1. Introduction
Respiratory syncytial virus (RSV) stands as a prominent contributor to lower respiratory tract ailments in both young children and the elderly [
1]. The initial identification of RSV traces back to 1955, when it was first isolated from chimpanzees displaying respiratory symptoms at the Walter Reed Army Institute of Research in the United States [
2]. Over subsequent years, the virus was also discovered in infants who suffered from severe lower respiratory illnesses [
3,
4]. Since that time, RSV has become known as a widespread pathogen, affecting almost every child by the age of two, with around half of them experiencing two infections within this period. [
5]. The primary modes of transmission involve respiratory droplets released during coughs or sneezes, as well as direct contact with contaminated surfaces. Infants, young children, and older adults, particularly those with chronic medical conditions, face an elevated risk of severe illness due to RSV infection [
6,
7]. People infected with RSV are usually contagious for a period ranging from 3 to 8 days, with the potential to spread the virus a day or two before showing any symptoms [
8]. However, some infants and individuals with weakened immune systems can remain contagious even after their symptoms have resolved, occasionally for as long as four weeks. [
8]. The typical symptoms of an RSV infection include a runny nose, decreased appetite, coughing, sneezing, fever, and wheezing, which tend to develop gradually rather than all at once [
9]. RSV is the most common cause of bronchiolitis and pneumonia in children under one year old [
7]. The CDC estimates that RSV causes approximately 58,000 to 80,000 hospitalizations and 100 to 300 deaths among children under five annually. Additionally, it results in 60,000 to 160,000 hospitalizations and 6000 to 10,000 deaths each year among adults aged 65 and older [
7]. RSV, which circulates during the winter months alongside influenza (flu) and other respiratory viruses, is frequently misdiagnosed due to its similar symptoms. Like the flu, its prevalence peaks between November and May [
10].
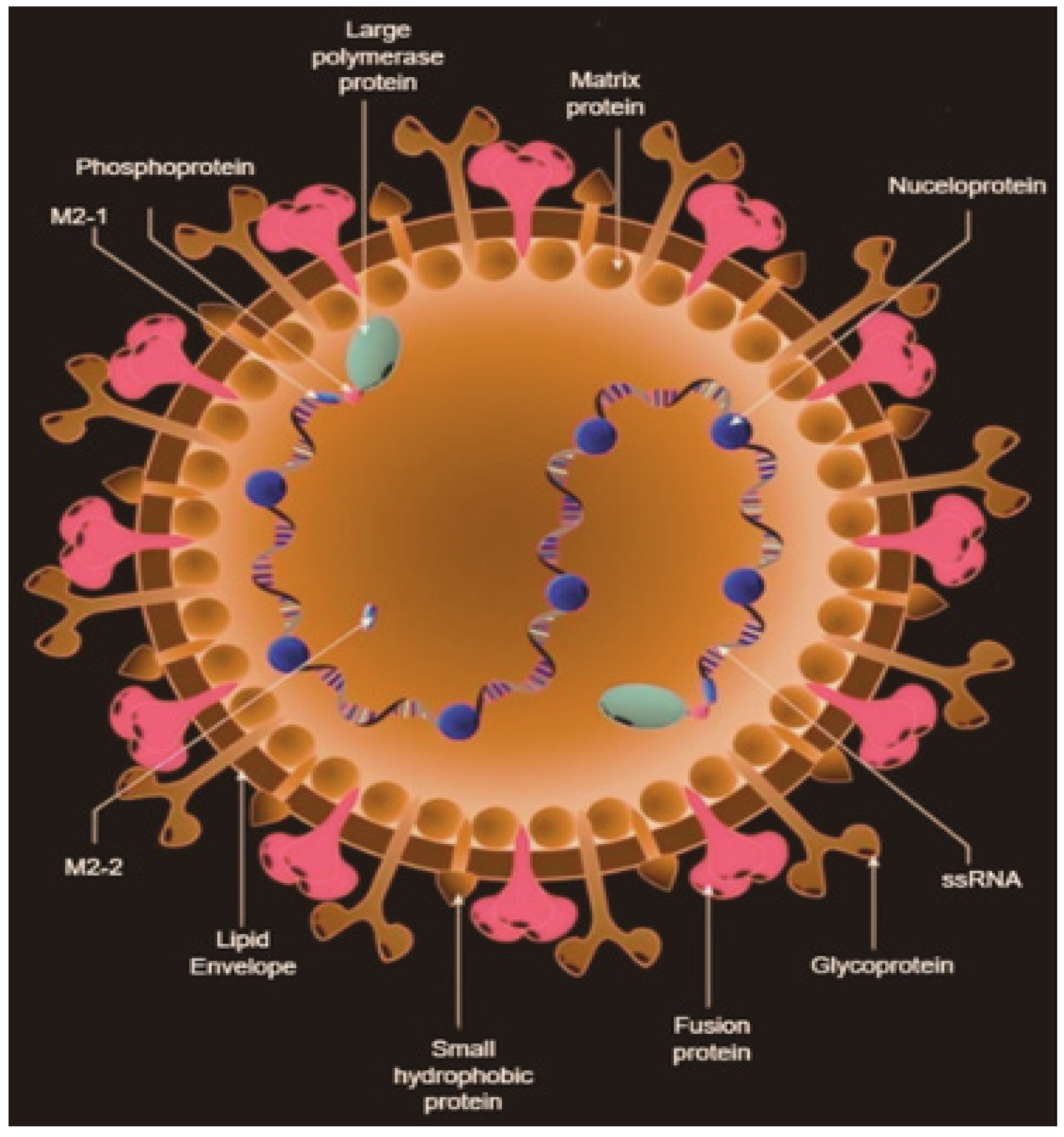
RSV is categorized as a filamentous enveloped virus and is part of the
Orthopneumovirus genus within the
Pneumoviridae family, under the order
Mononegavirales [
1]. This virus is characterized by its genetic structure, which consists of a single-stranded RNA genome with a negative sense. This genome includes 11 proteins encoded by a 15.2-kilobase (kb) RSV genome. Unlike influenza, RSV possesses a non-segmented genome, which means it lacks the capacity to re-assort genome segments. As a result, RSV cannot undergo the genetic rearrangements known as antigenic shifts, which can lead to major pandemics [
11]. RSV particles come in various shapes, including both spherical and filamentous forms of different sizes [
12]. These virions have three surface proteins: F, G, and SH (small hydrophobic), as shown in
Figure 1 [
13]. The G and F proteins are crucial for virion attachment and fusion, binding to specific carbohydrate structures known as GAGs and RhoA, respectively [
14,
15]. Once fusion takes place, the virion releases its nucleocapsid into the cytosol, permitting the RNA to enter the host cell. The M2 mRNA contains two overlapping open reading frames (ORFs) that code for M2-1 and M2-2. The M2-2 gene regulates the shift from transcription to genomic RNA production [
16]. The large (L) protein functions as a viral RNA-dependent RNA polymerase, encompassing multiple enzyme activities essential for RSV replication. This protein enters the genome, facilitating mRNA transcription. During replication, a complete positive-sense RNA complement of the genome called the antigenome is produced and serves as a template for further replication. Throughout this process, the N protein encapsulates the RNA, protecting it from degradation. The M protein plays a crucial role in coordinating the assembly of envelope proteins with nucleocapsid proteins (N, P, and M2-1). It also aids in the budding of new immature virions, a process that uses the host cell membrane. In filamentous virions, a helical arrangement of M (matrix) proteins is present, which is critical for forming infectious filamentous particles [
17].
As shown in
Figure 1, the RNA genome of RSV includes 10 genes that encode a total of 11 proteins. These proteins include two nonstructural proteins (NS1 and NS2). Additionally, there are four envelope proteins: the attachment glycoprotein (G), the fusion protein (F), the matrix protein (M), and the small hydrophobic protein (SH). Moreover, there are five ribonucleocapsid proteins: the nucleoprotein (N), phosphoprotein (P), large RNA polymerase (L), M2-1 (a transcription antiterminator that binds zinc), and M2-2 (a regulatory factor involved in balancing RNA replication and transcription) [
18]. In vaccine development, particular focus is given to the F protein. This is due to its presence on the outer envelope of the RSV virion and its high conservation across different RSV strains, making it a promising target for vaccine development. The F protein exists in two forms, prefusion and postfusion, with the prefusion form being less stable but more immunodominant compared to the postfusion form [
18].
To prevent disease outbreaks effectively, there is an urgent need to develop a safe and effective RSV vaccine that can induce immunological memory without causing immune-related complications following natural RSV infections [
19]. Current vaccine development efforts have emphasized whole-organism vaccines, including live attenuated and inactivated types. However, these vaccines can be costly to produce, require the cultivation of the infectious agent, and may cause vaccine-related illnesses in recipients [
20]. Additionally, they may not be suitable for individuals with compromised immune systems and require precise temperature control for storage [
21]. As a result, there has been a shift towards developing peptide-based vaccines (PBVs). PBVs involve identifying and chemically synthesizing immunodominant peptides, known as T-cell epitopes (TCEs), which can elicit specific immune responses against the pathogen [
22]. The design of PBVs focuses on removing unnecessary antigenic components, concentrating only on protein sections capable of triggering an immune response [
23,
24]. PBVs offer several advantages over traditional vaccines, including fewer side effects, simpler manufacturing processes, the absence of whole pathogen elements, increased specificity, greater stability, sustainability, and shorter production timelines [
25]. Despite these significant advantages, PBVs have received less attention, and their potential to enhance vaccine safety and immunogenicity remains largely unexplored [
26,
27].
It is important to emphasize the vital role of T cells in adaptive immunity, as they aid in various immune system functions and significantly contribute to the control, clearance, and protection against most viral infections. [
28]. Notably, CD8+ T cells are pivotal in the context of RSV pathogenesis, and there is a suggestion that RSV vaccines capable of inducing both antibodies and CD8+ T cells may prove effective [
29]. The consideration of vaccines that trigger CD8+ T-cell responses against both cancer and viruses is a promising avenue in vaccine design [
30,
31], underscoring the idea that T cells are well equipped to address evolving viral variants [
32,
33]. Identifying these immunodominant TCEs for PBV design through wet-lab experiments is challenging, costly, and time-consuming. However, the application of machine learning (ML) techniques can enable the accurate prediction of these epitopes, expediting vaccine development and making it more cost-effective compared to traditional wet-lab methods [
34]. This study presents a novel method for predicting the TCEs of RSV using a hybrid ML technique that leverages the physicochemical properties of peptides. The identified epitopes could be utilized as candidates in the development of PBVs against the RSV pathogen. The proposed model aims to aid the scientific community in identifying new and immunodominant TCEs specific to RSV.
Contributions
This study makes several significant contributions. Firstly, it involved the development and testing of eight hybrid ML predictive models created through various permutations and combinations of two classification techniques, two feature weighting methods, and two feature selection strategies, all aimed at predicting the TCEs of RSV. Secondly, an innovative feature extraction technique was introduced, capable of extracting the physicochemical properties of peptides at the amino acid level. Thirdly, the study employed heuristic and greedy search techniques to identify optimal features for model training after extracting features from peptide sequences. Fourthly, the research primarily focused on achieving high accuracy in TCE prediction, and the proposed hybrid techniques demonstrated promising results in terms of accuracy. These models were thoroughly evaluated using multiple parameters, including area under the curve (AUC), sensitivity, specificity, Gini, F-score, and MCC. The findings indicate that the combination of XGBoost with chi-squared and backward search is the most accurate and reliable predictive method for TCE prediction in the context of RSV. Finally, K-fold cross-validation (KFCV) was performed, demonstrating that the proposed model is reliable and consistent for TCE predictions across all folds.
2. Related Work
Considerable research has been conducted to identify the TCEs of RSV for the design of PBVs. Chen et al. predicted T-cell epitopes in RSV F and G proteins, finding three RSV-A and two RSV-B clusters, indicating diverse immunogenic profiles. Recent epidemic strains conserved more F protein epitopes but reduced G protein epitopes. This study offers a framework for studying RSV T-cell epitope evolution, crucial for vaccine design [
35]. The study [
36] aimed to identify RSV-specific T-cell epitopes in BALB/c mice. Novel CD8 T-cell epitopes in the F and G proteins and previously unknown CD4 T-cell epitopes in P, L, M2-1, and N proteins were discovered. Longer 17-mer CD4-T-cell epitopes proved more effective in stimulating CD4-T-cell responses compared to 15-mer peptides. This work addresses the lack of defined RSV-specific T-cell epitopes, enhancing our understanding of RSV-induced disease. Another study [
37] focused on designing a potential vaccine for RSV. Using reverse vaccinology, researchers predicted 95 cytotoxic T-lymphocyte (CTL) epitopes from the RSV proteome. After extensive screening for antigenicity, allergenicity, and toxicity, 70 epitopes with desirable properties were selected. Molecular docking identified stable binding in four epitopes, validating their potential as T-cell-specific RSV antigens. This approach provides an efficient method for screening immunogenic epitopes, offering promise for vaccine development against RSV. In [
38], the authors aimed to identify CD4+ and CD8+ T-cell epitopes in C57BL/6 mice infected with RSV. Using an overlapping peptide library encompassing the RSV proteome, researchers discovered two new CD4+ and three new CD8+ T-cell epitopes within various RSV proteins. Additionally, they characterized these newly identified epitopes, including their TCR Vb expression profiles and MHC restriction. These findings will advance future research on RSV-specific T-cell responses in C57BL/6 mice. Shah et al. [
39] focused on the potential use of epitope-based vaccines against RSV, which poses a significant threat to infants and the elderly. The study specifically targeted the fusion glycoprotein of RSV (RSV-FP) due to its conservation across strains and its ability to elicit cytotoxic T-cell (CTL) responses, crucial for viral clearance. Using immunoinformatics tools, the researchers identified seven 9-mer peptides within RSV-FP that strongly bind to 17 different HLA types, exhibit 100% sequence conservancy, and are estimated to provide a 76.03% population coverage worldwide. These findings hold promise for the development of effective RSV epitope-based vaccines. In this immunoinformatics study [
40], researchers aimed to design a multi-epitope vaccine against RSV. They identified eight CD8-T-cell and three CD4-T-cell epitopes from glycoproteins F and G, considering antigenicity and binding affinity. Molecular docking confirmed strong associations with HLA alleles. Using these epitopes, a stable, non-allergenic, and antigenic multi-epitope vaccine with a cholera toxin-derived adjuvant was designed. Computational simulations indicated the vaccine’s potential to generate antibodies and effector T cells. Codon optimization and in silico cloning ensured enhanced expression in Escherichia coli. Further experimental validation is expected to confirm the vaccine’s effectiveness against RSV infections. A study [
41] aimed to examine the role of vaccine-induced CD8+ T cells in protecting against RSV. Using a peptide vaccine (TriVax) in mice, researchers discovered that it induced strong anti-RSV CD8+ cytotoxic T lymphocytes. These vaccinated mice were protected against RSV infection, airway mucin expression, and lung inflammation when challenged six days post-vaccination. While effector CD8+ T cells exhibited strong cytokine expression and provided protection, memory CD8+ T cells, elicited 42 days post-vaccination, offered partial protection with lower cytokine expression, suggesting a link between protection and CD8+ T cell cytokine levels. Another study [
42] aimed to develop a vaccine against RSV that induces long-lasting immunological memory without causing immunopathology. Researchers used live attenuated influenza vaccine (LAIV) viruses with RSV epitopes integrated into the neuraminidase or NS1 genes. These chimeric vaccines protected against both influenza and RSV without causing harmful effects. The study focused on CD4- and CD8-T-cell responses, particularly lung tissue-resident memory T-cell subsets (TRM). The RSV epitopes did not impact influenza-specific CD4 memory T cells, and both LAIV+NA/RSV and LAIV+NS/RSV vaccines induced strong RSV-specific CD8 TRM cells in the lungs. This research indicates that LAIV-based vaccines can generate robust localized T-cell immunity against foreign pathogens without compromising the vaccine’s immunogenicity. The authors of [
43] reviewed computational tools for predicting T-cell epitopes, with a particular focus on neoepitopes relevant to cancer immunotherapy. They assessed various tools based on their methodologies, data utilization, and comparative advantages and disadvantages. The authors of [
44] investigated the impact of antigen processing on epitope immunogenicity. They developed an ML model to predict proteasomal degradation scores for peptides and experimentally tested peptides with varying scores. Their findings suggest a correlation between low degradation scores and enhanced T-cell activation, highlighting the potential for improving vaccine efficacy by optimizing antigen processing. The study [
45] addressed the challenge of epitope prediction for malaria due to the unique biology and evolving sequences of the parasite. The authors proposed an ML approach to develop a Plasmodium-specific epitope predictor. They built models using various ML algorithms trained on epitope data with sequence features and physicochemical properties. Their analysis suggests a model trained with specific classifiers after preprocessing outperforms others. This research represents the first in silico attempt to benchmark Plasmodium epitopes using ML and paves the way for peptide-based predictors in malaria vaccine development. The study [
46] reviewed various in silico methods for predicting SARS-CoV-2 T-cell epitopes, highlighting the importance of T-cell responses in COVID-19. The authors compared various ML-based approaches by evaluating their ability to identify experimentally validated immunogenic epitopes. This review provides insights into the performance of different prediction methods and suggests future research directions.
5. Results and Discussion
In this section, we present the results obtained from applying various hybrid techniques to a high-dimensional dataset, which comprises 108 features extracted from RSV peptide sequences. To determine the most effective hybrid technique, a comparative analysis was performed among the different hybrid methods used in this study, based on the evaluation parameters previously outlined.
Table 5 presents the accuracies achieved by the various hybrid approaches. It is evident from
Table 5 that XGBoost (XGB) demonstrates outstanding results, consistently exceeding 93% accuracy across all scenarios.
Notably, the hybrid approach combining ChST and BST achieves the highest accuracy of 97.29% for XGBoost (XGB) models. In terms of accuracy, random forest (RF) with various feature weighting (FW) and optimal feature selection techniques shows a range of accuracy from a low of 79.23% to a high of 94.19% with IGT and HCST among the different hybrid techniques used in this study. However, when evaluating the effectiveness of a hybrid model in a multiclass problem, accuracy alone does not suffice as the sole determining factor [
60]. Other crucial parameters such as recall, specificity, precision, negative predicted value of a particular class, AUROC, and F1 score of the predictive method must also be considered. To this end,
Table 6 presents a comprehensive comparison of these parameters for the best hybrid models achieved in this study. As depicted in
Table 6, the XGB model (Model 1) in combination with the chi-squared and backward search techniques demonstrates superior results across all parameters, boasting an impressive F1 score and AUROC value of 0.98 and 0.99, respectively.
Assessing the reliability of the technique is essential to determine whether the model is susceptible to overfitting or underfitting issues. Overfitting occurs when the model excels with training data but fails to generalize to testing data, while underfitting happens when the model performs poorly on both training and testing data. To verify the reliability and consistency of the hybrid techniques used in this study, 5-fold cross-validation (5 FCV) was performed on the top three hybrid methods. The accuracies achieved by these top-performing hybrid models across different folds are shown in
Table 7, and their accuracy is plotted in
Figure 4.
6. Conclusions
In conclusion, RSV poses a significant threat to individuals across all age groups, especially infants and young children, with seasonal outbreaks typically peaking during autumn and winter months. Vaccination remains the most effective strategy for managing viral disease outbreaks [
67]. While ongoing efforts aim to develop an RSV vaccine, many current methods involve using weakened forms of the entire pathogen to trigger an immune response. In contrast, the potential B-cell vaccine (PBV) concept emphasizes the identification and synthetic creation of specific immunodominant peptides, known as T-cell epitopes (TCEs), as potential components of a vaccine. Despite the many advantages of PBVs, such as enhanced safety, immunogenicity, and cost-effectiveness, they have not received widespread attention [
68]. Computational methods provide a quicker and more economical way to identify TCEs compared to traditional laboratory techniques. In this study, we developed and assessed eight hybrid predictive ML models for forecasting the TCEs of RSV [
69]. After extracting features from peptide sequences, we used heuristic and greedy search techniques to identify the most effective features for model training. Performance evaluation using various metrics, including accuracy, sensitivity, specificity, and AUROC curve, showed that the combination of XGBoost with ChST and BST was the most accurate and reliable predictive method. Our model provides deterministic TCE prediction, unlike other methods, such as NetMHC [
70] and CTLpred [
71], which only estimate binding potential. Furthermore, our model can predict peptides of various lengths, including those longer than 9-mers, addressing a limitation of CTLpred. However, it is crucial to validate model predictions through experimental methods (in vivo and in vitro) before considering them for vaccine development [
72]. In summary, the hybrid ML techniques proposed in this study demonstrated exceptional performance and surpassed current ML methods for predicting RSV TCEs. Future research should explore additional physicochemical properties and utilize advanced ML classifiers to further improve accuracy and other metrics. Overall, using computational methods to identify potential vaccine candidates could significantly impact global health by saving lives, preventing future outbreaks, and reducing the virus’s capacity to evade immunity through genetic mutations.








{kind=link}
{kind=link}
{kind=link}
{kind=link}