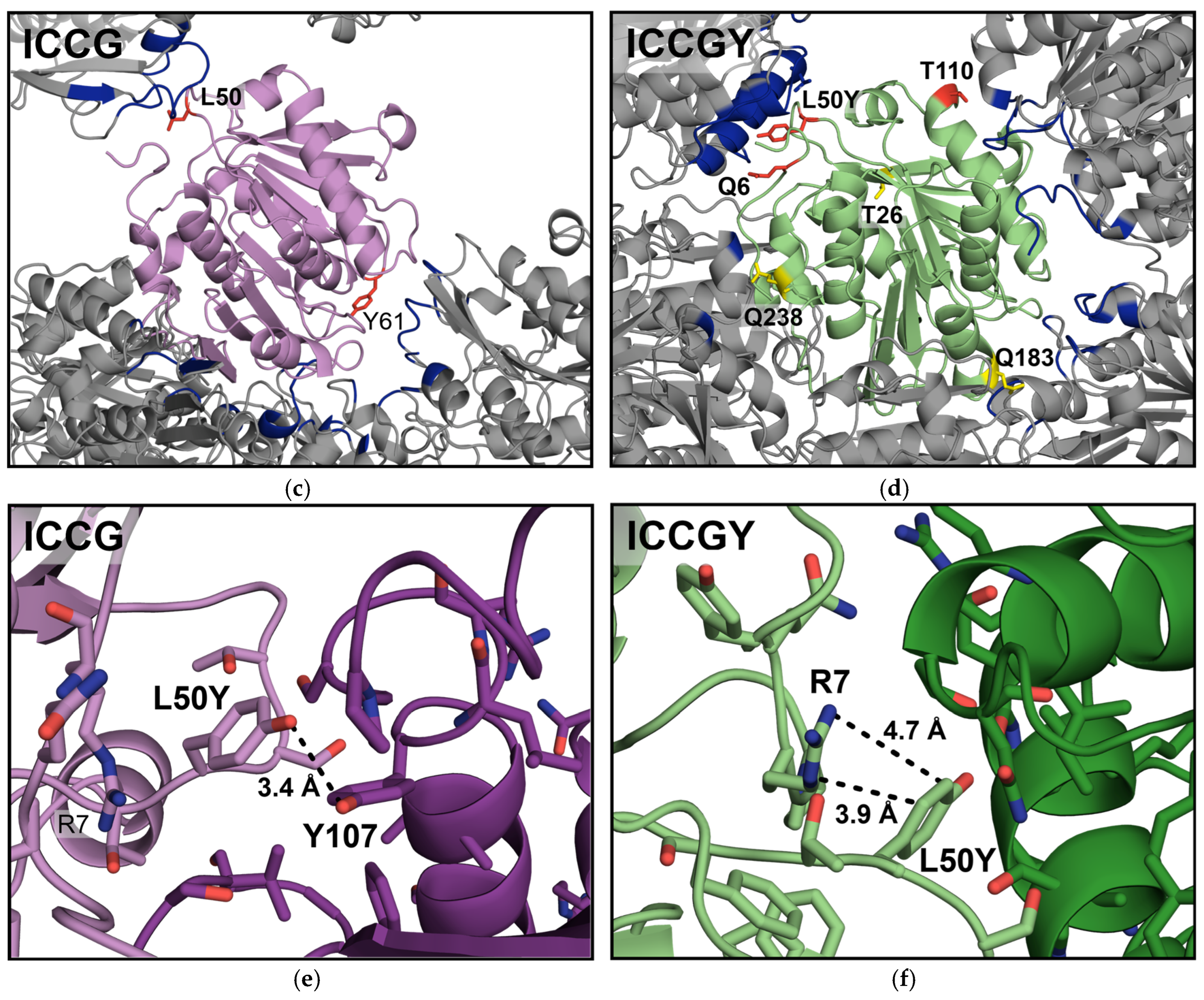
3. Results
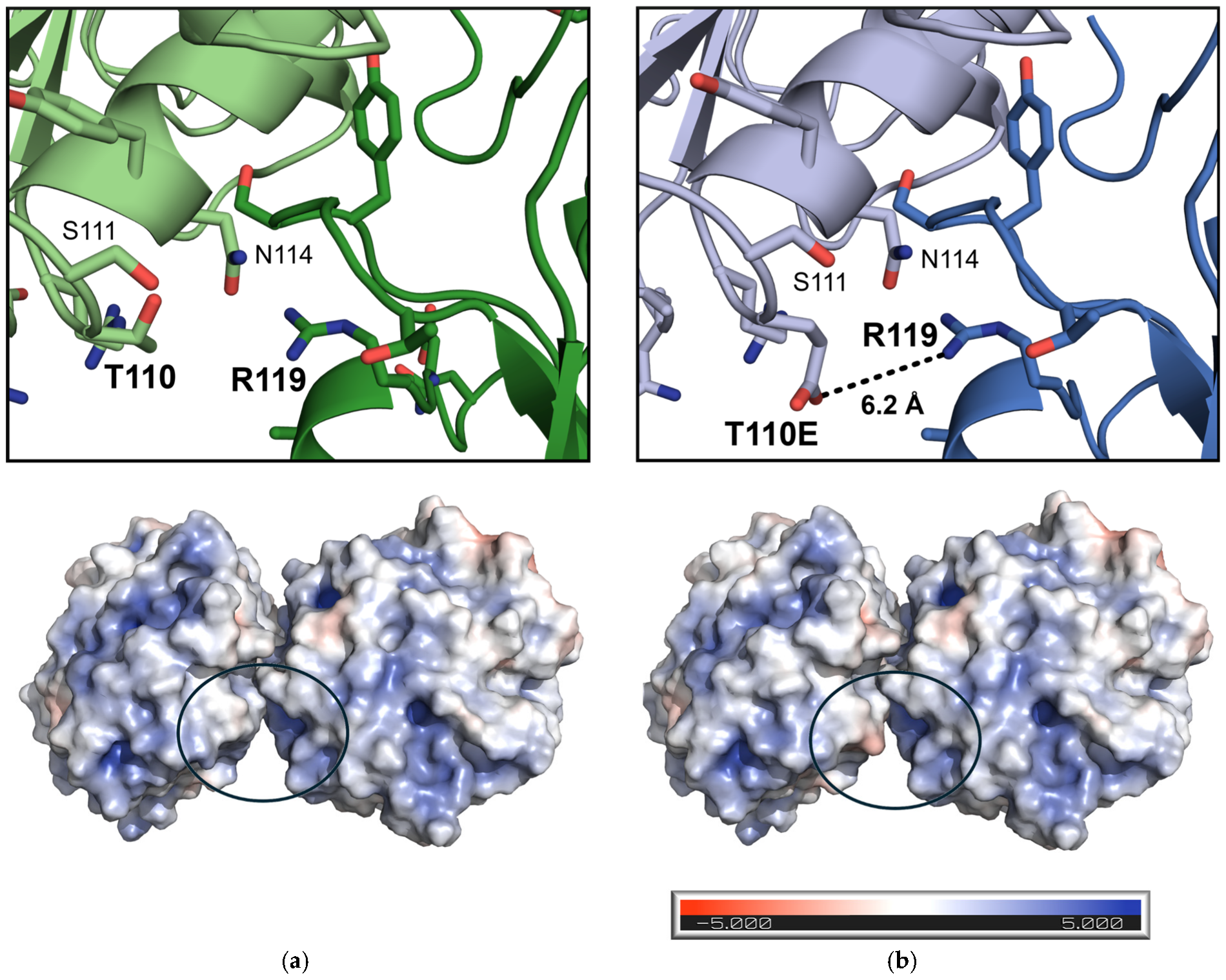
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| E (Glu) | Glutamic acid |
| EIA | Electrostatic interaction |
| HTP | High throughput |
| ICCG(Y) | LCC mutant: F243I/D238C/S283C/Y127G (L50Y) |
| IEX | Ion exchange chromatography |
| IMAC | Immobilized metal ion affinity chromatography |
| K (Lys) | Lysine |
| Lb/LkADH | Lactobacillus brevis/kefiri alcohol dehydrogenase |
| LCC | Leaf branch compost cutinase |
| NC | Negative control |
| NspER1-L1,5 | Nostoc sp. PCC 1720 ene reductase (with engineered Loop1,5) |
| PDB ID | Protein Data Bank Identification |
| PET | Poly(ethylene terephthalate) |
| Q (Gln) | Glutamine |
| R (Arg) | Arginine |
| RMSD | Root mean square deviation |
| SEC | Size exclusion chromatography |
| SDS-PAGE | Sodium dodecyl sulfate polyacrylamide gel electrophoresis |
| T (Thr) | Threonine |
| WT | Wild type |
References
- World Economic Forum. The New Plastics Economy—Rethinking the Future of Plastics. World Economic Forum. 2025. Available online: https://www3.weforum.org/docs/WEF_The_New_Plastics_Economy.pdf (accessed on 20 February 2025).
- Plastics Europe. Plastics—The Facts 2023. An Analysis of European Plastics Production, Demand and Waste Data. Plastics Europe. 2025. Available online: https://plasticseurope.org/wp-content/uploads/2024/11/PE_TheFacts_24_digital-1pager.pdf (accessed on 20 February 2025).
- PET Polymer: Chemical Economics Handbook. IHS Markit. 2018. Available online: https://ihsmarkit.com/products/pet-polymer-chemical-economics-handbook.html (accessed on 20 February 2025).
- Urbanek, A.K.; Kosiorowska, K.E.; Mirończuk, A.M. Current knowledge on polyethylene terephthalate degradation by genetically modified microorganisms. Front. Bioeng. Biotechnol. 2021, 9, 771133. [Google Scholar] [CrossRef] [PubMed]
- Ragaert, K.; Delva, L.; Van Geem, K. Mechanical and chemical recycling of solid plastic waste. Waste Manag. 2017, 69, 24–58. [Google Scholar] [CrossRef]
- Danso, D.; Chow, J.; Streit, W.R. Plastics: Environmental and biotechnological perspectives on microbial degradation. Appl. Environ. Microbiol. 2019, 85, e01095-19. [Google Scholar] [CrossRef]
- Sulaiman, S.; You, D.-J.; Kanaya, E.; Koga, Y.; Kanaya, S. Crystal structure and thermodynamic and kinetic stability of metagenome-derived LC-cutinase. Biochemistry 2014, 53, 1858–1869. [Google Scholar] [CrossRef]
- Chen, S.; Su, L.; Chen, J.; Wu, J. Cutinase: Characteristics, preparation, and application. Biotechnol. Adv. 2013, 31, 1754–1767. [Google Scholar] [CrossRef]
- Kawai, F.; Oda, M.; Tamashiro, T.; Waku, T.; Tanaka, N.; Yamamoto, M.; Mizushima, H.; Miyakawa, T.; Tanokura, M. A novel Ca2+-activated, thermostabilized polyesterase capable of hydrolyzing polyethylene terephthalate from Saccharomonospora viridis AHK190. Appl. Microbiol. Biotechnol. 2014, 98, 10053–10064. [Google Scholar] [CrossRef]
- Samak, N.A.; Jia, Y.; Sharshar, M.M.; Mu, T.; Yang, M.; Peh, S.; Xing, J. Recent advances in biocatalysts engineering for polyethylene terephthalate plastic waste green recycling. Environ. Int. 2020, 145, 106144. [Google Scholar] [CrossRef]
- Kawai, F.; Kawabata, T.; Oda, M. Current knowledge on enzymatic PET degradation and its possible application to waste stream management and other fields. Appl. Microbiol. Biotechnol. 2019, 103, 4253–4268. [Google Scholar] [CrossRef]
- Wei, R.; Zimmermann, W. Biocatalysis as a green route for recycling the recalcitrant plastic polyethylene terephthalate. Microb. Biotechnol. 2017, 10, 1302–1307. [Google Scholar] [CrossRef]
- Gao, R.; Pan, H.; Lian, J. Recent advances in the discovery, characterization, and engineering of poly(ethylene terephthalate) (PET) hydrolases. Enzym. Microb. Technol. 2021, 150, 109868. [Google Scholar] [CrossRef]
- Sulaiman, S.; Yamato, S.; Kanaya, E.; Kim, J.-J.; Koga, Y.; Takano, K.; Kanaya, S. Isolation of a novel cutinase homolog with polyethylene terephthalate-degrading activity from leaf-branch compost by using a metagenomic approach. Appl. Environ. Microbiol. 2012, 78, 1556–1562. [Google Scholar] [CrossRef] [PubMed]
- Tournier, V.; Topham, C.M.; Gilles, A.; David, B.; Folgoas, C.; Moya-Leclair, E.; Kamionka, E.; Desrousseaux, M.L.; Texier, H.; Gavalda, S.; et al. An engineered PET depolymerase to break down and recycle plastic bottles. Nature 2020, 580, 216–219. [Google Scholar] [CrossRef] [PubMed]
- Shirke, A.N.; White, C.; Englaender, J.A.; Zwarycz, A.; Butterfoss, G.L.; Linhardt, R.J.; Gross, R.A. Stabilizing leaf and branch compost cutinase (LCC) with glycosylation: Mechanism and effect on PET hydrolysis. Biochemistry 2018, 57, 1190–1200. [Google Scholar] [CrossRef] [PubMed]
- Ronkvist, Å.M.; Xie, W.; Lu, W.; Gross, R.A. Cutinase-catalyzed hydrolysis of poly(ethylene terephthalate). Macromolecules 2009, 42, 5128–5138. [Google Scholar] [CrossRef]
- Vertommen, M.; Nierstrasz, V.; van der Veer, M.; Warmoeskerken, M. Enzymatic surface modification of poly(ethylene terephthalate). J. Biotechnol. 2005, 120, 376–386. [Google Scholar] [CrossRef]
- Lalor, F.; Fitzpatrick, J.; Sage, C.; Byrne, E. Sustainability in the biopharmaceutical industry: Seeking a holistic perspective. Biotechnol. Adv. 2019, 37, 698–707. [Google Scholar] [CrossRef]
- Ho, S.V.; McLaughlin, J.M.; Cue, B.W.; Dunn, P.J. Environmental considerations in biologics manufacturing. Green Chem. 2010, 12, 755–766. [Google Scholar] [CrossRef]
- Hekmat, D. Large-scale crystallization of proteins for purification and formulation. Bioprocess Biosyst. Eng. 2015, 38, 1209–1231. [Google Scholar] [CrossRef]
- Roque, A.C.A.; Pina, A.S.; Azevedo, A.M.; Aires-Barros, R.; Jungbauer, A.; Di Profio, G.; Heng, J.Y.Y.; Haigh, J.; Ottens, M. Anything but conventional chromatography approaches in bioseparation. Biotechnol. J. 2020, 15, e1900274. [Google Scholar] [CrossRef]
- dos Santos, R.; Carvalho, A.L.; Roque, A.C.A. Renaissance of protein crystallization and precipitation in biopharmaceuticals purification. Biotechnol. Adv. 2017, 35, 41–50. [Google Scholar] [CrossRef]
- Smejkal, B.; Helk, B.; Rondeau, J.M.; Anton, S.; Wilke, A.; Scheyerer, P.; Fries, J.; Hekmat, D.; Weuster-Botz, D. Protein crystallization in stirred systems—Scale-up via the maximum local energy dissipation. Biotechnol. Bioeng. 2013, 110, 1956–1963. [Google Scholar] [CrossRef]
- Yang, H.; Belviso, B.D.; Li, X.; Chen, W.; Mastropietro, T.F.; Di Profio, G.; Caliandro, R.; Heng, J.Y.Y. Optimization of vapor diffusion conditions for anti-CD20 crystallization and scale-up to meso batch. Crystals 2019, 9, 230. [Google Scholar] [CrossRef]
- Shenoy, B.; Wang, Y.; Shan, W.; Margolin, A.L. Stability of crystalline proteins. Biotechnol. Bioeng. 2001, 73, 358–369. [Google Scholar] [CrossRef] [PubMed]
- Altan, I.; Charbonneau, P.; Snell, E.H. Computational crystallization. Arch. Biochem. Biophys. 2016, 602, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Abramson, J.; Adler, J.; Dunger, J.; Evans, R.; Green, T.; Pritzel, A.; Ronneberger, O.; Willmore, L.; Ballard, A.J.; Bambrick, J.; et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 2024, 630, 493–500. [Google Scholar] [CrossRef]
- Pak, M.A.; Markhieva, K.A.; Novikova, M.S.; Petrov, D.S.; Vorobyev, I.S.; Maksimova, E.S.; Kondrashov, F.A.; Ivankov, D.N. Using AlphaFold to predict the impact of single mutations on protein stability and function. PLoS ONE 2023, 18, e0282689. [Google Scholar] [CrossRef]
- Nowotny, P.; Hermann, J.; Li, J.; Krautenbacher, A.; Klöpfer, K.; Hekmat, D.; Weuster-Botz, D. Rational crystal contact engineering of Lactobacillus brevis alcohol dehydrogenase to promote technical protein crystallization. Cryst. Growth Des. 2019, 19, 2380–2387. [Google Scholar] [CrossRef]
- Grob, P.; Huber, M.; Walla, B.; Hermann, J.; Janowski, R.; Niessing, D.; Hekmat, D.; Weuster-Botz, D. Crystal contact engineering enables efficient capture and purification of an oxidoreductase by technical crystallization. Biotechnol. J. 2020, 15, e2000010. [Google Scholar] [CrossRef]
- Walla, B.; Bischoff, D.; Janowski, R.; von den Eichen, N.; Niessing, D.; Weuster-Botz, D. Transfer of a rational crystal contact engineering strategy between diverse alcohol dehydrogenases. Crystals 2021, 11, 975. [Google Scholar] [CrossRef]
- Derewenda, Z.S. Rational protein crystallization by mutational surface engineering. Structure 2004, 12, 529–535. [Google Scholar] [CrossRef] [PubMed]
- Cooper, D.R.; Boczek, T.; Grelewska, K.; Pinkowska, M.; Sikorska, M.; Zawadzki, M.; Derewenda, Z. Protein crystallization by surface entropy reduction: Optimization of the SER strategy. Acta Crystallogr. Sect. D Biol. Crystallogr. 2007, 63 Pt 5, 636–645. [Google Scholar] [CrossRef]
- Derewenda, Z.S.; Vekilov, P.G. Entropy and surface engineering in protein crystallization. Acta Crystallogr. Sect. D Biol. Crystallogr. 2006, 62 Pt 1, 116–124. [Google Scholar] [CrossRef]
- Walla, B.; Maslakova, A.; Bischoff, D.; Janowski, R.; Niessing, D.; Weuster-Botz, D. Rational introduction of electrostatic interactions at crystal contacts to enhance protein crystallization of an ene reductase. Biomolecules 2025, 15, 467. [Google Scholar] [CrossRef]
- Fritzsche, S.; Tischer, F.; Peukert, W.; Castiglione, K. You get what you screen for: A benchmark analysis of leaf branch compost cutinase variants for polyethylene terephthalate (PET) degradation. React. Chem. Eng. 2023, 8, 2156–2169. [Google Scholar] [CrossRef]
- Zheng, L.; Baumann, U.; Reymond, J.L. An efficient one-step site-directed and site-saturation mutagenesis protocol. Nucleic Acids Res. 2004, 32, e115. [Google Scholar] [CrossRef]
- Sambrook, J.; Russell, D.W. Preparation and transformation of competent E. coli using calcium chloride. Cold Spring Harb. Protoc. 2006, 2006, 3932. [Google Scholar] [CrossRef]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef]
- Fairbanks, G.; Steck, T.L.; Wallach, D.F.H. Electrophoretic analysis of the major polypeptides of the human erythrocyte membrane. Biochemistry 1971, 10, 2606–2617. [Google Scholar] [CrossRef]
- Bhunia, R.K.; Showman, L.J.; Jose, A.; Nikolau, B.J. Combined use of cutinase and high-resolution mass-spectrometry to query the molecular architecture of cutin. Plant Methods 2018, 14, 117. [Google Scholar] [CrossRef]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef] [PubMed]
- Murshudov, G.N.; Vagin, A.A.; Dodson, E.J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. Sect. D 1997, 53, 240–255. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of coot. Acta Crystallogr. Sect. D 2010, 66, 486–501. [Google Scholar] [CrossRef]
- Schrödinger, L.; DeLano, W. Pymol, Version 2.3. 2020. Available online: http://www.pymol.org/pymol (accessed on 15 January 2021).
- Buell, A.K.; Hung, P.; Salvatella, X.; Welland, M.E.; Dobson, C.M.; Knowles, T.P.J. Electrostatic effects in filamentous protein aggregation. Biophys. J. 2013, 104, 1116–1126. [Google Scholar] [CrossRef]
- Jiang, L.; Cao, S.; Cheung, P.P.-H.; Zheng, X.; Leung, C.W.T.; Peng, Q.; Shuai, Z.; Tang, B.Z.; Yao, S.; Huang, X. Real-time monitoring of hydrophobic aggregation reveals a critical role of cooperativity in hydrophobic effect. Nat. Commun. 2017, 8, 15639. [Google Scholar] [CrossRef]
- McPherson, A. Introduction to protein crystallization. Methods 2004, 34, 254–265. [Google Scholar] [CrossRef]
- Chruszcz, M.; Potrzebowski, W.; Zimmerman, M.D.; Grabowski, M.; Zheng, H.; Lasota, P.; Minor, W. Analysis of solvent content and oligomeric states in protein crystals—Does symmetry matter? Protein Sci. Publ. Protein Soc. 2008, 17, 623–632. [Google Scholar] [CrossRef]
- McPherson, A.; Gavira, J.A. Introduction to protein crystallization. Acta Crystallogr. F Struct. Biol. Commun. 2014, 70 Pt 1, 2–20. [Google Scholar] [CrossRef]
- Terwilliger, T.C.; Liebschner, D.; Croll, T.I.; Williams, C.J.; McCoy, A.J.; Poon, B.K.; Afonine, P.V.; Oeffner, R.D.; Richardson, J.S.; Read, R.J.; et al. AlphaFold predictions are valuable hypotheses and accelerate but do not replace experimental structure determination. Nat. Methods 2024, 21, 110–116. [Google Scholar] [CrossRef]
- Carugo, O. Participation of protein sequence termini in crystal contacts. Protein Sci. Publ. Protein Soc. 2011, 20, 2121–2124. [Google Scholar] [CrossRef]
- Zhou, H.X.; Pang, X. Electrostatic interactions in protein structure, folding, binding, and condensation. Chem. Rev. 2018, 118, 1691–1741. [Google Scholar] [CrossRef]
- Dasgupta, S.; Iyer, G.H.; Bryant, S.H.; Lawrence, C.E.; Bell, J.A. Extent and nature of contacts between protein molecules in crystal lattices and between subunits of protein oligomers. Proteins Struct. Funct. Bioinform. 1997, 28, 494–514. [Google Scholar] [CrossRef]
- Kissick, D.J.; Wanapun, D.; Simpson, G.J. Second-order nonlinear optical imaging of chiral crystals. Annu. Rev. Anal. Chem. 2011, 4, 419–437. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| LCC Variant, (PDB ID) | Protein Purification | Crystallization Method | Crystallization Conditions | Space Group |
|---|---|---|---|---|
| ICCG (6THT; [15]) | IEX, SEC | Vapor diffusion | 0.1 M imidazole, 1 M sodium citrate, pH 8.0 (12 °C) | P63 |
| ICCG WT (9QYP) | IMAC, SEC | Vapor diffusion | 0.8 M succinic acid pH 7.0 (20 °C) | I222 |
| ICCG L50Y (9QYU) | IMAC | Batch crystallization | 0.8 M succinic acid pH 7.0 (20 °C) | P41212 |
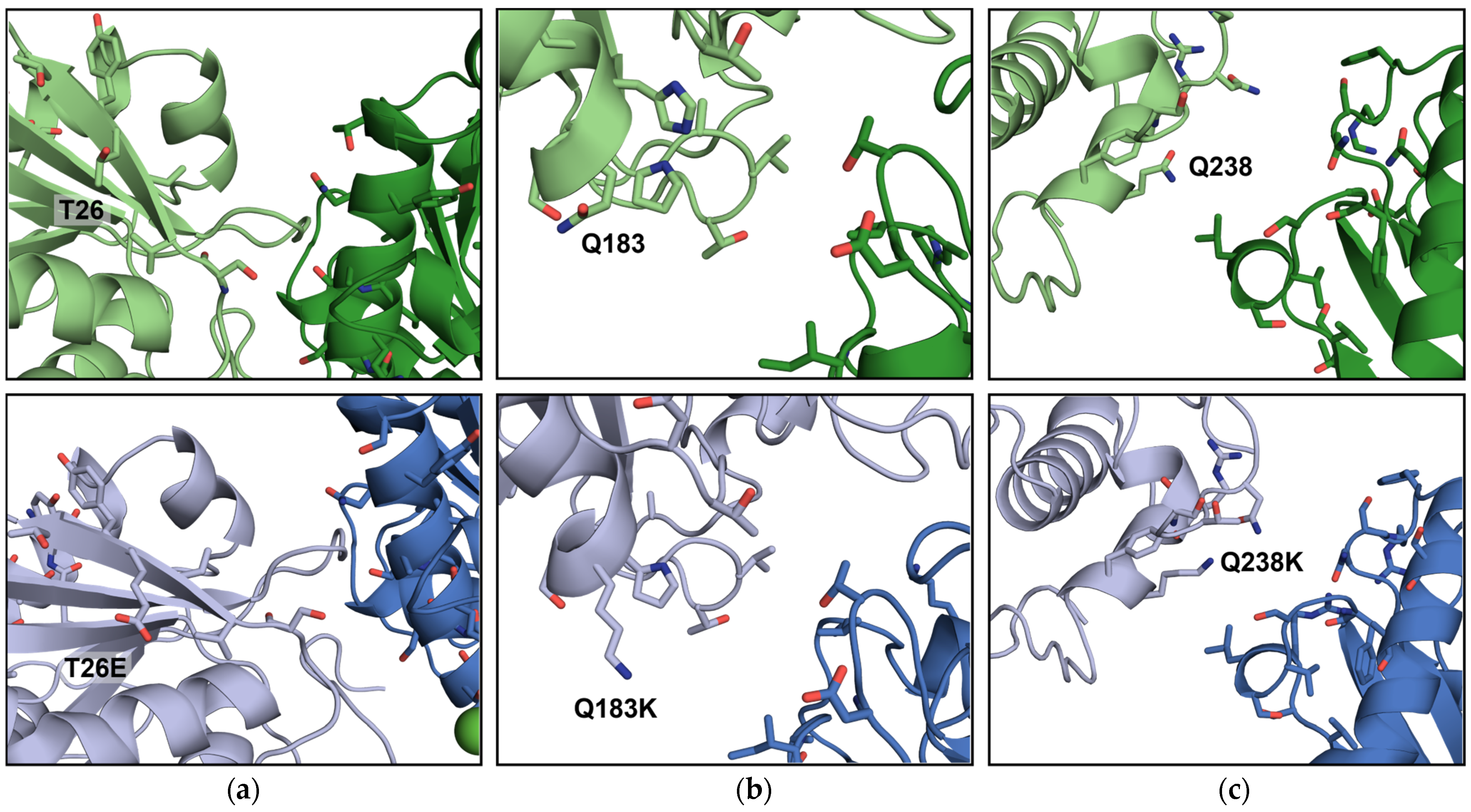
| ICCGY Mutant | Applied Strategy: Electrostatic Interaction (EIA), Negative Control (NC) | Potential Interaction Partner; Distance (In Silico), Å |
|---|---|---|
| Q6E | EIA | R139; 3.3 |
| T26E | NC | / |
| T110E | EIA | R119; 6.0 |
| Q183K | NC | / |
| Q238K | NC | / |
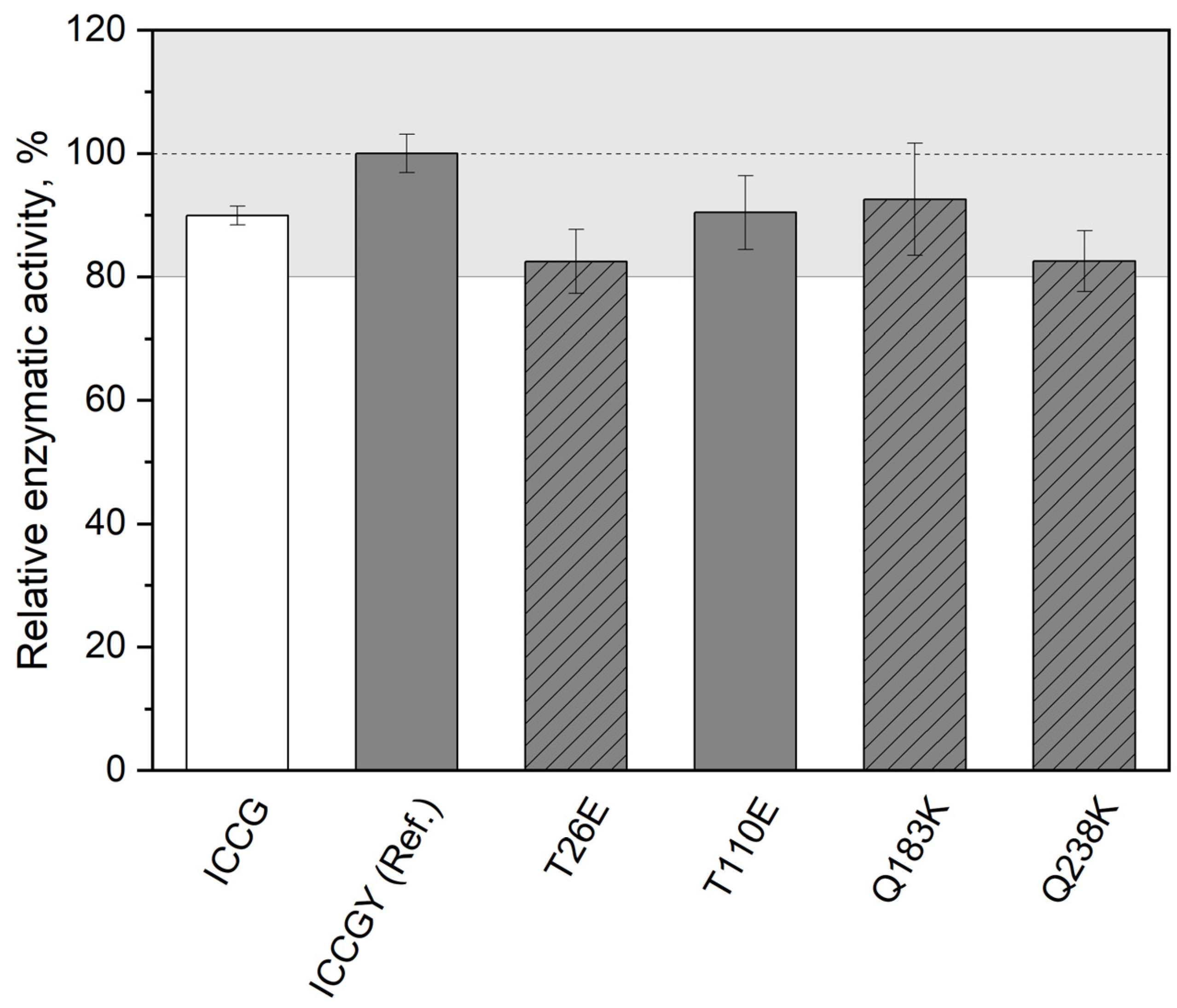
| ICCGY Variant | Protein Concentration, g L−1 | Crystallization Onset, h | Crystal Size, µm | Crystal Amount, - |
|---|---|---|---|---|
| WT | 10 | 26 | 247.8 ± 70.9 | 25 |
| 5 | / | / | / | |
| Q6E | / | / | / | / |
| T26E | 10 | 22 | 344.5 ± 63.2 (*) | 12 |
| T110E | 10 | 6 | 305.7 ± 78.2 | 21 |
| 5 | 20 | 188.8 ± 84.7 | 16 | |
| Q183K | 10 | 48 | 252.9 ± 97.2 | 12 |
| Q238K | / | / | / | / |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Walla, B.; Dietrich, A.-M.; Brames, E.; Bischoff, D.; Fritzsche, S.; Castiglione, K.; Janowski, R.; Niessing, D.; Weuster-Botz, D. Application of a Rational Crystal Contact Engineering Strategy on a Poly(ethylene terephthalate)-Degrading Cutinase. Bioengineering 2025, 12, 561. https://doi.org/10.3390/bioengineering12060561
Walla B, Dietrich A-M, Brames E, Bischoff D, Fritzsche S, Castiglione K, Janowski R, Niessing D, Weuster-Botz D. Application of a Rational Crystal Contact Engineering Strategy on a Poly(ethylene terephthalate)-Degrading Cutinase. Bioengineering. 2025; 12(6):561. https://doi.org/10.3390/bioengineering12060561
Chicago/Turabian StyleWalla, Brigitte, Anna-Maria Dietrich, Edwin Brames, Daniel Bischoff, Stefanie Fritzsche, Kathrin Castiglione, Robert Janowski, Dierk Niessing, and Dirk Weuster-Botz. 2025. "Application of a Rational Crystal Contact Engineering Strategy on a Poly(ethylene terephthalate)-Degrading Cutinase" Bioengineering 12, no. 6: 561. https://doi.org/10.3390/bioengineering12060561
APA StyleWalla, B., Dietrich, A.-M., Brames, E., Bischoff, D., Fritzsche, S., Castiglione, K., Janowski, R., Niessing, D., & Weuster-Botz, D. (2025). Application of a Rational Crystal Contact Engineering Strategy on a Poly(ethylene terephthalate)-Degrading Cutinase. Bioengineering, 12(6), 561. https://doi.org/10.3390/bioengineering12060561