



Deep Drug Discovery of Mac Domain of SARS-CoV-2 (WT) Spike Inhibitors: Using Experimental ACE2 Inhibition TR-FRET Assay, Screening, Molecular Dynamic Simulations and Free Energy Calculations


Abstract
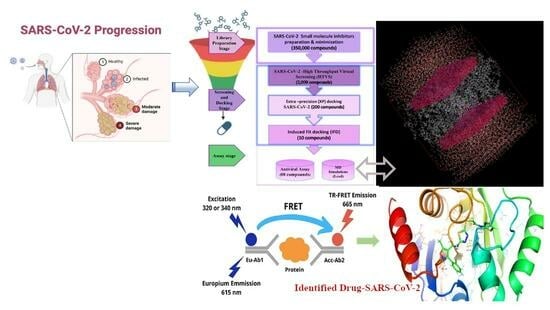
1. Introduction
2. Materials and Methods
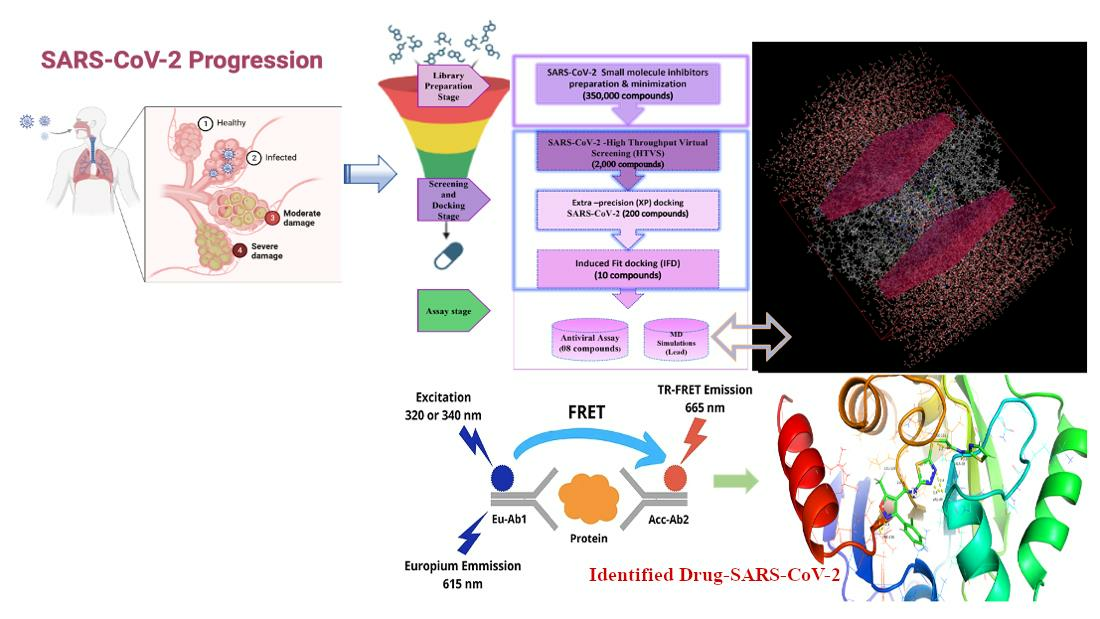
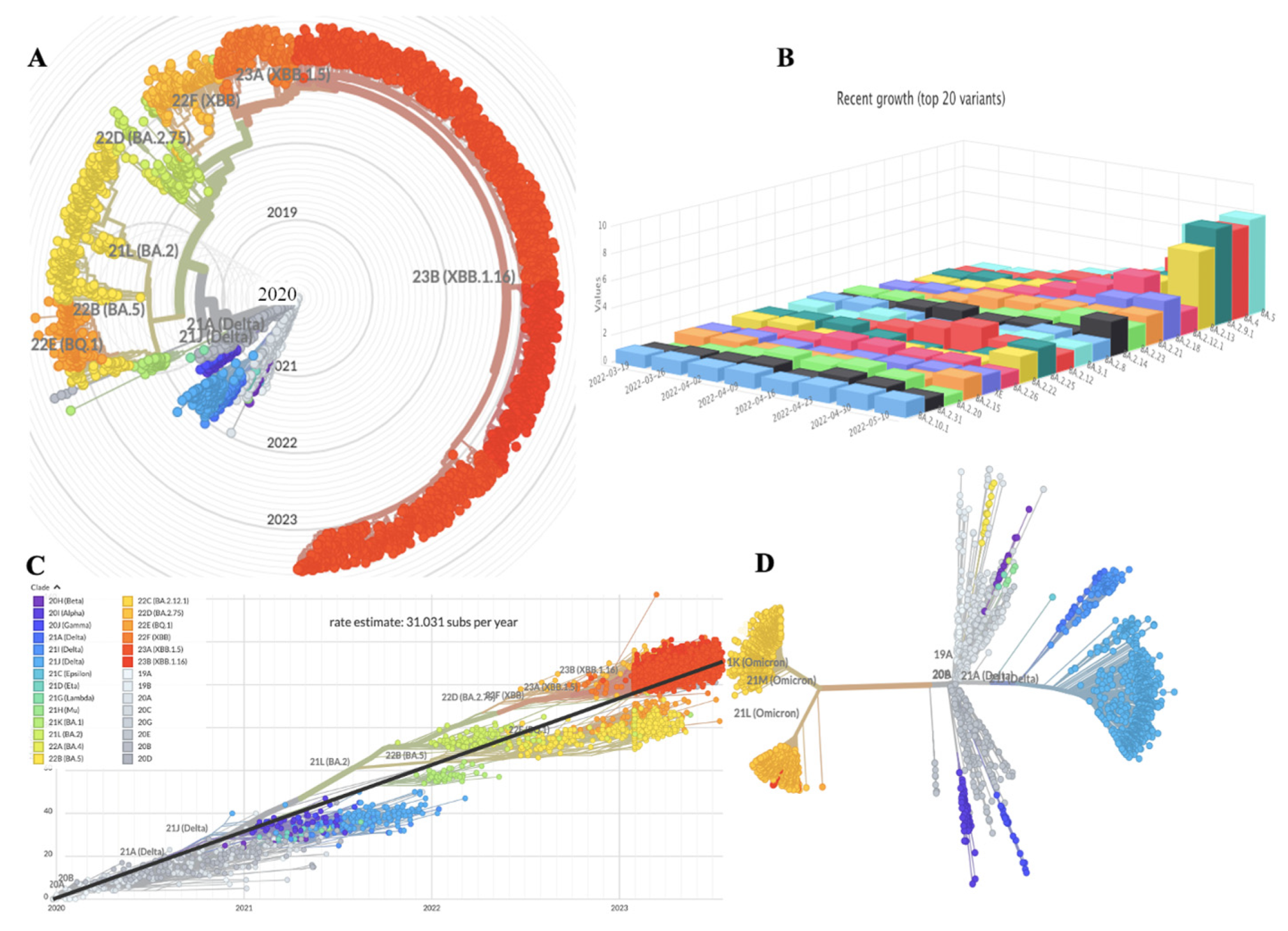
2.1. Mutational Landscape
2.2. Mac 1 Hydrolytic Activity and Cell Culture Experiments
2.3. TR-FRET (Time Resolved-Forster/Fluorescence Energy Transfer) Assay
2.4. Experimental Conditions
2.4.1. ACE2:SARS-CoV-2 Spike Inhibitor Screening
2.4.2. Assay Conditions
2.5. Data Analysis
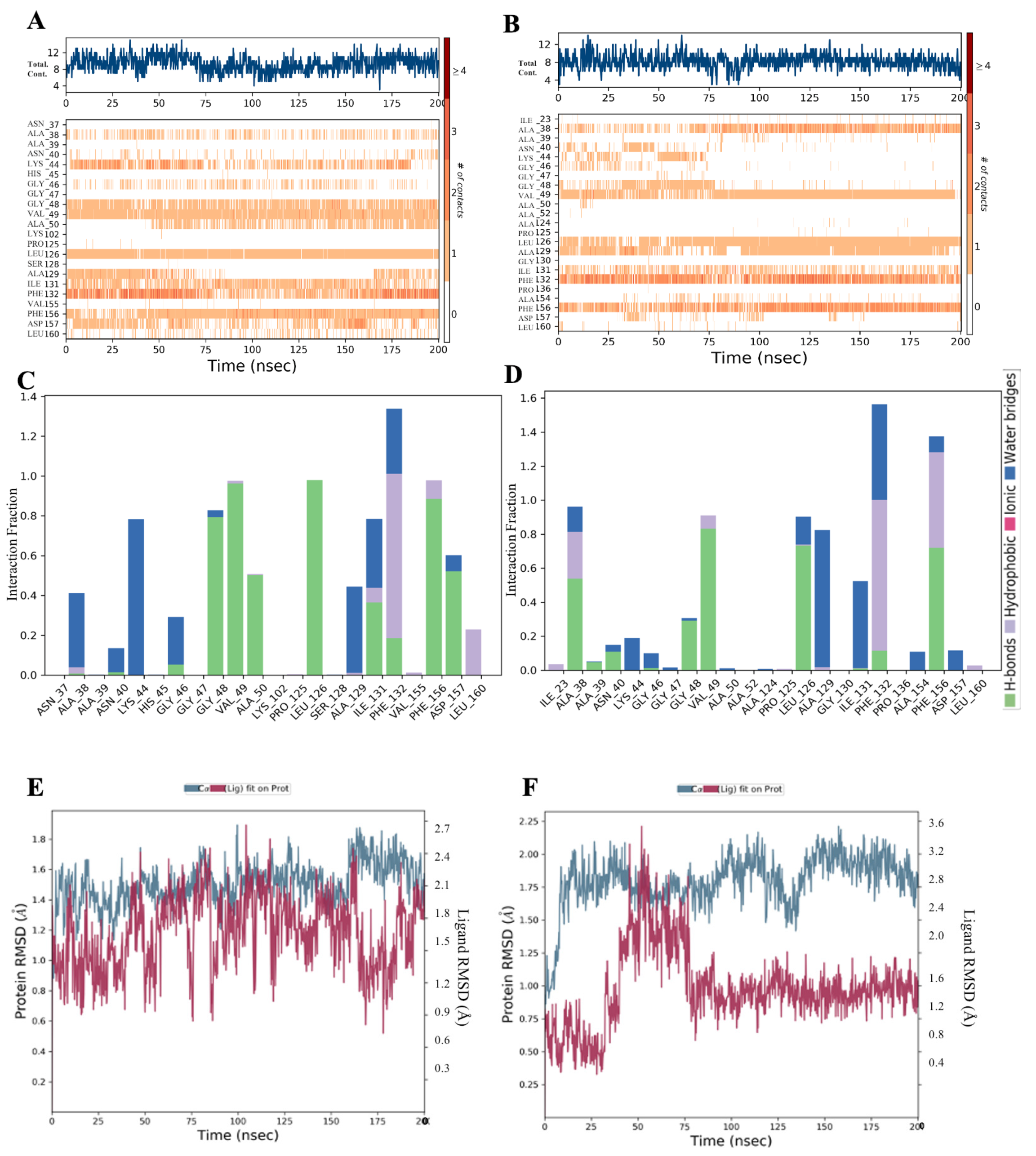
2.6. Molecular Dynamics Simulation (MDS)
2.7. Binding Affinity Analysis
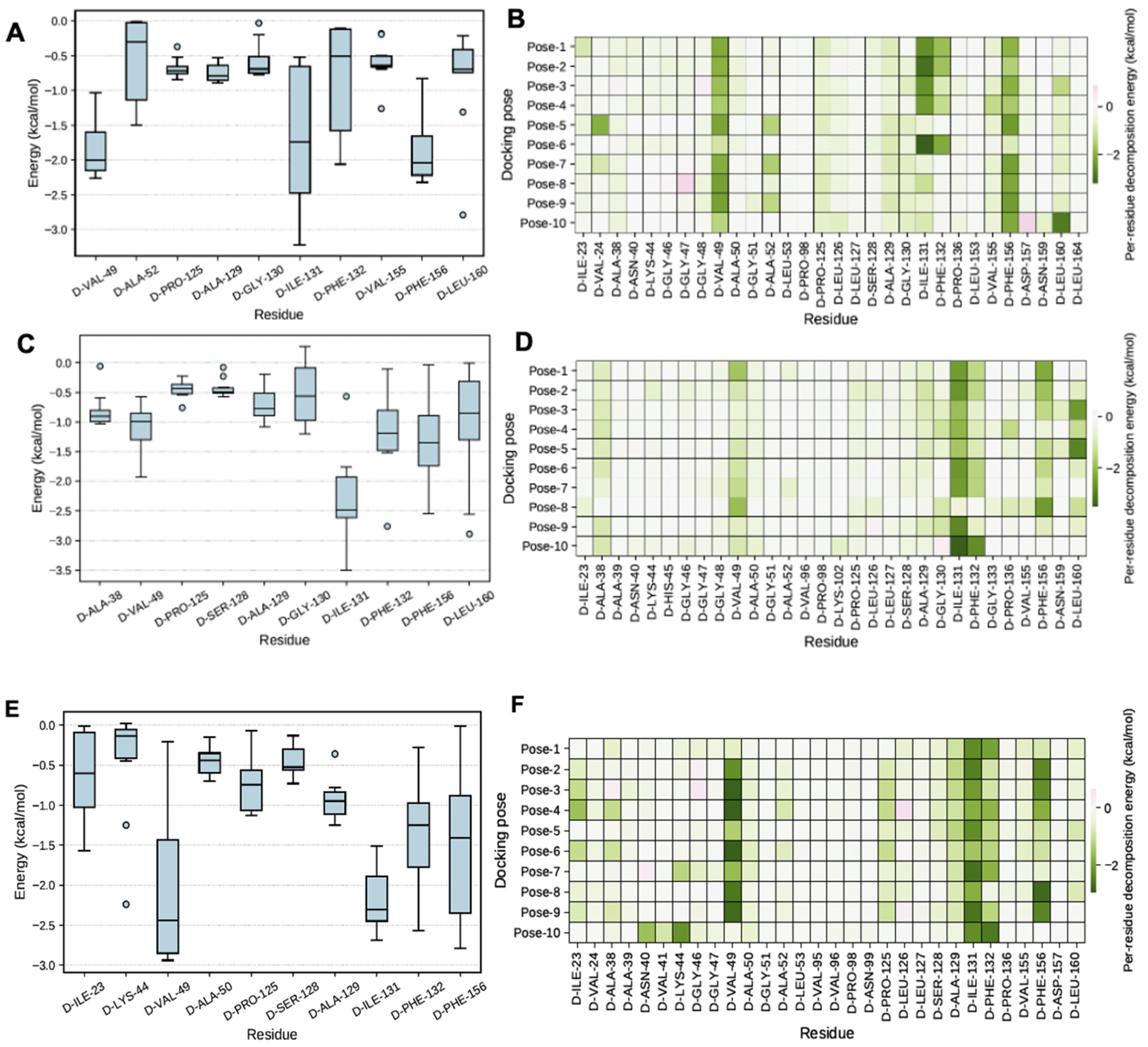
2.8. MMGBSA Calculations
3. Results
3.1. Binding of RBD of the SARS-CoV-2 Spike Protein to ACE2 Monitored by TR-FRET Assay
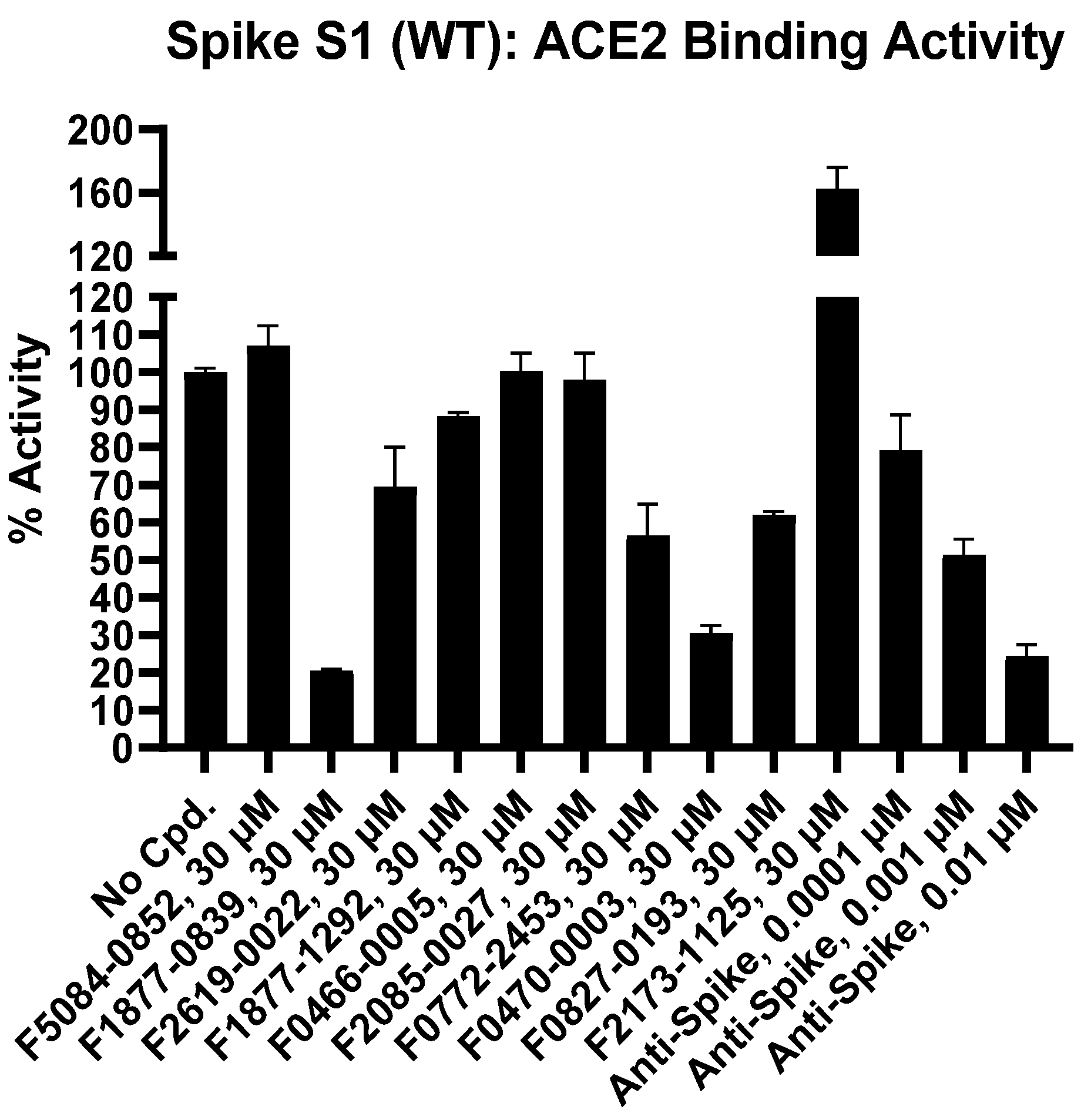
3.2. Inhibition of SARS-CoV-2 Spike RBD Binding to ACE2
4. Result and Discussion
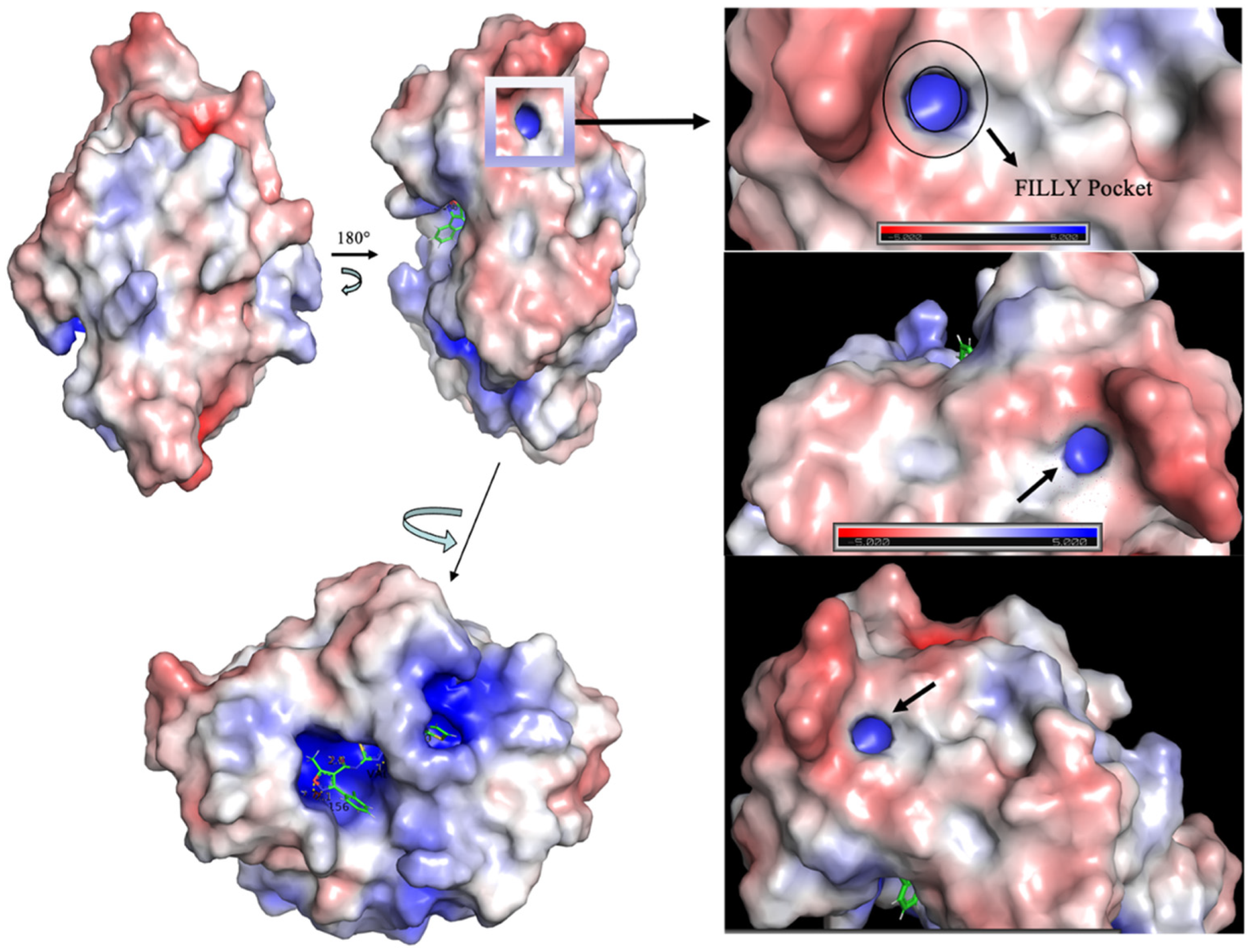
Spike Protein Has a Positive Electrostatic Surface That Promotes ACE2 Recognition
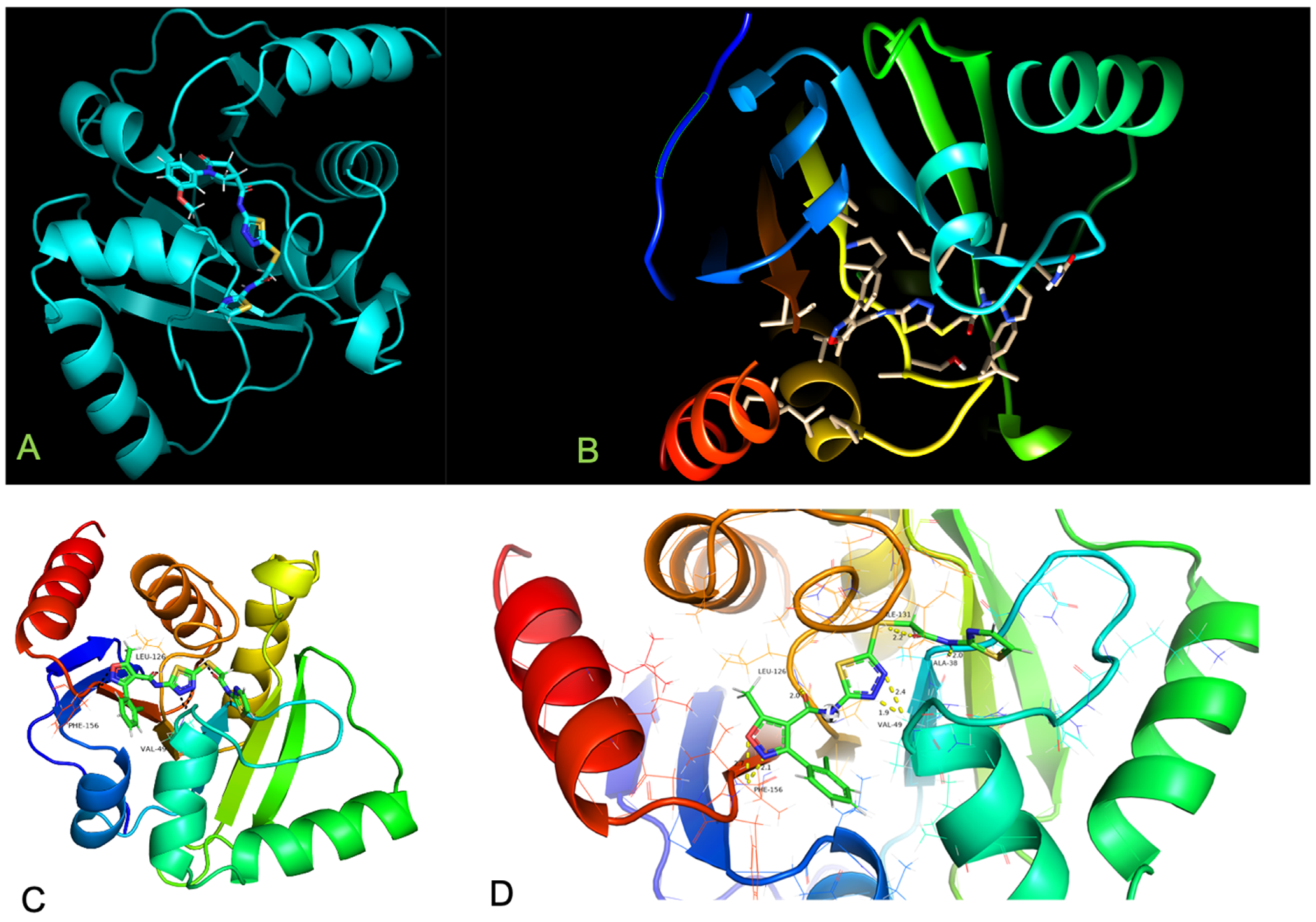
5. MMGBSA Calculations
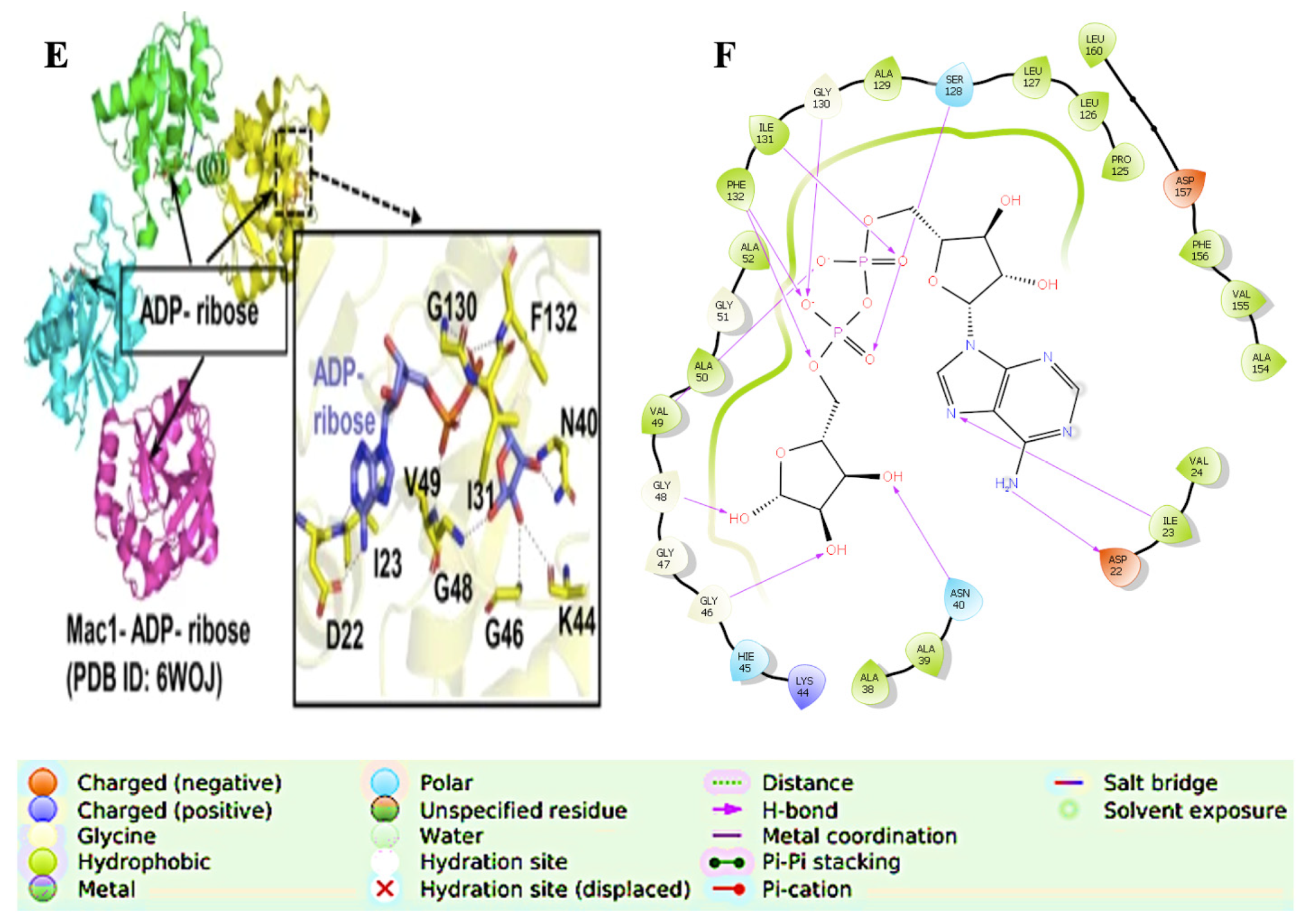
6. Catalytic Mechanism
Conformational Dynamics
7. Conclusions
8. Significance
9. Limitations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| MDS | Molecular Dynamics Simulations |
| ACE2 | Angiotensin-Converting Enzyme 2 |
| TR-FRET | Time Resolved Forster/Fluorescence energy transfer |
| HTVS | High-Throughput Virtual Screening |
| XP | Extra Precision |
| QPLD | Quantum Polarised Ligand Docking |
References
- Suzuki, R.; Yamasoba, D.; Kimura, I.; Wang, L.; Kishimoto, M.; Ito, J.; Morioka, Y.; Nao, N.; Nasser, H.; Uriu, K.; et al. Attenuated fusogenicity and pathogenicity of SARS-CoV-2 Omicron variant. Nature 2022, 603, 700–705. [Google Scholar] [CrossRef] [PubMed]
- WHO Coronavirus (COVID-19) Dashboard. Available online: https://covid19.who.int/ (accessed on 25 July 2023).
- Lv, M.; Luo, X.; Estill, J.; Liu, Y.; Ren, M.; Wang, J.; Wang, Q.; Zhao, S.; Wang, X.; Yang, S.; et al. On Behalf of the COVID-Evidence and Recommendations Working Group. Coronavirus disease (COVID-19): A scoping review. Eurosurveillance 2020, 25, 2000125. [Google Scholar] [CrossRef]
- Yang, X.; Yu, Y.; Xu, J.; Shu, H.; Xia, J.; Liu, H.; Wu, Y.; Zhang, L.; Yu, Z.; Fang, M.; et al. Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: A single-centered, retrospective, observational study. Lancet Respir. Med. 2020, 8, 475–481. [Google Scholar] [CrossRef]
- Yoshimoto, F.K. The Proteins of Severe Acute Respiratory Syndrome Coronavirus-2 (SARS CoV-2 or n-COV19), the Cause of COVID-19. Protein J. 2020, 39, 198–216. [Google Scholar] [CrossRef]
- Finkel, Y.; Mizrahi, O.; Nachshon, A.; Weingarten-Gabbay, S.; Morgenstern, D.; Yahalom-Ronen, Y.; Tamir, H.; Achdout, H.; Stein, D.; Israeli, O.; et al. The coding capacity of SARS-CoV-2. Nature 2020, 589, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.F.-W.; Kok, K.-H.; Zhu, Z.; Chu, H.; To, K.K.-W.; Yuan, S.; Yuen, K.-Y. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg. Microbes Infect. 2020, 9, 221–236. [Google Scholar] [CrossRef] [PubMed]
- Cecon, E.; Burridge, M.; Cao, L.; Carter, L.; Ravichandran, R.; Dam, J.; Jockers, R. SARS-CoV-2 spike binding to ACE2 in living cells monitored by TR-FRET. Cell Chem. Biol. 2021, 29, 74–83.e4. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef]
- Letko, M.; Marzi, A.; Munster, V. Functional assessment of cell en- try and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat. Microbiol. 2020, 5, 562–569. [Google Scholar] [CrossRef]
- Hulswit, R.J.G.; De Haan, C.A.M.; Bosch, B.J. Coronavirus Spike Protein and Tropism Changes. Adv. Virus Res. 2016, 96, 29–57. [Google Scholar] [CrossRef]
- Carabelli, A.M.; Peacock, T.P.; Thorne, L.G.; Harvey, W.T.; Hughes, J.; COVID-19 Genomics UK Consortium; Peacock, S.J.; Barclay, W.S.; de Silva, T.I.; Towers, G.J.; et al. SARS-CoV-2 variant biology: Immune escape, transmission and fitness. Nat. Rev. Microbiol. 2023, 21, 162–177. [Google Scholar] [CrossRef] [PubMed]
- Tamura, T.; Ito, J.; Uriu, K.; Zahradnik, J.; Kida, I.; Anraku, Y.; Nasser, H.; Shofa, M.; Oda, Y.; Lytras, S.; et al. Virological characteristics of the SARS-CoV-2 XBB variant derived from recombination of two Omicron subvariants. Nat. Commun. 2023, 14, 2800. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Lai, S.; Gao, G.F.; Shi, W. The emergence, genomic diversity and global spread of SARS-CoV-2. Nature 2021, 600, 408–418. [Google Scholar] [CrossRef]
- Yurkovetskiy, L.; Wang, X.; Pascal, K.E.; Tomkins-Tinch, C.; Nyalile, T.P.; Wang, Y.; Baum, A.; Diehl, W.E.; Dauphin, A.; Carbone, C.; et al. Structural and functional analysis of the D614G SARS-CoV-2 spike protein variant. Cell 2020, 183, 739–751.e738. [Google Scholar] [CrossRef] [PubMed]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef]
- Imbert, I.; Snijder, E.J.; Dimitrova, M.; Guillemot, J.-C.; Lécine, P.; Canard, B. The SARS-Coronavirus PLnc domain of nsp3 as a replication/transcription scaffolding protein. Virus Res. 2008, 133, 136–148. [Google Scholar] [CrossRef]
- Shan, H.; Liu, J.; Shen, J.; Dai, J.; Xu, G.; Lu, K.; Han, C.; Wang, Y.; Xu, X.; Tong, Y.; et al. Development of potent and selective inhibitors targeting the papain-like protease of SARS-CoV-2. Cell Chem. Biol. 2021, 28, 855–865.e9. [Google Scholar] [CrossRef]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Guo, Y.R.; Cao, Q.D.; Hong, Z.S.; Chen, S.D.; Jin, H.J.; Tan, K.S.; Wang, D.Y.; Yan, Y. The origin, transmission and clinical therapies on coronavirus disease 2019 (COVID-19) outbreak—An update on the status. Mil. Med. Res. 2020, 7, 11. [Google Scholar] [CrossRef]
- Wu, A.; Peng, Y.; Huang, B.; Ding, X.; Wang, X.; Niu, P.; Meng, J.; Zhu, Z.; Zhang, Z.; Wang, J.; et al. Genome Composition and Divergence of the Novel Coronavirus (2019-nCoV) Originating in China. Cell Host Microbe 2020, 27, 325–328. [Google Scholar] [CrossRef]
- Srinivasan, S.; Cui, H.; Gao, Z.; Liu, M.; Lu, S.; Mkandawire, W.; Narykov, O.; Sun, M.; Korkin, D. Structural genomics of SARS-CoV-2 indicates evolutionary conserved functional regions of viral proteins. Viruses 2020, 12, 360. [Google Scholar] [CrossRef] [PubMed]
- Alhammad, Y.M.O.; Kashipathy, M.M.; Roy, A.; Gagné, J.-P.; McDonald, P.; Gao, P.; Nonfoux, L.; Battaile, K.P.; Johnson, D.K.; Holmstrom, E.D.; et al. The SARS-CoV-2 Conserved Macrodomain Is a Mono-ADP-Ribosylhydrolase. J. Virol. 2021, 95, e01969-20. [Google Scholar] [CrossRef] [PubMed]
- Egloff, M.-P.; Malet, H.; Putics, A.; Heinonen, M.; Dutartre, H.; Frangeul, A.; Gruez, A.; Campanacci, V.; Cambillau, C.; Ziebuhr, J.; et al. Structural and Functional Basis for ADP-Ribose and Poly(ADP-Ribose) Binding by Viral Macro Domains. J. Virol. 2006, 80, 8493–8502. [Google Scholar] [CrossRef]
- Putics, A.; Gorbalenya, A.E.; Ziebuhr, J. Identification of protease and ADP-ribose 1’’-monophosphatase activities associated with transmissible gastroenteritis virus non-structural protein 3. J. Gen. Virol. 2006, 87, 651–656. [Google Scholar] [CrossRef] [PubMed]
- Saikatendu, K.S.; Joseph, J.S.; Subramanian, V.; Clayton, T.; Griffith, M.; Moy, K.; Velasquez, J.; Neuman, B.W.; Buchmeier, M.J.; Stevens, R.C.; et al. Structural Basis of Severe Acute Respiratory Syndrome Coronavirus ADP-Ribose-1″-Phosphate Dephosphorylation by a Conserved Domain of nsP3. Structure 2005, 13, 1665–1675. [Google Scholar] [CrossRef] [PubMed]
- Cho, C.C.; Lin, M.H.; Chuang, C.Y.; Hsu, C.H. Macro domain from Middle East respiratory syndrome coronavirus (MERS-CoV) is an efficient ADP-ribose binding module: Crystal structure and biochemical studies. J. Biol. Chem. 2016, 291, 4894–4902. [Google Scholar] [CrossRef]
- Xu, Y.; Cong, L.; Chen, C.; Wei, L.; Zhao, Q.; Xu, X.; Ma, Y.; Bartlam, M.; Rao, Z. Crystal Structures of Two Coronavirus ADP-Ribose-1″-Monophosphatases and Their Complexes with ADP-Ribose: A Systematic Structural Analysis of the Viral ADRP Domain. J. Virol. 2009, 83, 1083–1092. [Google Scholar] [CrossRef]
- Rack, J.G.M.; Perina, D.; Ahel, I. Macrodomains: Structure, Function, Evolution, and Catalytic Activities. Annu. Rev. Biochem. 2016, 85, 431–454. [Google Scholar] [CrossRef]
- Neuman, B.W. Bioinformatics and functional analyses of coronavirus nonstructural proteins involved in the formation of replicative organelles. Antivir. Res. 2016, 135, 97–107. [Google Scholar] [CrossRef]
- Lei, J.; Kusov, Y.; Hilgenfeld, R. Nsp3 of coronaviruses: Structures and functions of a large multi-domain protein. Antivir. Res. 2017, 149, 58–74. [Google Scholar] [CrossRef]
- Perina, D.; Mikoč, A.; Ahel, J.; Ćetković, H.; Žaja, R.; Ahel, I. Distribution of protein poly(ADPribosyl)ation systems across all domains of life. DNA Repair. 2014, 23, 4–16. [Google Scholar] [CrossRef] [PubMed]
- Claverie, J.-M. A Putative Role of de-Mono-ADP-Ribosylation of STAT1 by the SARS-CoV-2 Nsp3 Protein in the Cytokine Storm Syndrome of COVID-19. Viruses 2020, 12, 646. [Google Scholar] [CrossRef] [PubMed]
- Dhankhar, P.; Dalal, V.; Kumar, V. Screening of Severe Acute Respiratory Syndrome Coronavirus 2 RNA-Dependent RNA Polymerase Inhibitors Using Computational Approach. J. Comput. Biol. 2021, 28, 1228–1247. [Google Scholar] [CrossRef] [PubMed]
- Ghufran, M.; Ullah, M.; Khan, H.A.; Ghufran, S.; Ayaz, M.; Siddiq, M.; Abbas, S.Q.; Hassan, S.S.U.; Bungau, S. In-Silico Lead Druggable Compounds Identification against SARS COVID-19 Main Protease Target from In-House, Chembridge and Zinc Databases by Structure-Based Virtual Screening, Molecular Docking and Molecular Dynamics Simulations. Bioengineering 2023, 10, 100. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Terashi, G.; Christoffer, C.W.; Zhu, M.; Kihara, D. Protein docking model evaluation by 3D deep convolutional neural networks. Bioinformatics 2019, 36, 2113–2118. [Google Scholar] [CrossRef]
- Laurini, E.; Marson, D.; Aulic, S.; Fermeglia, M.; Pricl, S. Computational Alanine Scanning and Structural Analysis of the SARS-CoV-2 Spike Protein/Angiotensin-Converting Enzyme 2 Complex. ACS Nano 2020, 14, 11821–11830. [Google Scholar] [CrossRef]
- Han, P.; Li, L.; Liu, S.; Wang, Q.; Zhang, D.; Xu, Z.; Han, P.; Li, X.; Peng, Q.; Su, C.; et al. Receptor binding and complex structures of human ACE2 to spike RBD from omicron and delta SARS-CoV-2. Cell 2022, 185, 630–640.e10. [Google Scholar] [CrossRef]
- Willett, B.J.; Grove, J.; MacLean, O.A.; Wilkie, C.; De Lorenzo, G.; Furnon, W.; Cantoni, D.; Scott, S.; Logan, N.; Ashraf, S.; et al. SARS-CoV-2 Omicron is an immune escape variant with an altered cell entry pathway. Nat. Microbiol. 2022, 7, 1161–1179. [Google Scholar] [CrossRef]
- Li, Q.; Wu, J.; Nie, J.; Zhang, L.; Hao, H.; Liu, S.; Zhao, C.; Zhang, Q.; Liu, H.; Nie, L.; et al. The Impact of Mutations in SARS-CoV-2 Spike on Viral Infectivity and Antigenicity. Cell 2020, 182, 1284–1294.e9. [Google Scholar] [CrossRef]
- Fehr, A.R.; Channappanavar, R.; Jankevicius, G.; Fett, C.; Zhao, J.; Athmer, J.; Meyerholz, D.K.; Ahel, I.; Perlman, S. The Conserved Coronavirus Macrodomain Promotes Virulence and Suppresses the Innate Immune Response during Severe Acute Respiratory Syndrome Coronavirus Infection. mBio 2016, 7, e01721-16. [Google Scholar] [CrossRef]
- Eriksson, K.K.; Cervantes-Barragán, L.; Ludewig, B.; Thiel, V. Mouse hepatitis virus liver pathology is dependent on ADP-ribose-1″-phosphatase, a viral function conserved in the alpha-like supergroup. J. Virol. 2008, 82, 12325–12334. [Google Scholar] [CrossRef]
- Putics, A.; Filipowicz, W.; Hall, J.; Gorbalenya, A.E.; Ziebuhr, J. ADP-Ribose-1″-Monophosphatase: A Conserved Coronavirus Enzyme That Is Dispensable for Viral Replication in Tissue Culture. J. Virol. 2005, 79, 12721–12731. [Google Scholar] [CrossRef] [PubMed]
- Fehr, A.R.; Athmer, J.; Channappanavar, R.; Phillips, J.M.; Meyerholz, D.K.; Perlman, S. The nsp3 Macrodomain Promotes Virulence in Mice with Coronavirus-Induced Encephalitis. J. Virol. 2014, 89, 1523–1536. [Google Scholar] [CrossRef] [PubMed]
- Fehr, A.R.; Singh, S.A.; Kerr, C.M.; Mukai, S.; Higashi, H.; Aikawa, M. The impact of PARPs and ADP-ribosylation on inflammation and host–pathogen interactions. Genes Dev. 2020, 34, 341–359. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Bowers, K.J.; Chow, D.E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In Proceedings of the SC’06: 2006 ACM/IEEE Conference on Supercomputing, Tampa, FL, USA, 11–17 November 2006; p. 43. [Google Scholar] [CrossRef]
- Iqbal, S.; Potharaju, R.; Naveen, S.; Lokanath, N.K.; Mohanakrishnan, A.K.; Gunasekaran, K. Design, crystal structure determination, molecular dynamic simulation and MMGBSA calculations of novel p38-alpha MAPK inhibitors for combating Alzheimer’s disease. J. Biomol. Struct. Dyn. 2021, 40, 6114–6127. [Google Scholar] [CrossRef]
- Case, D.A.; Darden, T.; Cheatham, T.E., III; Simmerling, C.; Wang, J.; RDuke, E.; Luo, R.; Crowley, M.; Walker, R.; Zhang, W.; et al. Amber. Constant Press. Mol. Dyn. Algorithms 2008, 10, 1–304. [Google Scholar]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Miller, B.R.; McGee, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, V.; Dhankhar, P.; Dalal, V. Promising antivirals for PLpro of SARS-CoV-2 using virtual screening, molecular docking, dynamics, and MMPBSA. J. Biomol. Struct. Dyn. 2022, 41, 4650–4666. [Google Scholar] [CrossRef]
- Kalathiya, U.; Padariya, M.; Mayordomo, M.; Lisowska, M.; Nicholson, J.; Singh, A.; Baginski, M.; Fahraeus, R.; Carragher, N.; Ball, K.; et al. Highly conserved homotrimer cavity formed by the SARS-CoV-2 spike glycoprotein: A novel binding site. J. Clin. Med. 2020, 9, 1473. [Google Scholar] [CrossRef]
- Weiner, P.K.; Langridge, R.; Blaney, J.M.; Schaefer, R.; Kollman, P.A. Electrostatic potential molecular surfaces. Proc. Natl. Acad. Sci. USA 1982, 79, 3754–3758. [Google Scholar] [CrossRef] [PubMed]
- McCoy, A.J.; Epa, V.C.; Colman, P.M. Electrostatic complementarity at protein/protein interfaces. J. Mol. Biol. 1997, 268, 570–584. [Google Scholar] [CrossRef] [PubMed]
- Gan, H.H.; Zinno, J.; Piano, F.; Gunsalus, K.C. Omicron Spike Protein Has a Positive Electrostatic Surface That Promotes ACE2 Recognition and Antibody Escape. Front. Virol. 2022, 2, 894531. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand–protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibitor, Conc. | Percent Inhibition |
|---|---|
| F5084-0852, 30 µM | 0 |
| F1877-0839, 30 µM | 79 |
| F2619-0022, 30 µM | 30 |
| F1877-1292, 30 µM | 12 |
| F0466-0005, 30 µM | 0 |
| F2085-0027, 30 µM | 2 |
| F0772-2453, 30 µM | 43 |
| F0470-0003, 30 µM | 69 |
| F0827-0193, 30 µM | 38 |
| F2173-1125, 30 µM | 0 |
| Anti-Spike, 0.0001 µM | 21 |
| Anti-Spike, 0.001 µM | 49 |
| Anti-Spike, 0.01 µM | 76 |
| Compound I.D. | TR-FRET Ratio | % Activity | % Inhibition | ||
|---|---|---|---|---|---|
| Repeat 1 | Repeat 2 | Repeat 1 | Repeat 2 | ||
| No Compound | 0.99 | 1.00 | 99 | 101 | 0 |
| F5084-0852, 30 µM | 1.00 | 1.03 | 103 | 111 | 0 |
| F1877-0839, 30 µM | 0.77 | 0.77 | 20 | 21 | 79 |
| F2619-0022, 30 µM | 0.89 | 0.93 | 62 | 77 | 30 |
| F1877-1292, 30 µM | 0.96 | 0.96 | 88 | 89 | 12 |
| F0466-0005, 30 µM | 1.01 | 0.99 | 104 | 97 | 0 |
| F2085-0027, 30 µM | 0.97 | 1.00 | 93 | 103 | 2 |
| F0772-2453, 30 µM | 0.89 | 0.86 | 62 | 51 | 43 |
| F0470-0003, 30 µM | 0.80 | 0.79 | 32 | 29 | 69 |
| F0827-0193, 30 µM | 0.89 | 0.89 | 63 | 61 | 38 |
| F2173-1125, 30 µM * | 1.15 | 1.20 | 153 | 172 | 0 |
| Anti-Spike, 0.0001 µM | 0.92 | 0.95 | 72 | 86 | 21 |
| Anti-Spike, 0.001 µM | 0.87 | 0.85 | 54 | 49 | 49 |
| Anti-Spike, 0.01 µM | 0.79 | 0.77 | 27 | 22 | 76 |
| Background | 0.71 | 0.71 | |||
| Name of Compound | Solv GB | vdW | Coulomb | Covalent | Hbond | ∆GTotal (kcal/mol) |
|---|---|---|---|---|---|---|
| F1877-0839 | 29.66 ± 6.24 | −72.14 ± 2.96 | −233.35 ± 2.03 | 14.97 ± 0.02 | −0.86 ± 0.14 | −106.38 ± 1.56 |
| F0470-0003 | 48.16 ± 5.16 | −54.17 ± 2.89 | −40.41 ± 2.46 | 1.23 ± 0.30 | −0.78 ± 0.11 | −99.68 ± 1.52 |
| Cocrystal | 23.75 ± 5.09 | −62.55 ± 2.78 | −31.65 ±2.17 | 6.7 ± 0.27 | −7.78 ± 0.10 | −92.01 ± 1.58 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iqbal, S.; Lin, S.-X. Deep Drug Discovery of Mac Domain of SARS-CoV-2 (WT) Spike Inhibitors: Using Experimental ACE2 Inhibition TR-FRET Assay, Screening, Molecular Dynamic Simulations and Free Energy Calculations. Bioengineering 2023, 10, 961. https://doi.org/10.3390/bioengineering10080961
Iqbal S, Lin S-X. Deep Drug Discovery of Mac Domain of SARS-CoV-2 (WT) Spike Inhibitors: Using Experimental ACE2 Inhibition TR-FRET Assay, Screening, Molecular Dynamic Simulations and Free Energy Calculations. Bioengineering. 2023; 10(8):961. https://doi.org/10.3390/bioengineering10080961
Chicago/Turabian StyleIqbal, Saleem, and Sheng-Xiang Lin. 2023. "Deep Drug Discovery of Mac Domain of SARS-CoV-2 (WT) Spike Inhibitors: Using Experimental ACE2 Inhibition TR-FRET Assay, Screening, Molecular Dynamic Simulations and Free Energy Calculations" Bioengineering 10, no. 8: 961. https://doi.org/10.3390/bioengineering10080961
APA StyleIqbal, S., & Lin, S.-X. (2023). Deep Drug Discovery of Mac Domain of SARS-CoV-2 (WT) Spike Inhibitors: Using Experimental ACE2 Inhibition TR-FRET Assay, Screening, Molecular Dynamic Simulations and Free Energy Calculations. Bioengineering, 10(8), 961. https://doi.org/10.3390/bioengineering10080961