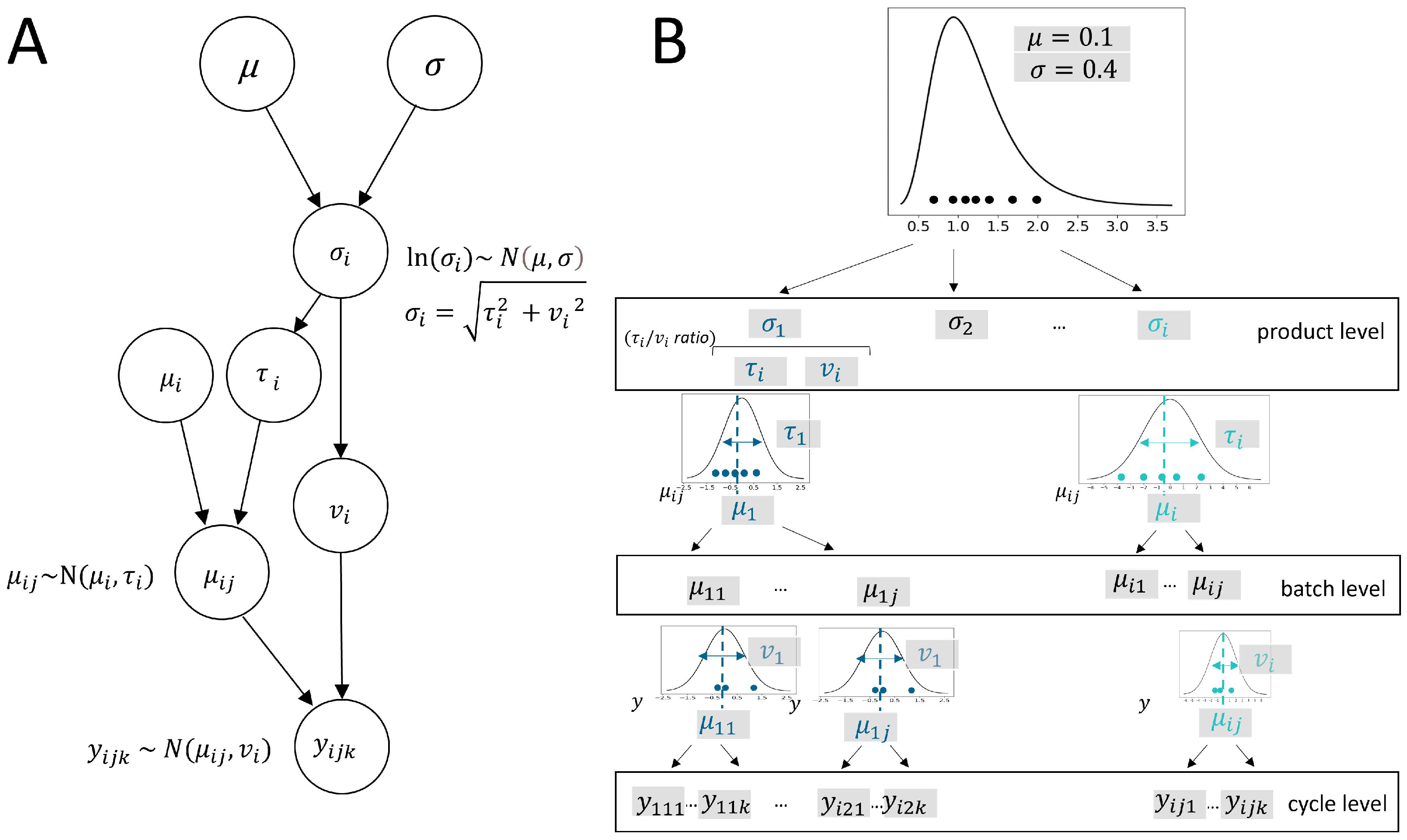
1. Introduction
In the development of new biopharmaceutical drugs, the urgency to accelerate the process for earlier patient access is accompanied by the need to save on large-scale batch runs during the developmental phase. While these savings are crucial in reducing the time and high costs required for batch production, they lead to data scarcity, posing a significant challenge for CMC (Chemistry, Manufacturing, and Controls) bioprocess development. A critical aspect of biopharmaceutical development is establishing comprehensive process knowledge based on the determination of the variability of critical quality attributes (CQAs). Understanding and accounting for process variability at defined input-parameter set points is fundamental. It ensures regulatory compliance, product quality, process robustness, cost efficiency, and effective risk management. Providing accurate process variability estimates is essential for statistical analysis during the developmental phase, ultimately determining the specifications of future products.
However, traditional statistical methods for estimating process variability often fall short when data are limited [
1,
2], leading to biased or inaccurate variance estimates. Currently, no alternative statistical approach exists that enables the meta-analysis of hierarchical process data as a predictive model for new products while accounting for product variance as a random effect. This study primarily focuses on addressing this issue by demonstrating a novel methodological approach designed to handle small sample sizes in bioprocess development. It provides a description of the hierarchical model, which is designed to be applicable to a multilevel data structure arising from batch and cycle (i.e., measures per batch) dependencies.
Data scarcity often presents significant challenges, highlighting the need for innovative statistical methods to enhance reliability [
3]. This issue is also prevalent in clinical statistics. Borrowing information from historical data is a currently discussed topic that is particularly beneficial in scenarios with limited patient numbers [
4]; in the context of early-phase clinical trials; or in rare diseases where patient numbers are limited and, thus, only small sample sizes are available [
5]. This scarcity can lead to unreliable estimates and hinder the ability to draw meaningful conclusions. Empirical Bayesian borrowing methods address this issue by incorporating information from observed data into the analysis as a prior, allowing for more robust statistical inferences. The concept of Meta-Analytic-Predictive (MAP) priors [
6,
7,
8] is a key tool in this context. MAP prior approaches can inform a current study by leveraging data from multiple historical studies. MAP priors combine data to create a prior distribution that reflects accumulated knowledge. This prior distribution represents the initial belief about a parameter based on meta-analytic data (e.g., from previous products) before observing any current data. This prior belief is then updated with new data (e.g., from a new product), supporting the analysis as an informative prior. In this way, Bayesian borrowing techniques can lead to more accurate statistical inferences [
7]. They can improve the reliability of clinical trial outcomes, especially when historical and current data are similar, such as clinical control group data, leading to better-informed decisions. Furthermore, an empirical Bayesian prior approach (e.g., power prior approach [
9]) allows for a more flexible and data-driven way to determine how much weight should be given to historical information from previous projects, ensuring that the prior information is neither overemphasized nor underutilized.
Particularly common in the context of clinical drug development, meta-analysis methodologies allow for the combination of results from multiple clinical trials to draw more robust conclusions. A critical assumption in meta-analysis is the homogeneity of effect sizes, which underpins statistical testing for between-study heterogeneity. This testing assesses whether the included studies are sufficiently similar to be combined. Various methodological approaches exist to estimate the variance between studies. Variability among studies is often captured by random-effect modeling, incorporating a random-effect variance component (
), assuming that all studies are sampled from the same population. Estimates of heterogeneity variance and its confidence interval can be provided through several methods, including the DerSimonian and Laird method or variants such as the Paule–Mandel method and approaches by Hartung and Makambi and Sidik and Jonkman. Langan et al. [
10,
11] and Veroniki et al. [
12] provided a comprehensive review of these methods. However, such methods perform poor on a small number of studies [
13]. Bayesian approaches, as suggested by Bodnar et al. [
14], offer a promising alternative, providing more accurate interval estimates.
In a meta-analysis setting, when pooling data with clustered samples, such as those from the same groups—for instance, studies and treatment groups within the context of clinical trials analog to products and batches within the context of CMC manufacturing—it is crucial to account for these dependencies. Ignoring such dependencies can lead to flawed inferences due to the underestimation of standard errors, as noted in [
1,
2]. To address this, a nested variance model can be employed, allowing for the estimation of variance components at each level within the hierarchy [
15]. This approach effectively manages heteroscedasticity and ensures more accurate results. Such a nested multilevel model design considers variances both within groups and between groups as random effects, thereby partitioning the total variance and offering insights into the contribution of each hierarchical level to the overall variability. Nested random-effect models are powerful tools for understanding hierarchical data structures. However, they do not provide variance estimates across groups, which limits their use as predictive models. Assuming that all groups have the same variance (homoscedasticity), the model is essentially averaging the variances, which can obscure the true variability when violating this assumption [
16]. The hierarchical nature of these models can lead to either wider or narrower confidence intervals for variances due to the additional levels of uncertainty, i.e., less precise estimates of the variance components at multiple levels (within-group and between-group variability).
Location-scale models [
17,
18,
19,
20] offer a robust framework for understanding and modeling data variability. In the context of meta-analysis, location-scale models are particularly valuable, as they account for variability both within and between groups. These models are especially useful when data exhibit differences in both location (mean) and scale (variance) across groups. The location parameter represents the central tendency, while the scale parameter captures dispersion or variability. By estimating these parameters, location-scale models effectively manage heterogeneity, providing a probability distribution for the variance of a response variable. This approach is well-suited to the hierarchical nature of meta-analytic data, where individual studies (or batches in bioprocess development) may have different mean values and variability.
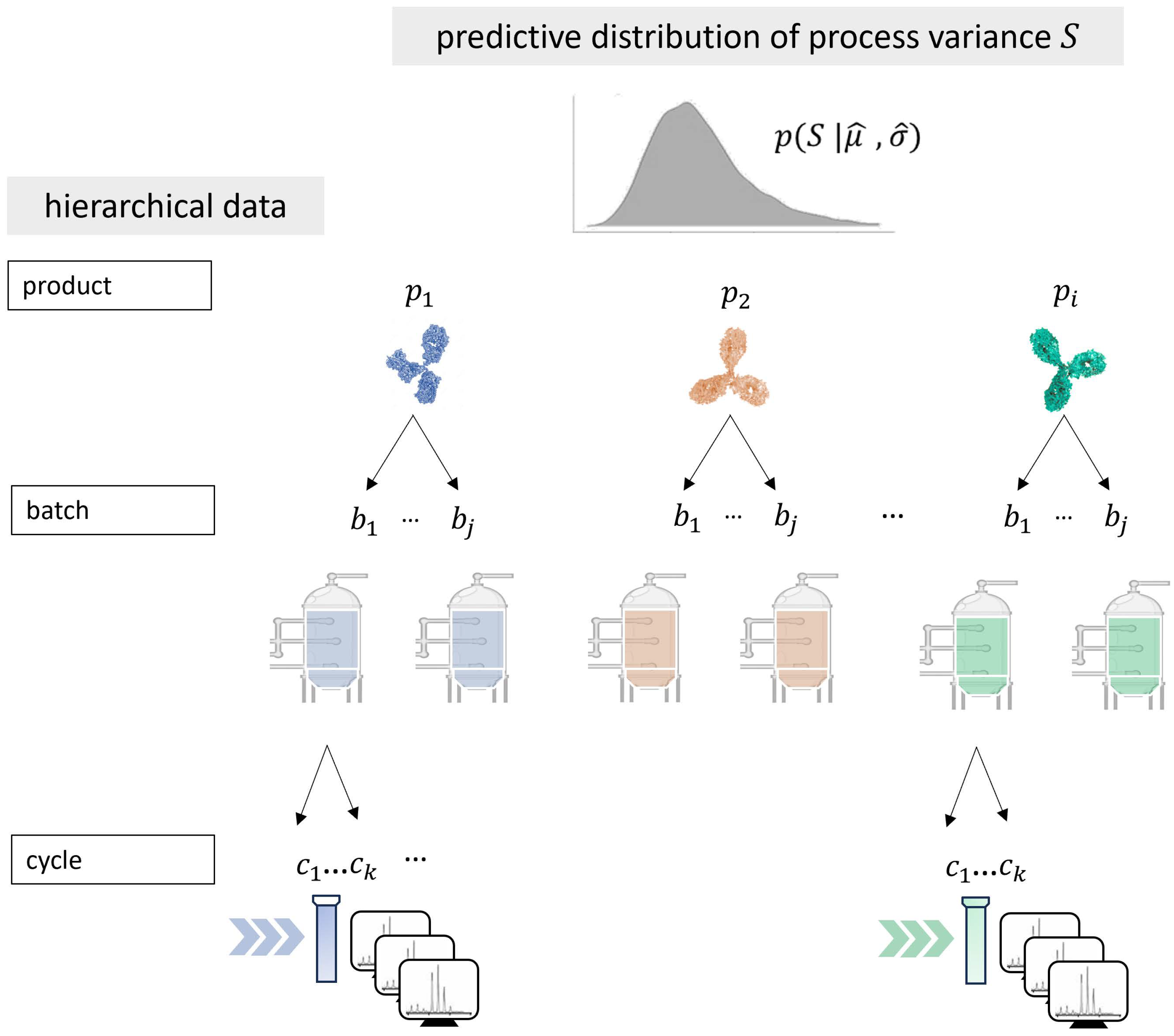
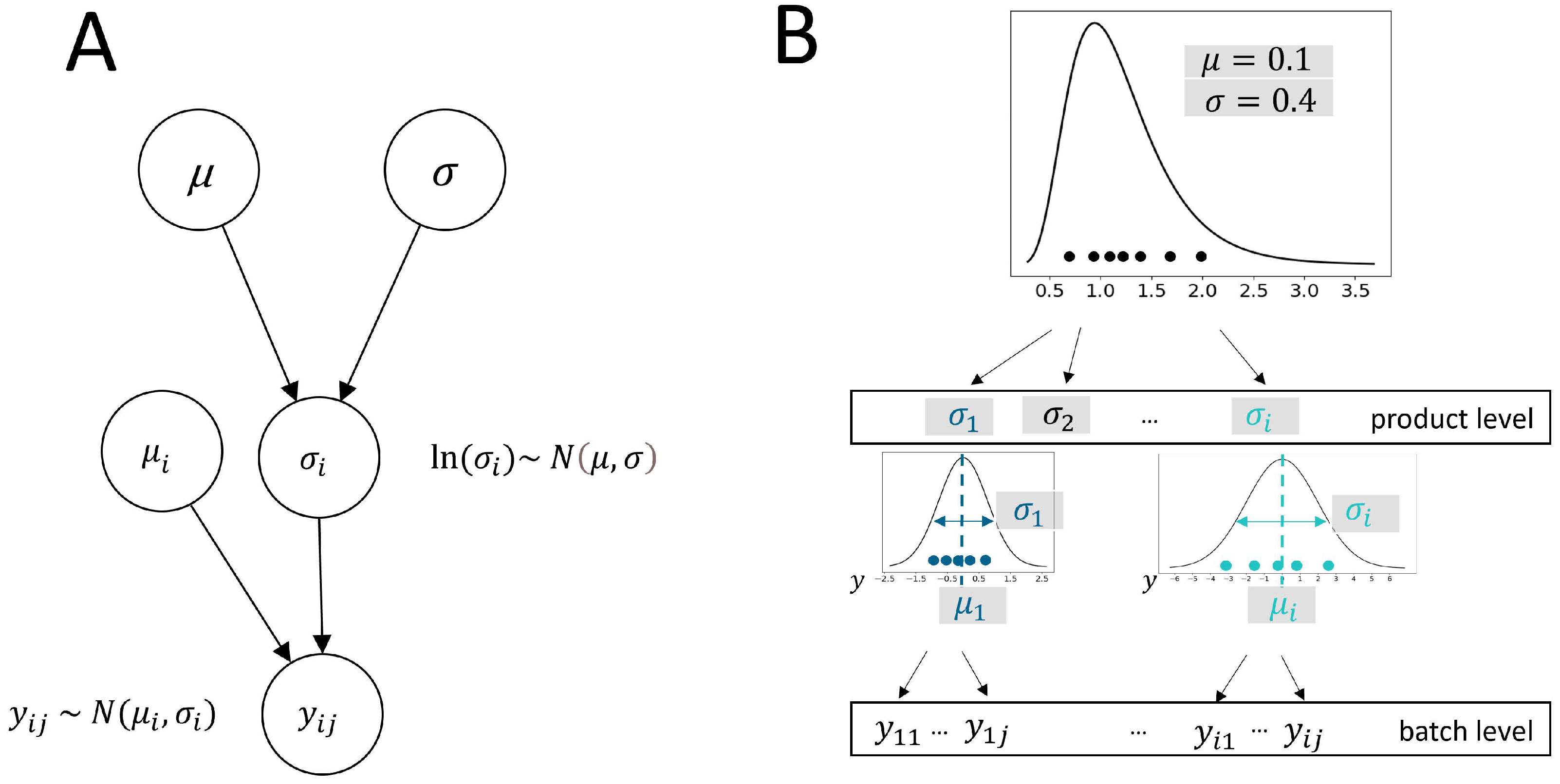
This integration of location-scale modeling assumptions within a new multilevel meta-analysis model is seen to have great potential in the development and manufacturing of biopharmaceuticals to enhance the ability to draw meaningful conclusions from small sample sizes. It allows for the pooling of information across processes being developed on a comparable platform. A model that appropriately considers the data hierarchy can thereby improve the robustness of statistical inferences to process variance estimated based on limited data. In this paper, we present a Bayesian hierarchical model that extends the principles of location-scale modeling to accommodate multilevel data structures and allows for the prediction of population variance distribution. The model is able to describe heterogeneity of variances across products as random effects considering both between-batch variance (similar to between-study variance in meta-analysis) and within-batch variance. In this way, the total product variance, as the sum of both variance components, is described as originating from a lognormal distribution.
This has significant relevance for biopharmaceutical process development.
Currently, there is no other robust statistical method that can fully address these challenges of small sample sizes, making this an ongoing area of investigation. By incorporating both location and scale parameters, our model can better capture the inherent variability in biopharmaceuticalplatform processes, leading to more reliable and precise estimates and providing a more accurate representation of the data. The general model structure is introduced, facilitating Bayesian inference on model parameters. In the presented simulation study, we systematically examined the impact of dataset sample sizes and ratios of variance components. By doing so, we validated the model and demonstrated the robustness and accuracy of model predictions. By leveraging Bayesian hierarchical modeling, this approach offers the novel advantage of allowing for meta-analytic study of variance of CQA measures. The resulting model prediction is proposed to serve as a meta-analytic predictive prior for new products, informing early phases of development where only a few large-scale runs are available. In this way, the method aims to enhance the efficiency of bioprocess development, enabling the faster delivery of new therapies to patients while ensuring quality and efficacy. Thus, it provides a promising solution to the industry’s pressing need for cost-effective and timely data analysis.
4. Discussion
In this study, we introduced a novel Bayesian hierarchical model designed to estimate process variability in biopharmaceutical data. This model framework facilitates the analysis of variances within a meta-analysis, assuming a global distribution at the population level. We developed a multilevel model that employs Bayesian statistical inference to derive population parameters, describing platform process variability across historical products. The Bayesian framework is particularly suitable for our objective, as it is highly effective in characterizing variability from limited data [
25].
Our approach expands traditional meta-analysis approaches used for between-study variance estimation in clinical trials. The modeling framework shares similarities with clinical trial meta-analysis, where heterogeneity (
), as a scale parameter, relates to between-study differences [
26], which can be estimated and, thus, inform future predicted variability. This can be seen as analogous to the product-to-product variability within our CMC biopharmaceutical application case. However, our proposed hierarchical model provides a predictive distribution of variances, from which prediction intervals can be directly derived. Unlike conventional methods that often assume the homogeneity of effect sizes (i.e., the magnitude of the difference between treatment and control) and, thus, comparable variability between groups (for a summary and comparison of estimators, see Veroniki et al. [
12]), our random-effect model accounts for heteroscedasticity and dependencies between measures of the same batches. This adds an additional hierarchical level that accounts for within-batch variance. The multilevel structure could be modeled within frequentist hierarchical mixed-effect models [
27]. These approaches are particularly useful when dealing with nested data but do not inherently provide a predictive distribution in the way Bayesian models such as our random-effect model do. Furthermore, for our intended statistical model application and purpose, a significant advantage of the Bayesian approach is seen in its ability to integrate the predicted distribution of process variance directly as prior information. Bayesian non-parametric methods, such as Dirichlet process mixtures or Gaussian process regression [
28,
29], can account for variability across groups. While they share similarities with random-effect models, their advantage lies in their ability to grow in complexity by adapting the flexible number of parameters. This feature is particularly useful when the number of groups is unknown, although it is not the case in our application. Similar to our newly proposed approach that provides a hierarchical framework, other Bayesian random-effect models, such as network meta-analysis [
30], also account for heterogeneity between groups. These model may be applicable to the hierarchical data in our application scenario. However, these models do not share the same objective as ours and are more readily used for determining effect sizes rather than providing predictive variance distribution for future products.
In comparison to these random-effect models that focus on between-group variance, our approach differs in that it includes the total variances of products as random effects. The model enables prediction of cross-product variance, making it applicable to new drug products. This represents a real advantage of our random-effect model of variance over fixed-effect models, which only provide estimates for individual products.
Our newly developed method shares foundational similarities with location-scale modeling [
17,
18,
19,
20,
31].
Importantly, our approach extends existing models to handle hierarchical data structures. Our simulation study demonstrates that the model can manage both single-cycle and multi-cycle data—and, crucially for real-case meta-analysis, a mixture of both. This introduces a novel meta-analysis approach, broadening the applicability of location-scale models to multilevel meta-analysis within the biopharmaceutical context. This new approach is highly relevant because current CMC statistics face the challenge of scarce data, which introduces uncertainty to variability estimates of CQAs measured across unit operations in both upstream and downstream processes. An important aspect frequently discussed in the context of clinical statistics is the number of samples (in our case, the number of products and number of batches). This is particularly crucial when dealing with scarce data, as in our application. In such cases, a point estimate for the variance is affected by high uncertainty. To account for this uncertainty, several methods have been proposed [
32,
33,
34], which provide confidence intervals and facilitate variance interpretation [
35]. Unlike our approach, these methods do not consider the variance itself as a random effect.
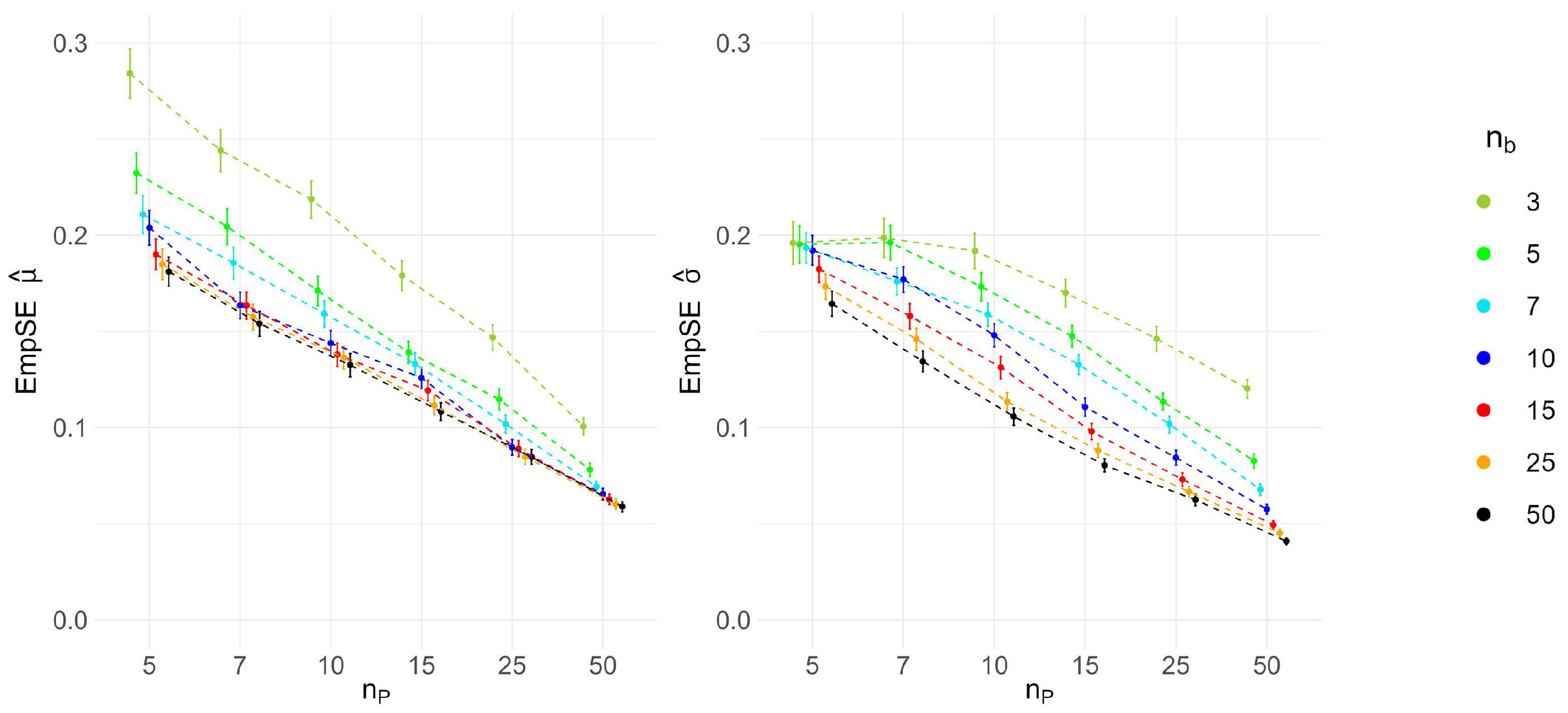
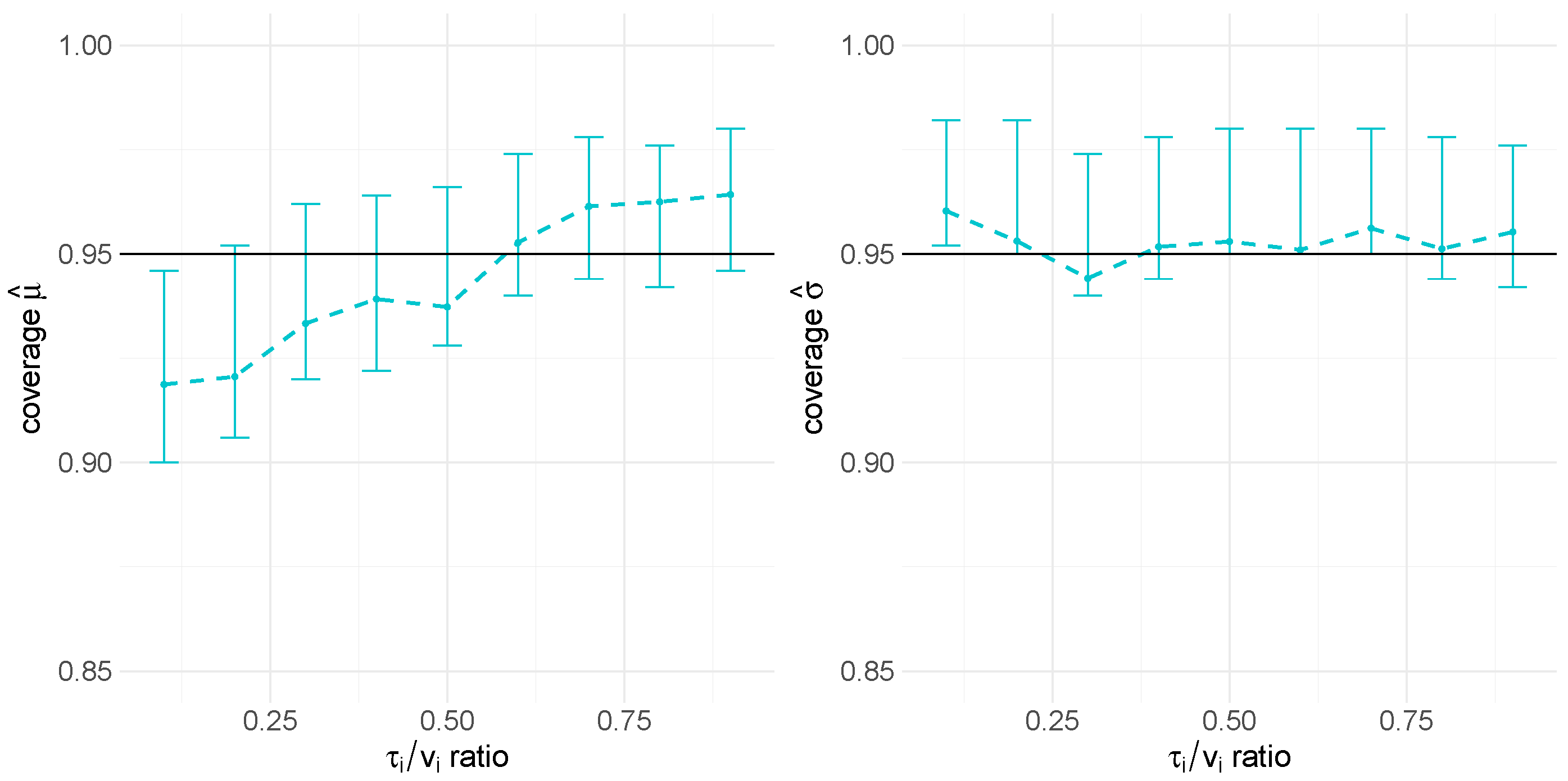
The simulation study results for the multilevel data revealed a bias in the mean parameter estimate and the coverage of the CI for high between-batch variance (i.e., low ratio). Although the bootstrap intervals for the coverage CI include the target 95% value, the resulting posterior mean of the mean parameter of the lognormal distribution is increased. However, this overestimation bias is not reflected within the predictive distribution. Therefore, the results still support the validity of our random-variance model for multilevel data.
In the simulation study, we generated data based on the model, assuming as the total variance of products follow a lognormal distribution. As an alternative, the variance components at the level of batches () and cycles () could also be assumed to follow this non-negative distribution. However, we are not interested in their distributions because only the distribution of the total variance is of interest for biopharmaceutical applications. Thus, we do not demand them to be random but fixed effects, eliminating the need for additional distributional assumptions for both parameters, as well as their correlation. This means that for our simulation, as well as real-world data, no test for correlation is necessary.
To assess the robustness of the model inference, we can compare the quantiles of the mean posterior estimates of from the hierarchical model to the theoretical quantiles of a lognormal distribution using a QQ plot. A close alignment of the points along the reference line indicates that the assumption of being lognormally distributed is well-supported by the simulation study results. In real data analysis, it is crucial to visually check the plausibility of the population distribution assumption to verify whether the products are suitable for evaluation together in the meta-analysis. To check the plausibility of the assumption that is lognormally distributed, standard deviations calculated for individual products can be evaluated using a QQ plot. This visual confirmation is important in real data analysis before evaluating model inference results and can support the model’s underlying assumption. Within a real-world data analysis, it would be beneficial to use goodness-of-fit tests, such as the Kolmogorov–Smirnov test or the Anderson–Darling test, to further validate the distributional assumption.
Another assumption made by the model is that variance components are uncorrelated. This assumption is plausible within the context of CMC in biopharmaceutical processes. Batch variability in biopharmaceutical manufacturing can arise from several sources, particularly differences in seed trains. When comparing different seed trains, variability can stem from factors such as raw materials, medium composition, culture conditions, and process parameter settings. Although these factors are maintained within strict and controlled ranges, small differences can lead to variations in CQA measurements. Within-batch variance, which can be assessed by measuring multiple cycles per batch, has different sources. It encompasses the measurement of various parts of the overall material and pure analytical variance, such as equipment performance and operator techniques.
As mentioned before, the proposed model has several practical implications for CMC biopharmaceutical drug development and manufacturing. Upstream and downstream processing of biopharmaceuticals involves multiple steps, including cascaded cell cultivation, harvest, and purification. During these unit operations, CQAs define the required product quality. Variance estimation for analytical measurements of a CQA at a corresponding operating unit is typically performed and used, for example, to derive specification limits. Our simulation study’s prediction interval coverage results demonstrate that the model provides valid results for both single-cycle and multi-cycle data, as well as for different ratios of variance components. By incorporating historical data, the Bayesian model can be applied, for instance, within an MAP prior approach, where the predicted distribution of historical variances could be included as prior knowledge for inference on new data. Integrating the predictive distribution in future statistical analyses would appropriately account for differences between products while borrowing information to infer variance parameters for individual products. This way, the approach allows for more accurate variance estimates of CQAs in platform processes, which is crucial for quality control and process optimization. Establishing a comprehensive methodological workflow that applies the proposed meta-analysis framework can provide significant support for future product development. Leveraging existing platform process data minimizes the need for extensive new experiments, saving both time and resources and thereby promoting sustainability. More accurate variance estimates enable better-informed decisions during the early phases of process development, potentially accelerating the overall development timeline. Variance estimation can also be a useful tool to evaluate models, such as for the design of experiments, providing references for expected variances and ensuring process modeling quality.
Our simulation studies covered a range of sample sizes. However, real-world data may present additional complexities. In a real-data meta-analysis, products should be critically assessed with regard to their similarity (e.g., molecule types) beforehand. This pre-assessment of a dataset prevents the distortion of variance estimation due to outliers that do not reflect the true variance of the platform process.
The model assumes that the data follow a lognormal distribution, which may not always hold true in practice. Future research could explore the applicability of other distributions.
In future studies, various methodological approaches to applying the model could be explored. The model can directly predict process variance across products based on meta-analytic data. The simulation study introduced and explained the model as a hierarchical random-variance model, which is highly valuable for CMC in biopharmaceutical applications. Additionally, the inferred distribution could be employed as an empirical MAP prior, as mentioned in the
Section 1. This allows for a detailed comparison of different Bayesian borrowing approaches, which are well-known in clinical statistics [
36]. For biopharmaceutical applications, the new model approach presented here can support predictions for process variance in individual unit operations across the production chain of a drug product. A further direction for future work would be the extension of our hierarchical random-variance model to time-series data. Location-scale models of hierarchical time series data would bring additional benefits for upstream process development and manufacturing.
The work presented in this study is an important step in these directions. By leveraging our historical platform knowledge, we can highlight the value of our data in advancing CMC’s future biopharmaceutical drug development and ensuring quality for patients.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}