
Use of Stromal Intervention and Exogenous Neoantigen Vaccination to Boost Pancreatic Cancer Chemo-Immunotherapy by Nanocarriers



Abstract
:1. Introductory Statement
2. Nano-Enabled Chemo-Immunotherapy in PDAC
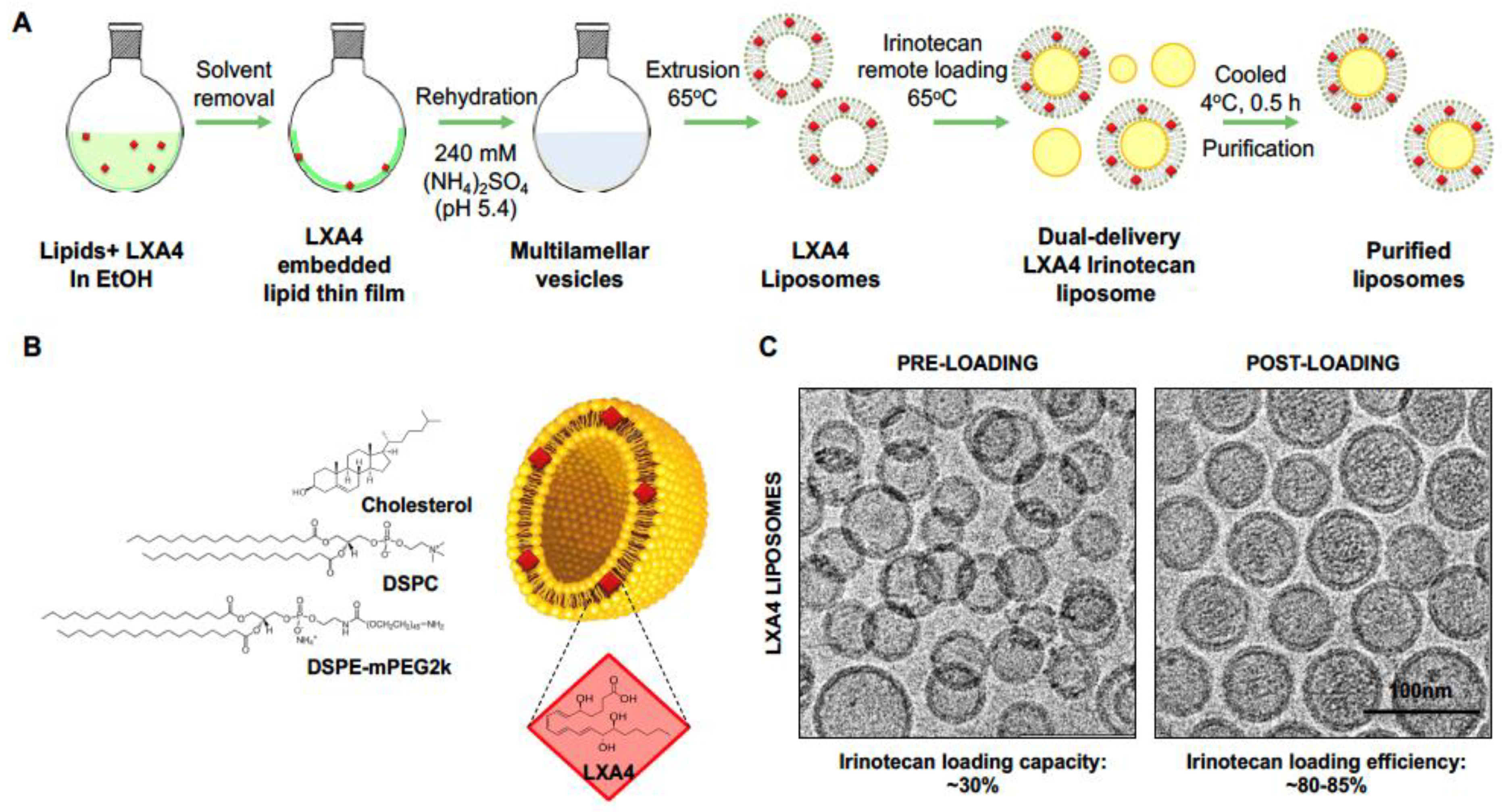
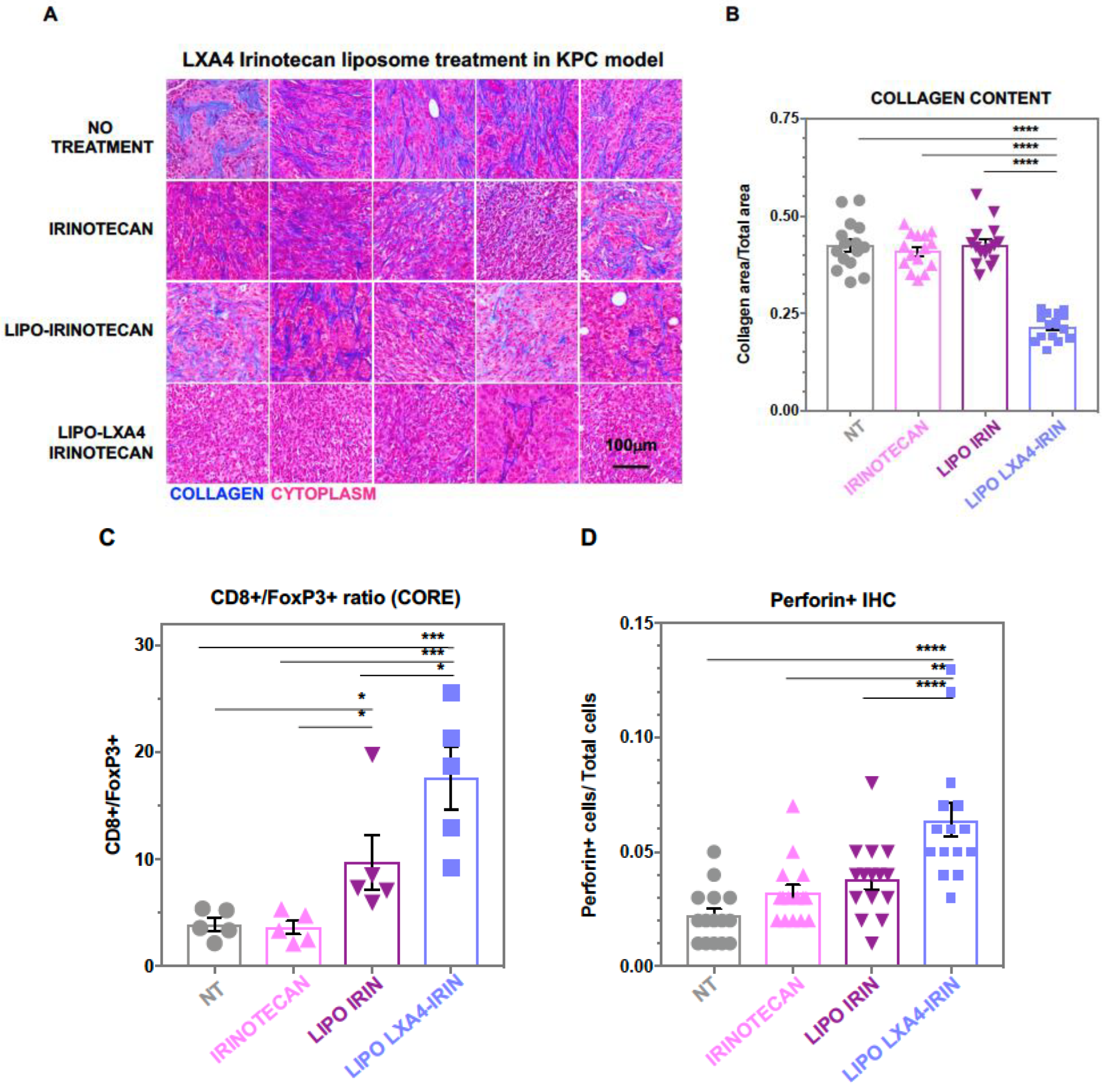
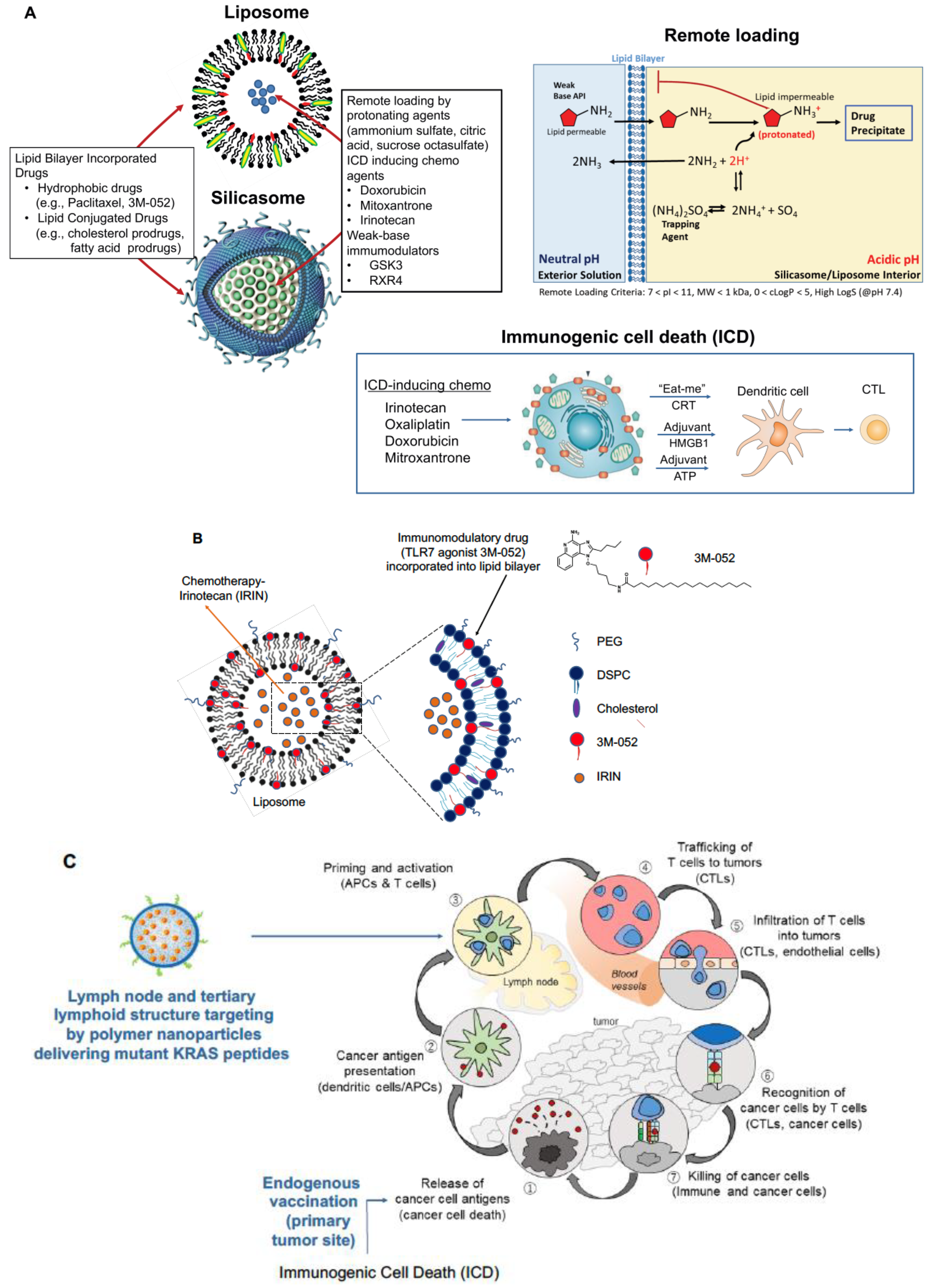
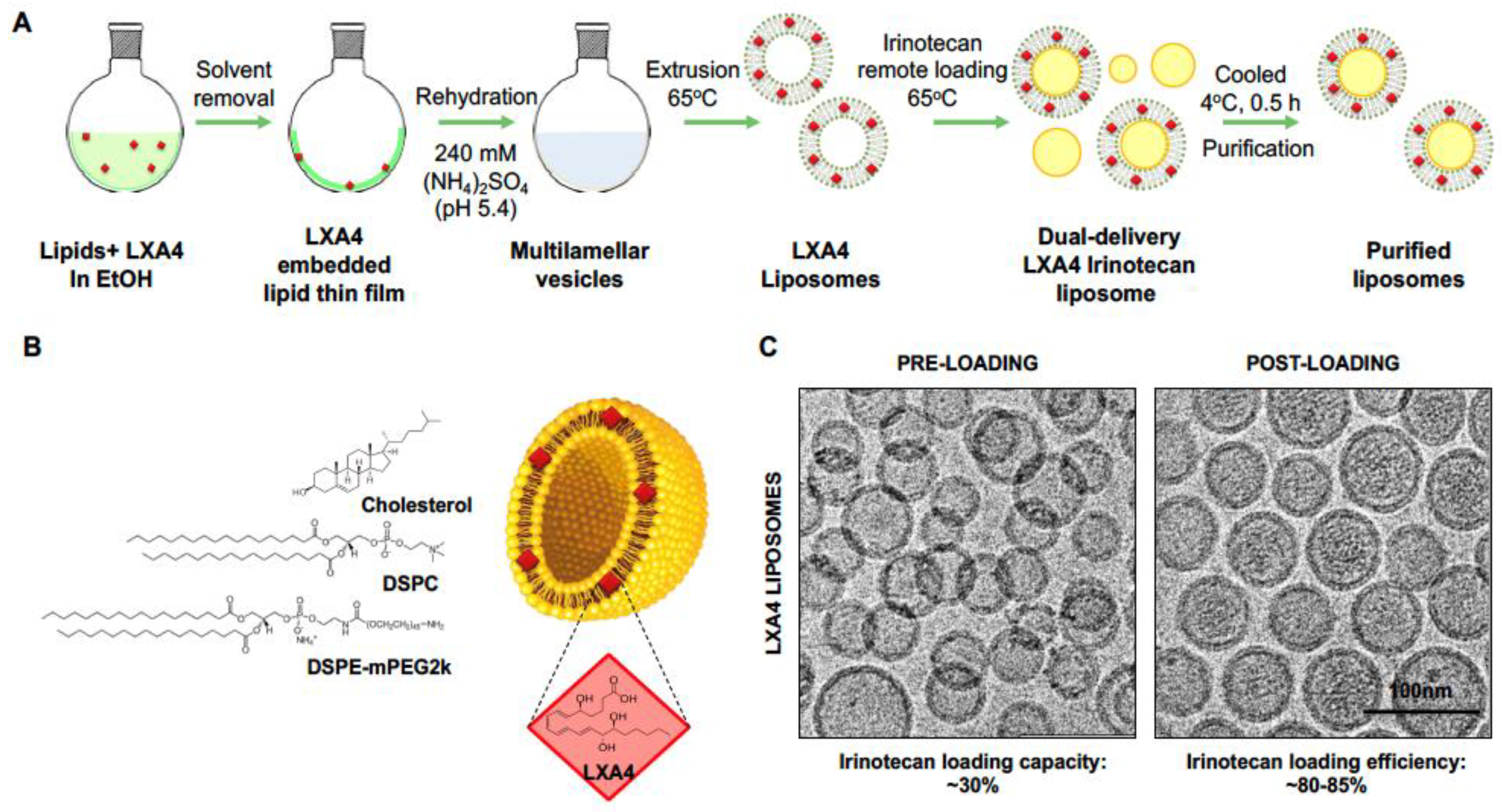
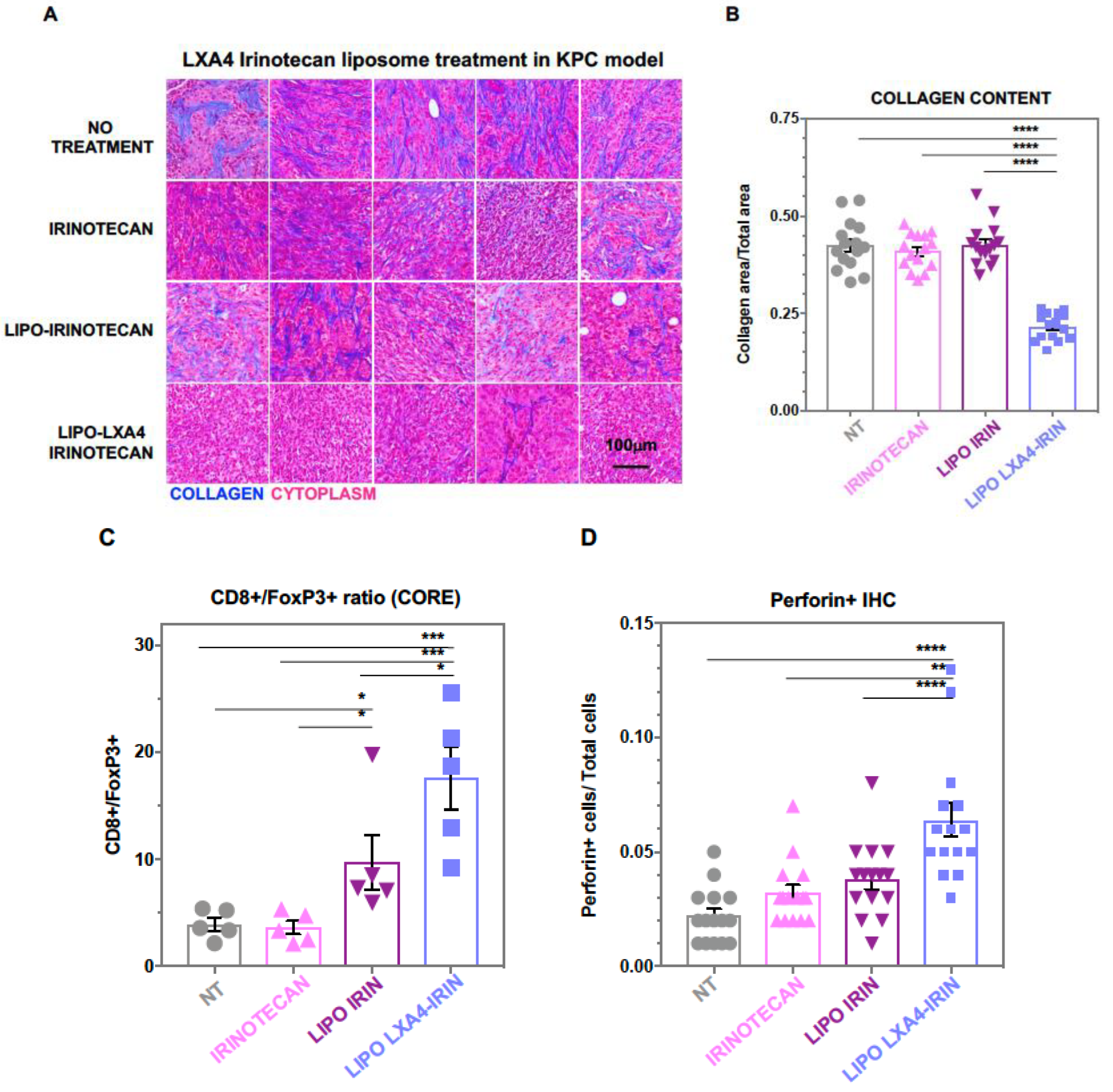
2.1. Development of a Dual-Delivery Irinotecan/Lipoxin A4 Liposome to Target the Stroma and Improve PDAC Immunotherapy
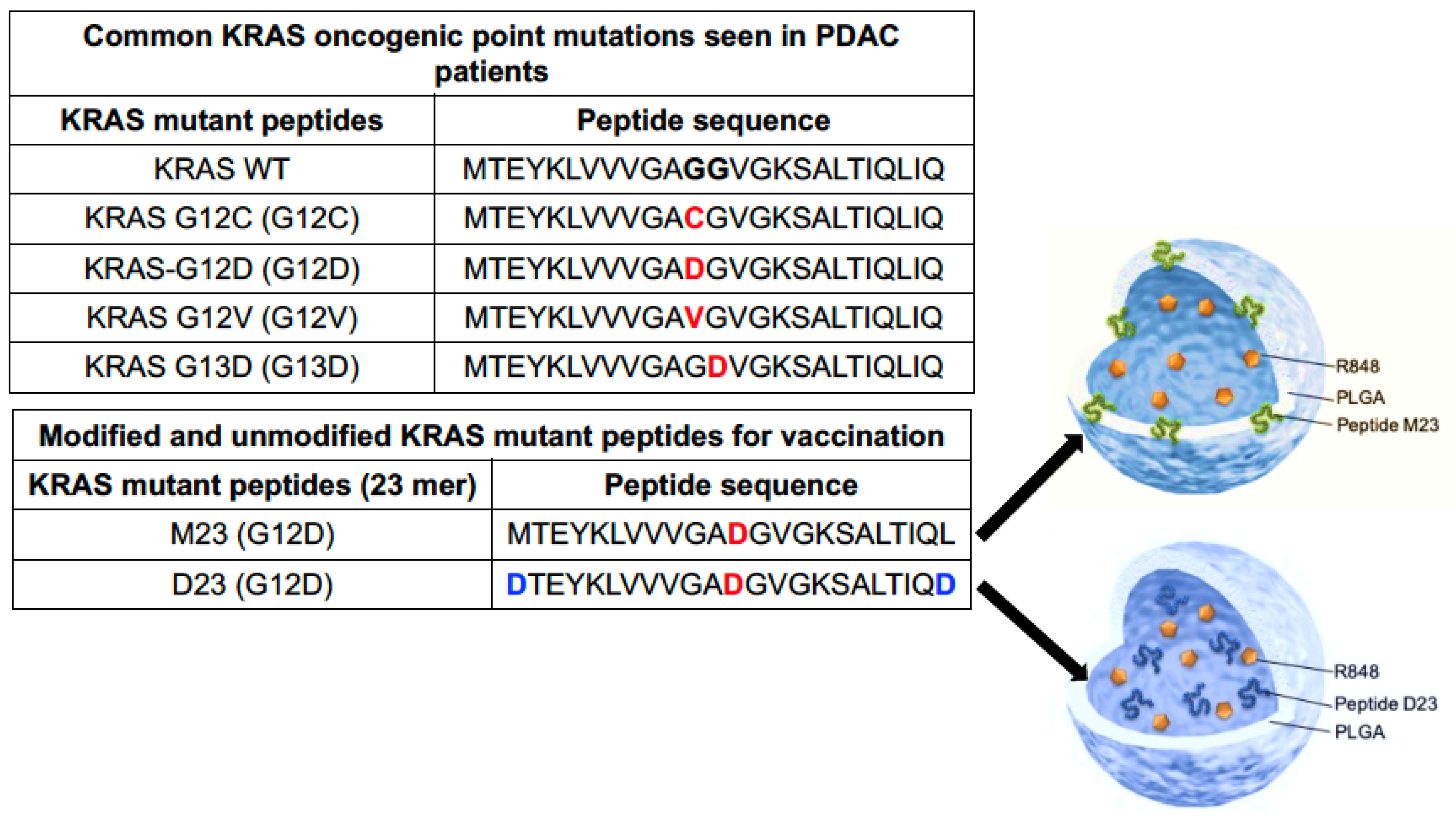
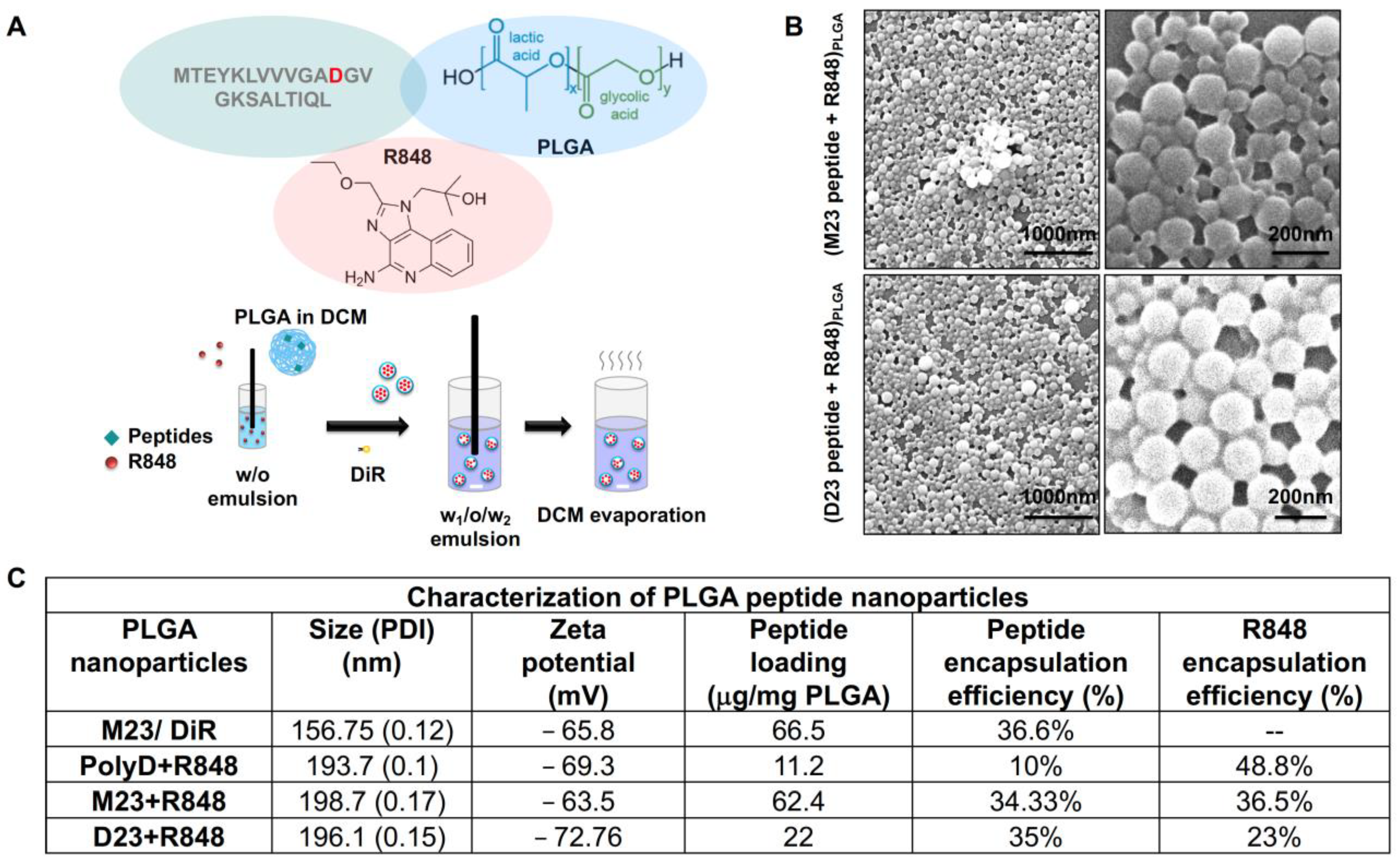
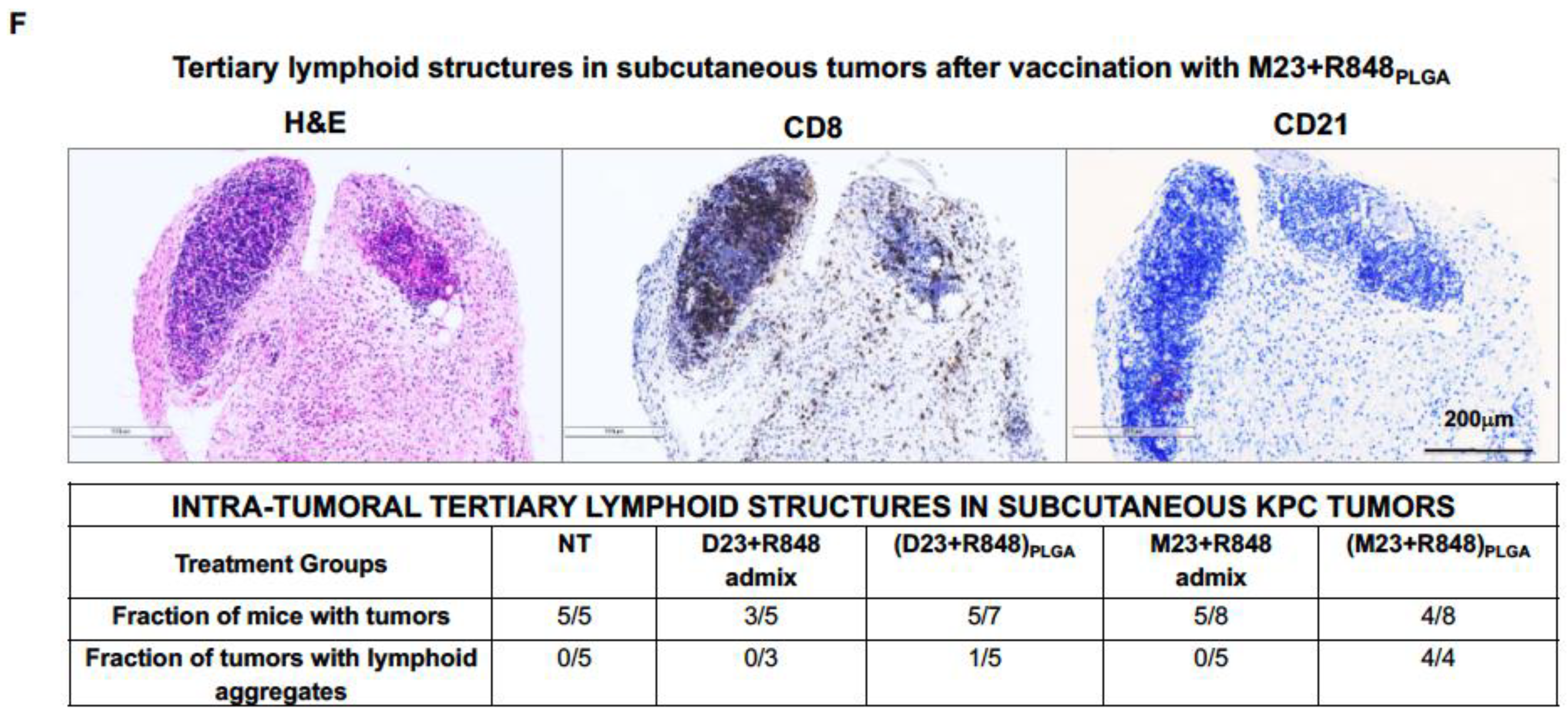
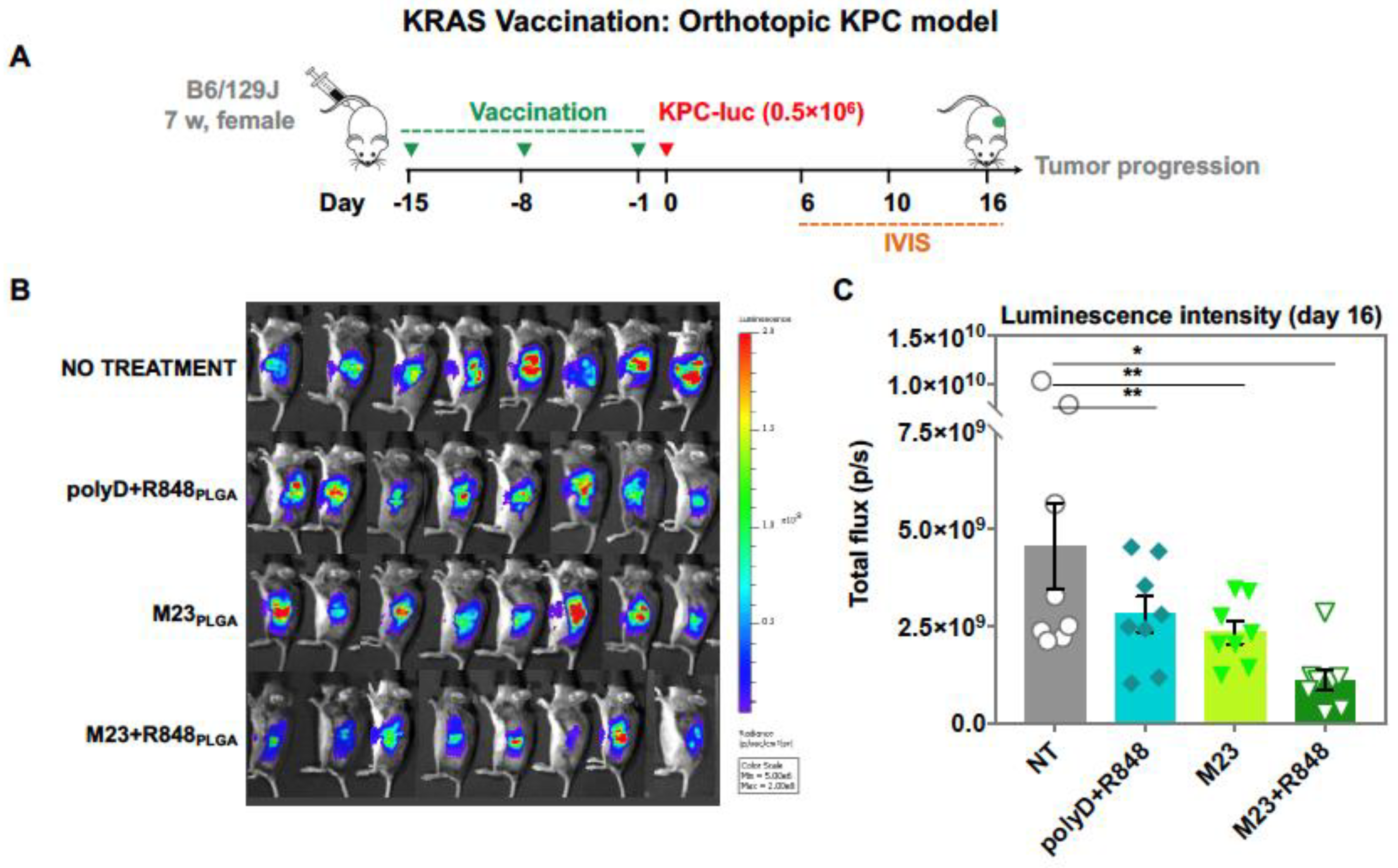
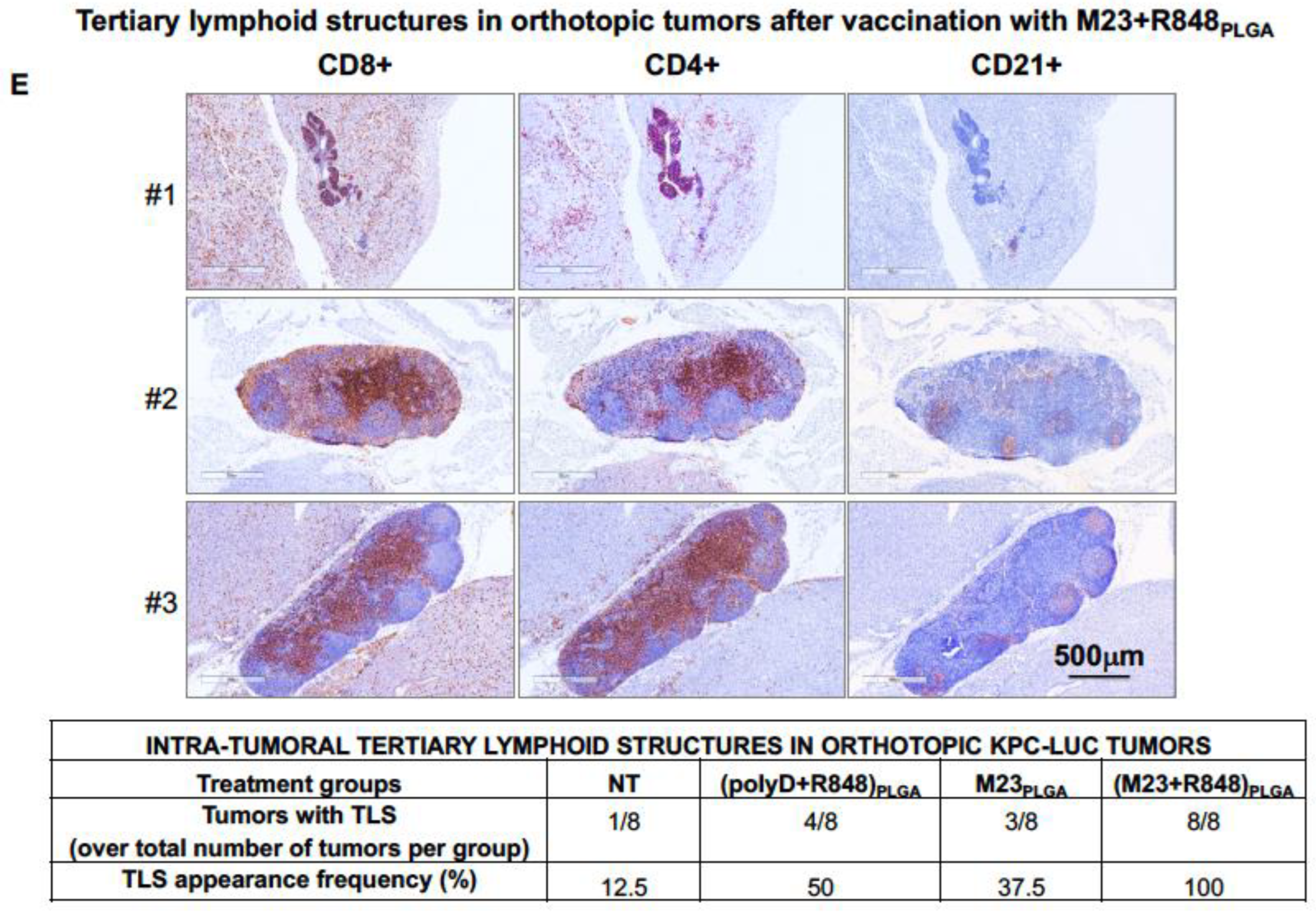
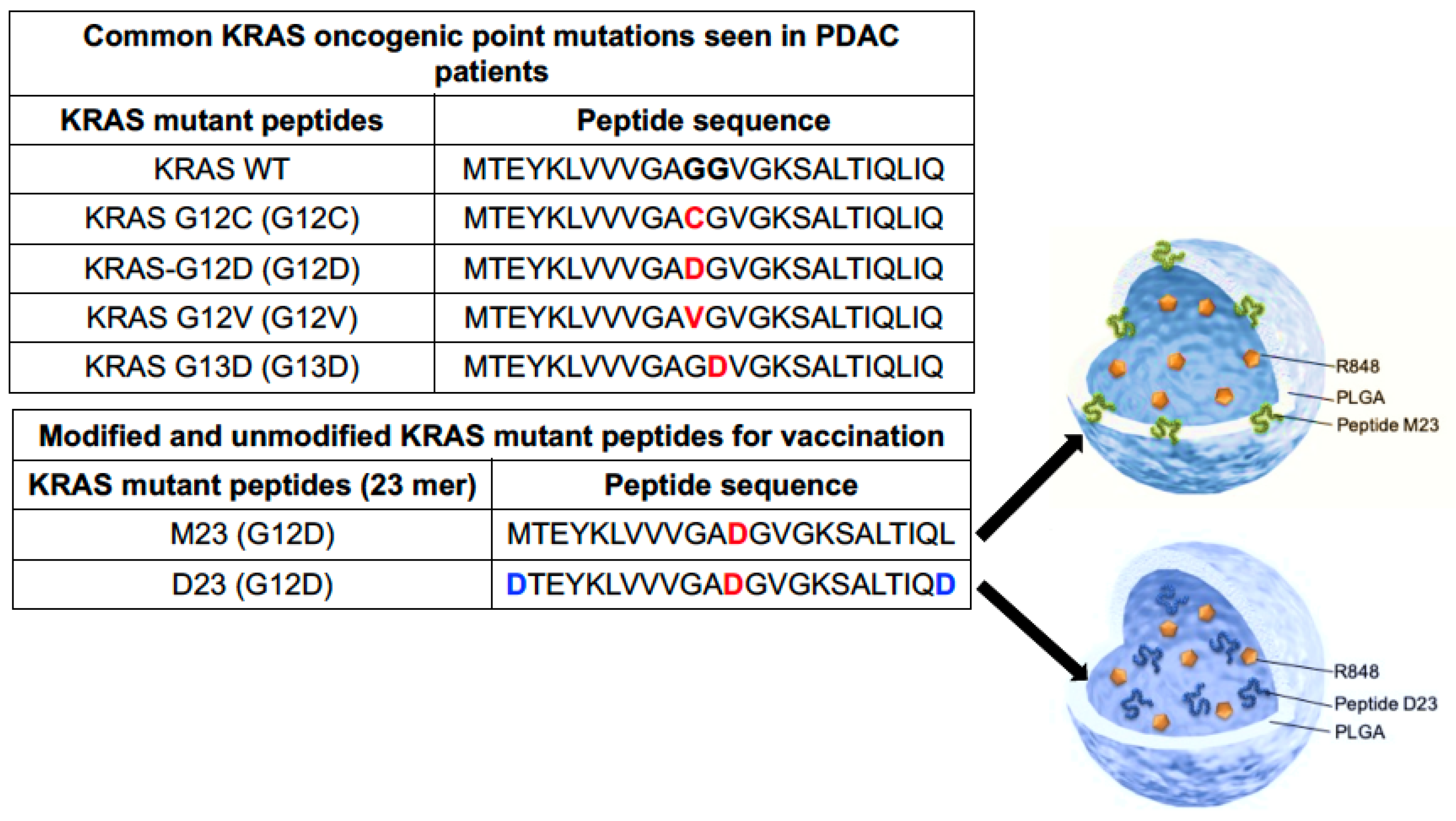
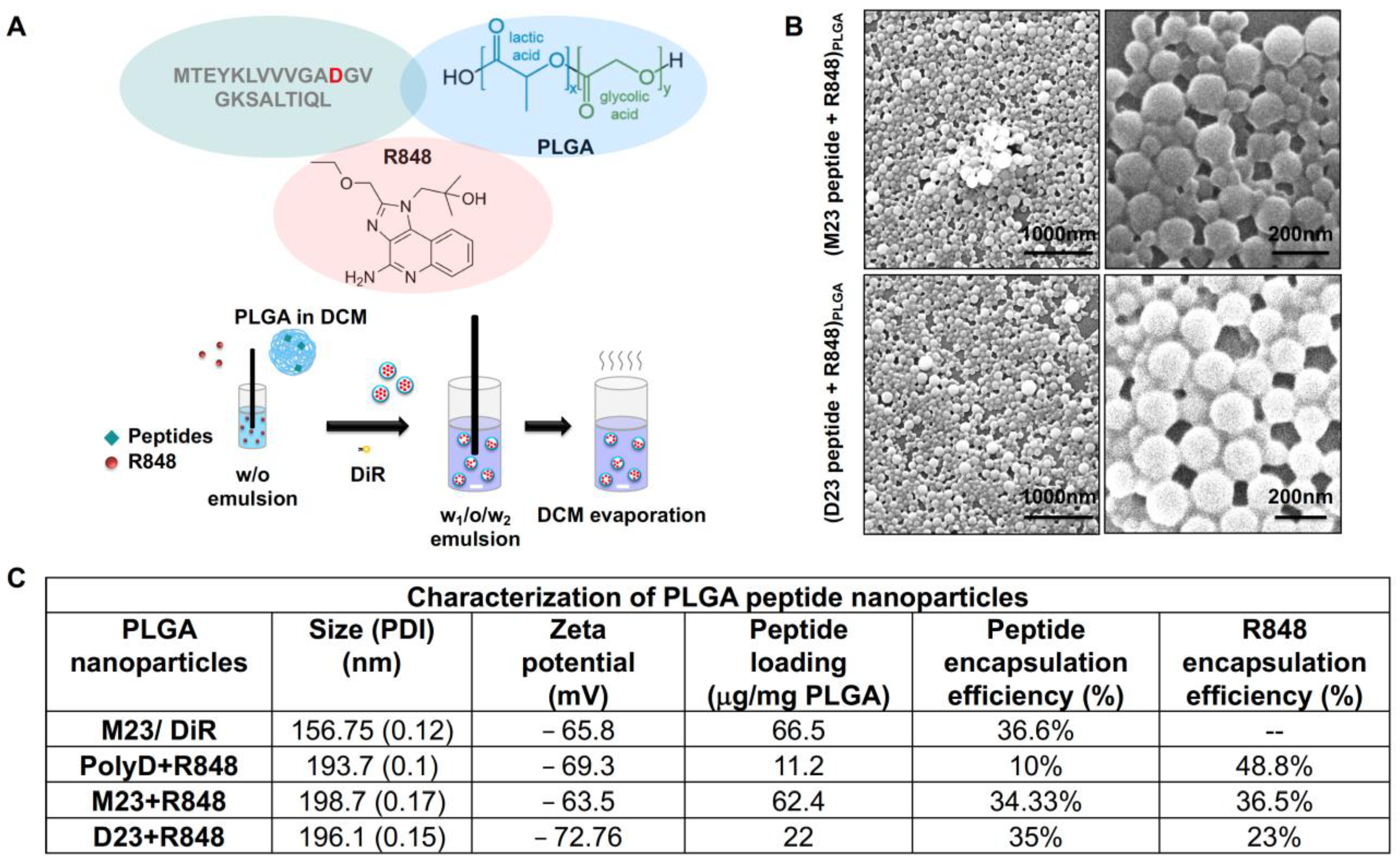
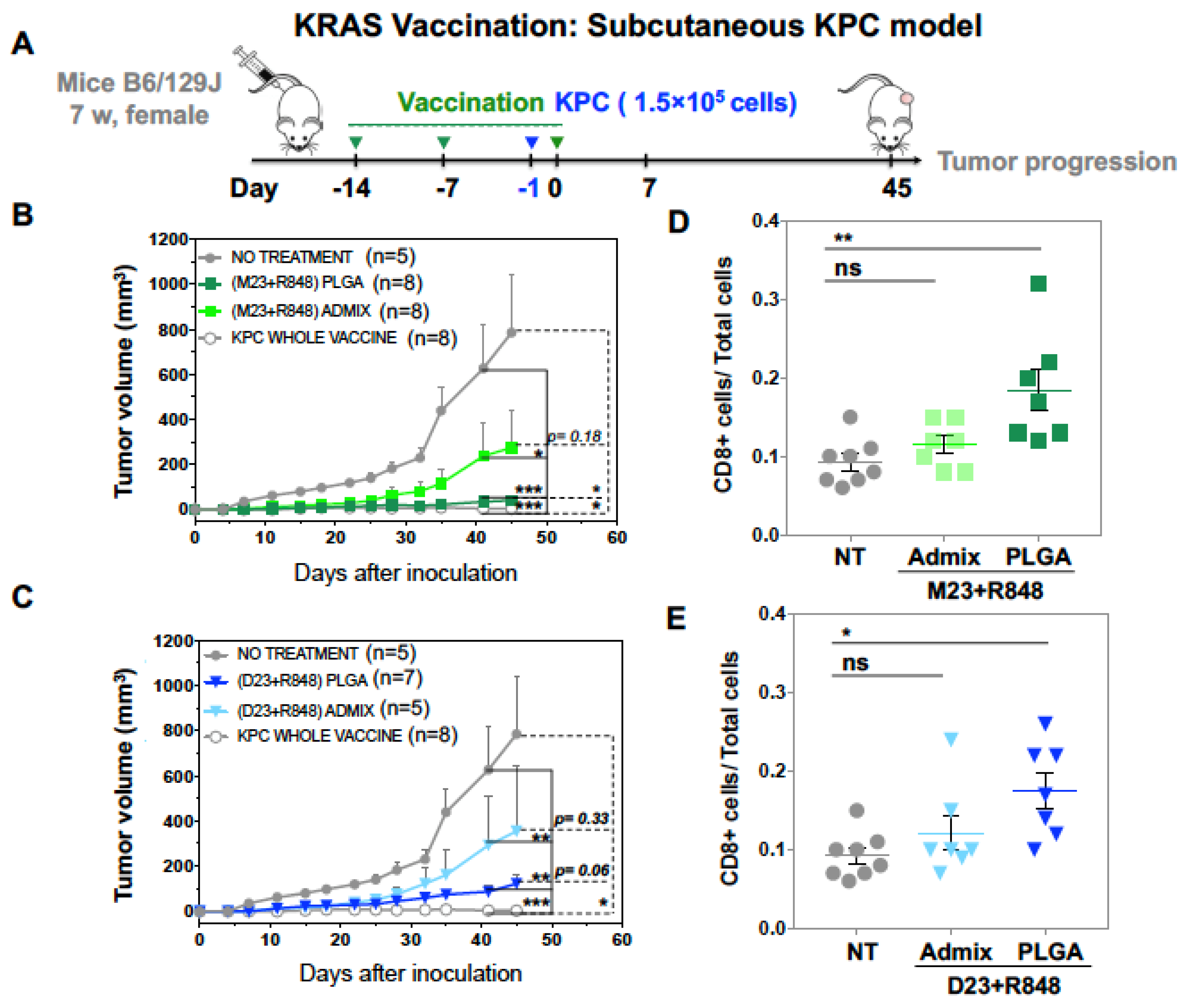
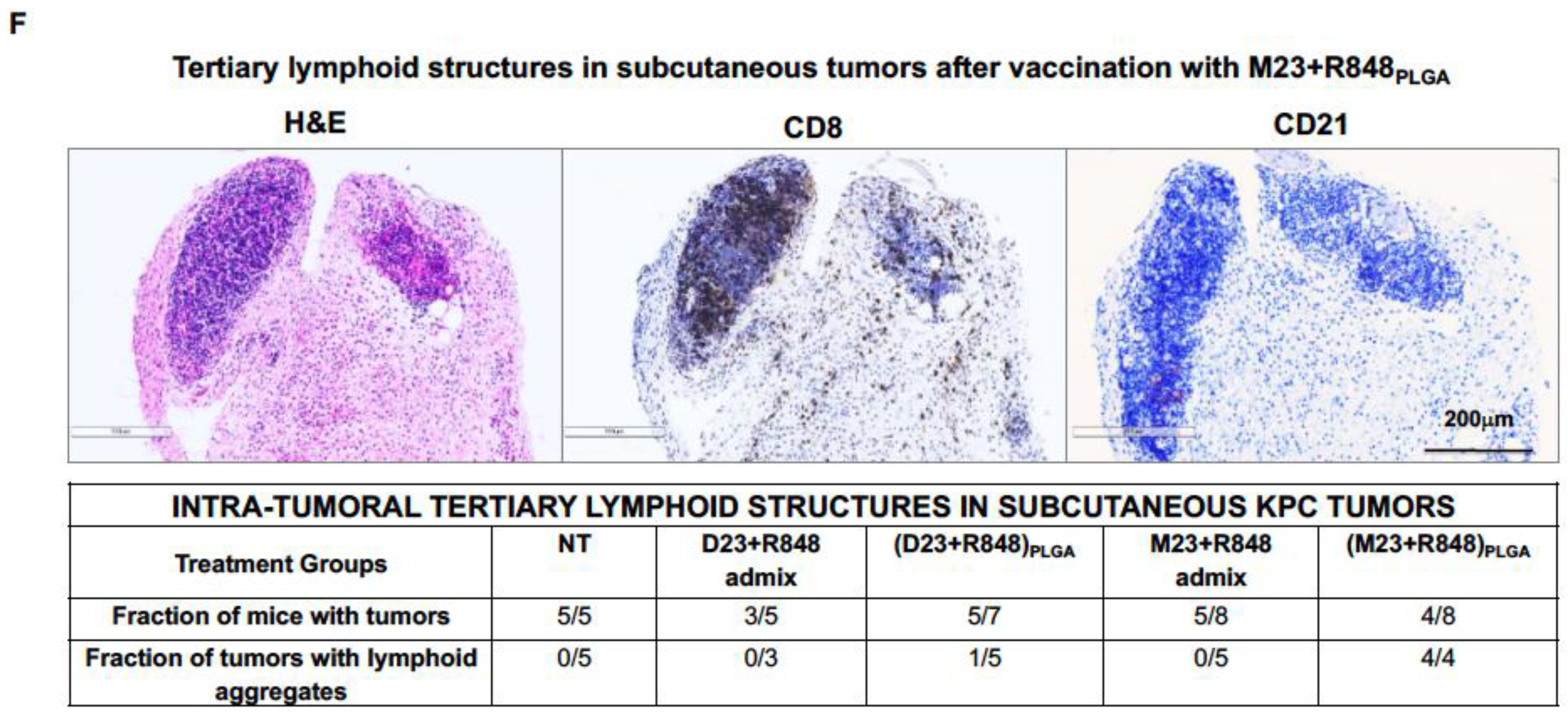
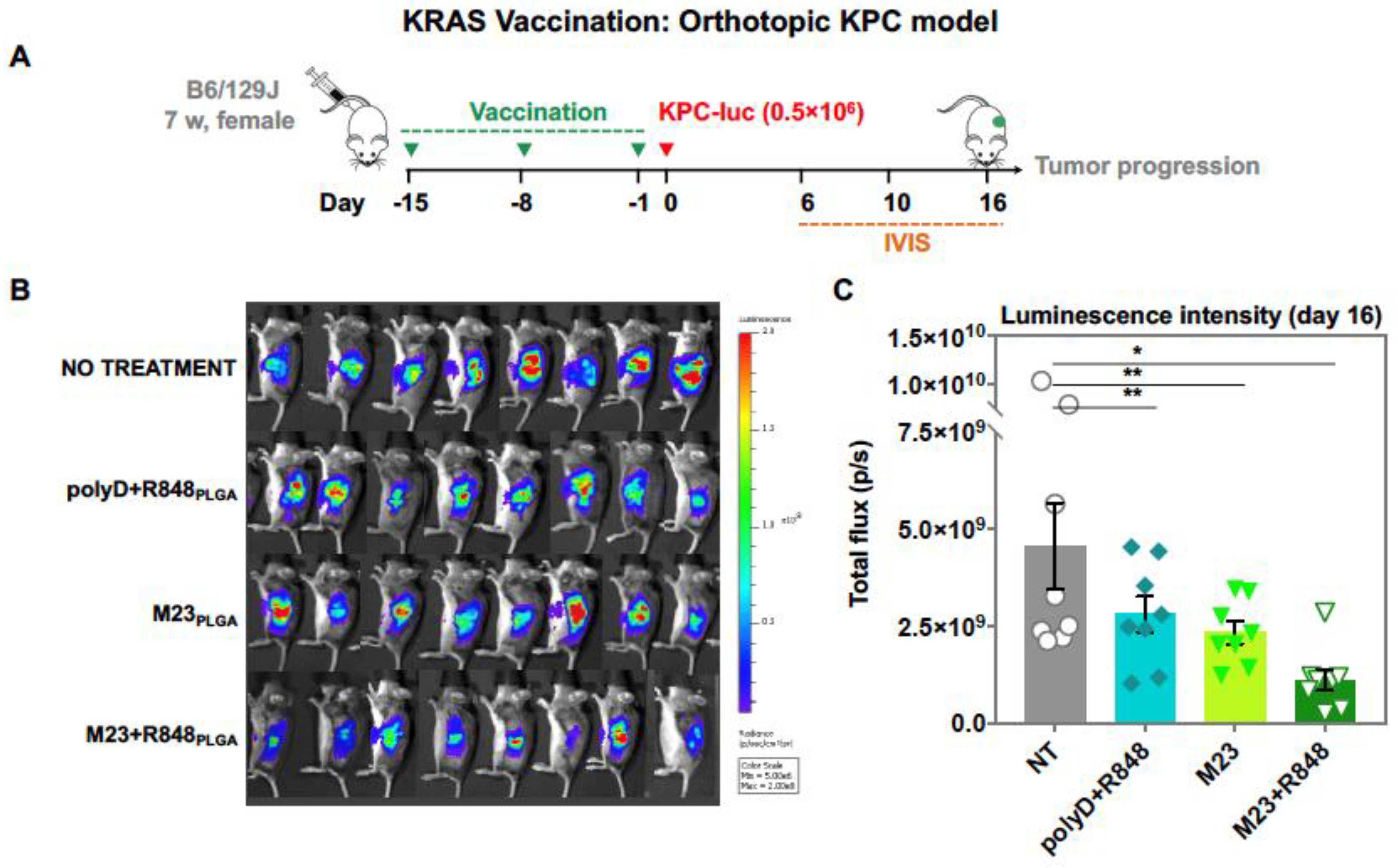
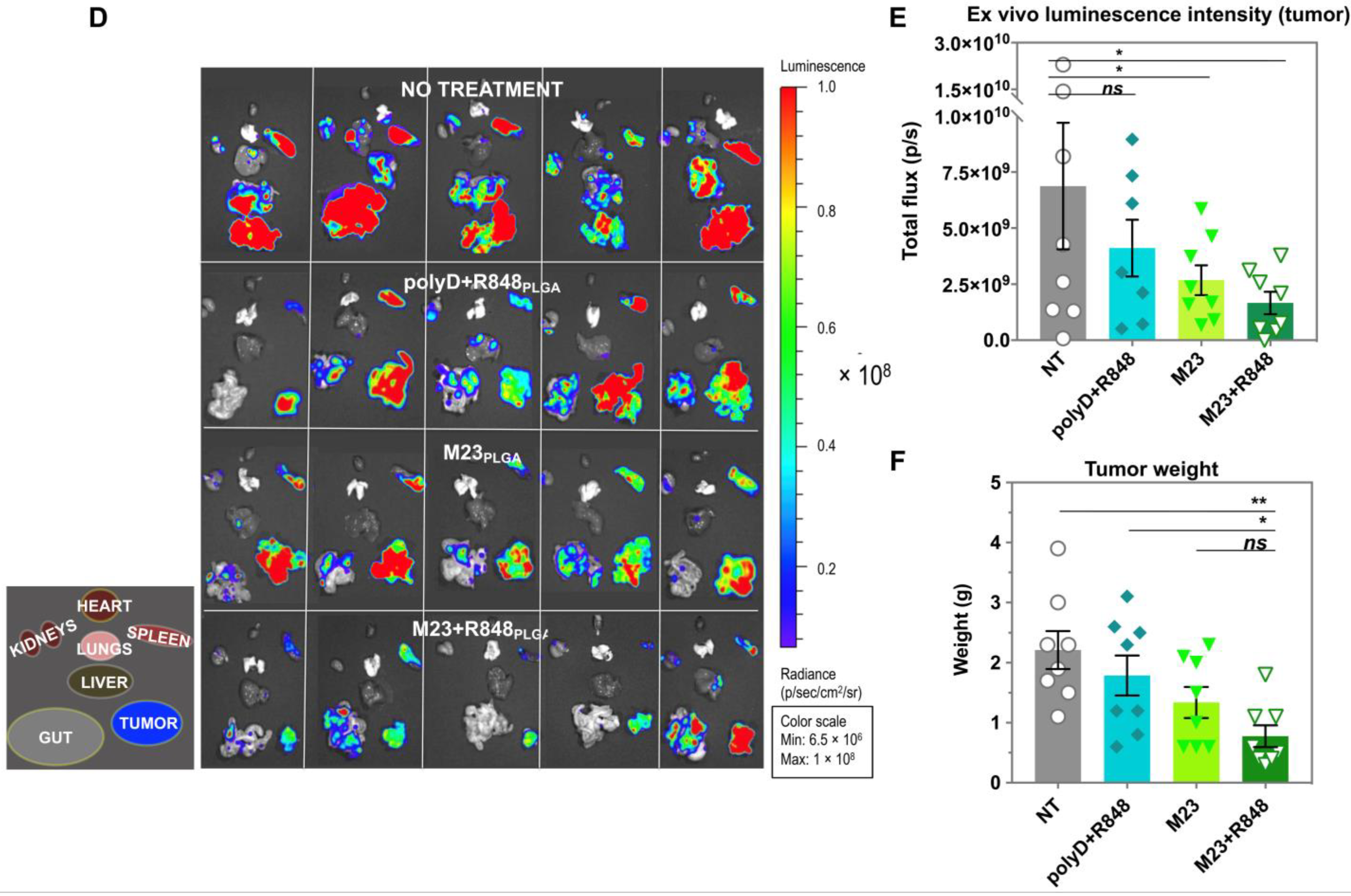
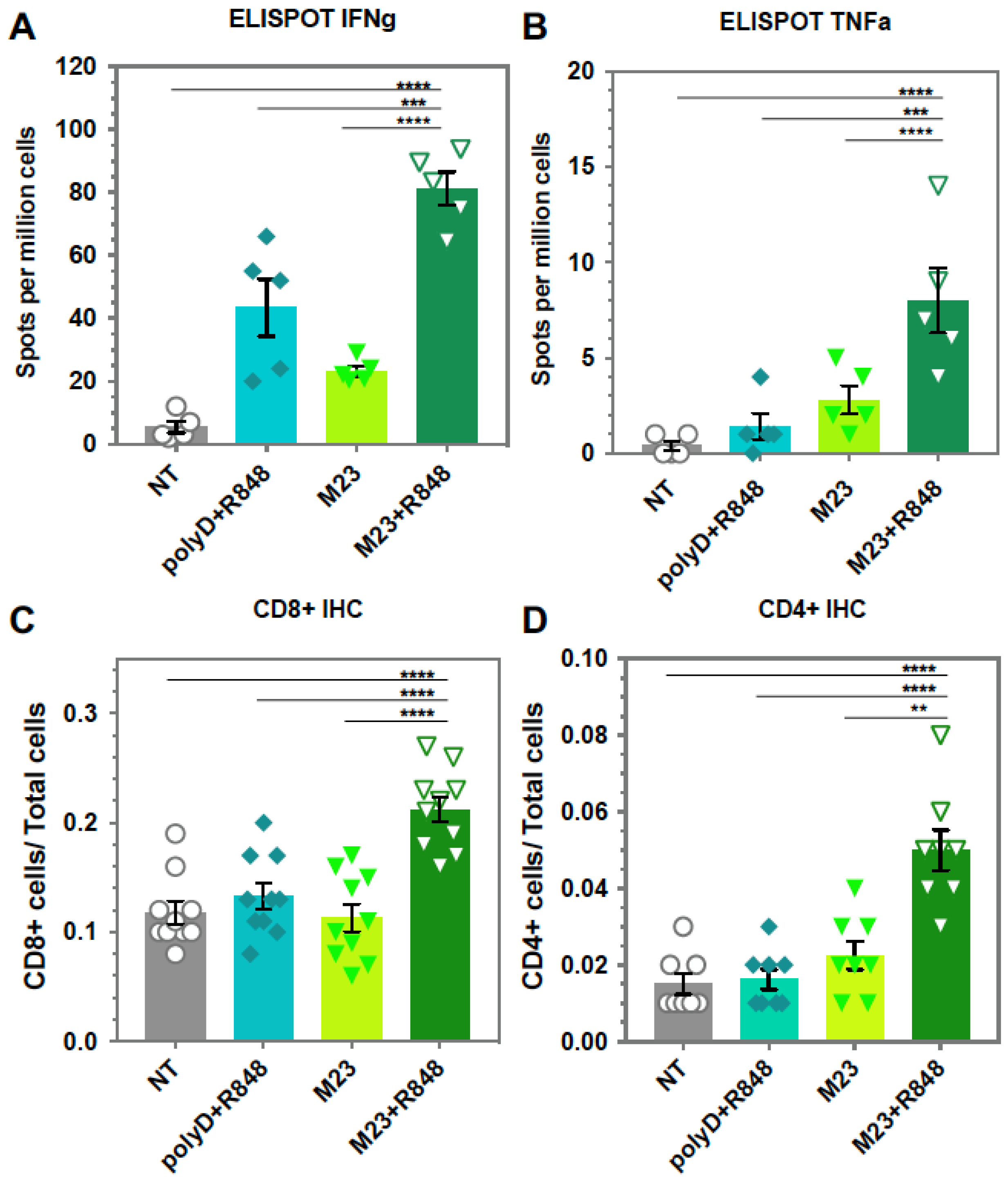
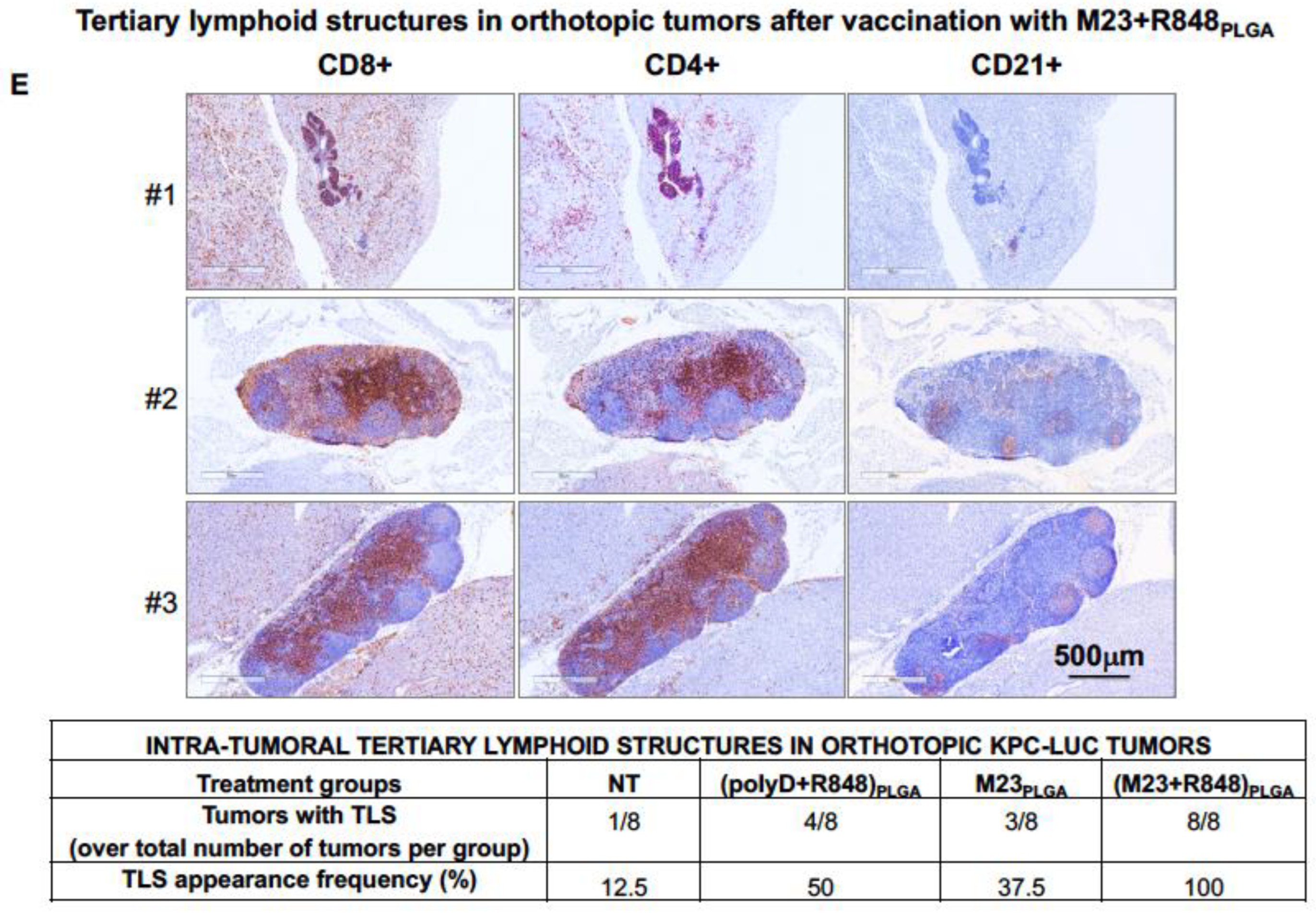
2.2. Combined Use of Mutant KRAS Vaccination Approach to Boost the Endogenous Vaccination Response to ICD in PDAC
3. Conclusions and Future Prospects
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Observatory, T.G.C. Pancreas (Source: Globocan 2020). 2020. Available online: https://gco.iarc.fr (accessed on 1 October 2023).
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef]
- Li, Y.; Xiang, S.; Pan, W.; Wang, J.; Zhan, H.; Liu, S. Targeting tumor immunosuppressive microenvironment for pancreatic cancer immunotherapy: Current research and future perspective. Front. Oncol. 2023, 13, 1166860. [Google Scholar] [CrossRef] [PubMed]
- Nel, A.E.; Mei, K.-C.; Liao, Y.-P.; Liu, X. Multifunctional Lipid Bilayer Nanocarriers for Cancer Immunotherapy in Heterogeneous Tumor Microenvironments, Combining Immunogenic Cell Death Stimuli with Immune Modulatory Drugs. ACS Nano 2022, 16, 5184–5232. [Google Scholar] [CrossRef] [PubMed]
- Noubissi Nzeteu, G.A.; Gibbs, B.F.; Kotnik, N.; Troja, A.; Bockhorn, M.; Meyer, N.H. Nanoparticle-based immunotherapy of pancreatic cancer. Front. Mol. Biosci. 2022, 9, 948898. [Google Scholar] [CrossRef] [PubMed]
- Green, M.R.; Manikhas, G.M.; Orlov, S.; Afanasyev, B.; Makhson, A.M.; Bhar, P.; Hawkins, M.J. Abraxane®, a novel Cremophor®-free, albumin-bound particle form of paclitaxel for the treatment of advanced non-small-cell lung cancer. Ann. Oncol. 2006, 17, 1263–1268. [Google Scholar] [CrossRef] [PubMed]
- Meng, H.; Wang, M.; Liu, H.; Liu, X.; Situ, A.; Wu, B.; Ji, Z.; Chang, C.H.; Nel, A.E. Use of a lipid-coated mesoporous silica nanoparticle platform for synergistic gemcitabine and paclitaxel delivery to human pancreatic cancer in mice. ACS Nano 2015, 9, 3540–3557. [Google Scholar] [CrossRef]
- Zhang, H. Onivyde for the therapy of multiple solid tumors. Onco Targets Ther. 2016, 9, 3001–3007. [Google Scholar] [CrossRef] [PubMed]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouché, O.; Guimbaud, R.; Bécouarn, Y.; Adenis, A.; Raoul, J.-L.; Gourgou-Bourgade, S.; de la Fouchardière, C.; et al. FOLFIRINOX versus Gemcitabine for Metastatic Pancreatic Cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef]
- Passero, F.C., Jr.; Grapsa, D.; Syrigos, K.N.; Saif, M.W. The safety and efficacy of Onivyde (irinotecan liposome injection) for the treatment of metastatic pancreatic cancer following gemcitabine-based therapy. Expert. Rev. Anticancer Ther. 2016, 16, 697–703. [Google Scholar] [CrossRef]
- Wang-Gillam, A.; Hubner, R.A.; Siveke, J.T.; Von Hoff, D.D.; Belanger, B.; de Jong, F.A.; Mirakhur, B.; Chen, L.-T. NAPOLI-1 phase 3 study of liposomal irinotecan in metastatic pancreatic cancer: Final overall survival analysis and characteristics of long-term survivors. Eur. J. Cancer 2019, 108, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Wang, X.; Liao, Y.P.; Chang, C.H.; Nel, A.E. Nanocarrier Co-formulation for Delivery of a TLR7 Agonist plus an Immunogenic Cell Death Stimulus Triggers Effective Pancreatic Cancer Chemo-immunotherapy. ACS Nano 2022, 16, 13168–13182. [Google Scholar] [CrossRef]
- Liu, X.; Situ, A.; Kang, Y.; Villabroza, K.R.; Liao, Y.; Chang, C.H.; Donahue, T.; Nel, A.E.; Meng, H. Irinotecan Delivery by Lipid-Coated Mesoporous Silica Nanoparticles Shows Improved Efficacy and Safety over Liposomes for Pancreatic Cancer. ACS Nano 2016, 10, 2702–2715. [Google Scholar] [CrossRef]
- Allen, S.D.; Liu, X.; Jiang, J.; Liao, Y.P.; Chang, C.H.; Nel, A.E.; Meng, H. Immune checkpoint inhibition in syngeneic mouse cancer models by a silicasome nanocarrier delivering a GSK3 inhibitor. Biomaterials 2021, 269, 120635. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef]
- Liu, X.; Jiang, J.; Liao, Y.-P.; Tang, I.; Zheng, E.; Qiu, W.; Lin, M.; Wang, X.; Ji, Y.; Mei, K.-C.; et al. Combination Chemo-Immunotherapy for Pancreatic Cancer Using the Immunogenic Effects of an Irinotecan Silicasome Nanocarrier Plus Anti-PD-1. Adv. Sci. 2021, 8, 2002147. [Google Scholar] [CrossRef]
- Liu, X.; Jiang, J.; Chang, C.H.; Liao, Y.-P.; Lodico, J.J.; Tang, I.; Zheng, E.; Qiu, W.; Lin, M.; Wang, X.; et al. Development of Facile and Versatile Platinum Drug Delivering Silicasome Nanocarriers for Efficient Pancreatic Cancer Chemo-Immunotherapy. Small 2021, 17, 2005993. [Google Scholar] [CrossRef] [PubMed]
- Balachandran, V.P.; Łuksza, M.; Zhao, J.N.; Makarov, V.; Moral, J.A.; Remark, R.; Herbst, B.; Askan, G.; Bhanot, U.; Senbabaoglu, Y.; et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature 2017, 551, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Balli, D.; Rech, A.J.; Stanger, B.Z.; Vonderheide, R.H. Immune Cytolytic Activity Stratifies Molecular Subsets of Human Pancreatic Cancer. Clin. Cancer Res. 2017, 23, 3129–3138. [Google Scholar] [CrossRef] [PubMed]
- Stromnes, I.M.; Hulbert, A.; Pierce, R.H.; Greenberg, P.D.; Hingorani, S.R. T-cell Localization, Activation, and Clonal Expansion in Human Pancreatic Ductal Adenocarcinoma. Cancer Immunol. Res. 2017, 5, 978–991. [Google Scholar] [CrossRef]
- Michael, A.J.; Derek, S.C.; Albrecht, N.; Tashinga, E.B.; Natalie, C.; Kristopher, K.F.; Christine, F.; Tomoaki, N.; Meredith, E.C.; Heather, I.Z.; et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut 2013, 62, 112. [Google Scholar] [CrossRef]
- Hingorani, S.R.; Zheng, L.; Bullock, A.J.; Seery, T.E.; Harris, W.P.; Sigal, D.S.; Braiteh, F.; Ritch, P.S.; Zalupski, M.M.; Bahary, N.; et al. HALO 202: Randomized Phase II Study of PEGPH20 Plus Nab-Paclitaxel/Gemcitabine Versus Nab-Paclitaxel/Gemcitabine in Patients With Untreated, Metastatic Pancreatic Ductal Adenocarcinoma. J. Clin. Oncol. 2017, 36, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Ijichi, H.; Chytil, A.; Gorska, A.E.; Aakre, M.E.; Bierie, B.; Tada, M.; Mohri, D.; Miyabayashi, K.; Asaoka, Y.; Maeda, S.; et al. Inhibiting Cxcr2 disrupts tumor-stromal interactions and improves survival in a mouse model of pancreatic ductal adenocarcinoma. J. Clin. Investig. 2011, 121, 4106–4117. [Google Scholar] [CrossRef] [PubMed]
- Neesse, A.; Frese, K.K.; Bapiro, T.E.; Nakagawa, T.; Sternlicht, M.D.; Seeley, T.W.; Pilarsky, C.; Jodrell, D.I.; Spong, S.M.; Tuveson, D.A. CTGF antagonism with mAb FG-3019 enhances chemotherapy response without increasing drug delivery in murine ductal pancreas cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 12325–12330. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.W.; Morton, J.P.; Pinese, M.; Saturno, G.; Jamieson, N.B.; McGhee, E.; Timpson, P.; Leach, J.; McGarry, L.; Shanks, E.; et al. Targeting the LOX/hypoxia axis reverses many of the features that make pancreatic cancer deadly: Inhibition of LOX abrogates metastasis and enhances drug efficacy. EMBO Mol. Med. 2015, 7, 1063–1076. [Google Scholar] [CrossRef]
- Chauhan, V.P.; Martin, J.D.; Liu, H.; Lacorre, D.A.; Jain, S.R.; Kozin, S.V.; Stylianopoulos, T.; Mousa, A.S.; Han, X.; Adstamongkonkul, P.; et al. Angiotensin inhibition enhances drug delivery and potentiates chemotherapy by decompressing tumour blood vessels. Nat. Commun. 2013, 4, 2516. [Google Scholar] [CrossRef]
- Xiao, Y.; Qin, T.; Sun, L.; Qian, W.; Li, J.; Duan, W.; Lei, J.; Wang, Z.; Ma, J.; Li, X.; et al. Resveratrol Ameliorates the Malignant Progression of Pancreatic Cancer by Inhibiting Hypoxia-induced Pancreatic Stellate Cell Activation. Cell Transplant. 2020, 29, 963689720929987. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Jia, F.; Huang, Y.; Jin, Q.; Ji, J. Cancer-Associated Fibroblast-Targeted Delivery of Captopril to Overcome Penetration Obstacles for Enhanced Pancreatic Cancer Therapy. ACS Appl. Bio Mater. 2022, 5, 3544–3553. [Google Scholar] [CrossRef]
- Schnittert, J.; Heinrich, M.A.; Kuninty, P.R.; Storm, G.; Prakash, J. Reprogramming tumor stroma using an endogenous lipid lipoxin A4 to treat pancreatic cancer. Cancer Lett. 2018, 420, 247–258. [Google Scholar] [CrossRef]
- Pei, Y.; Chen, L.; Huang, Y.; Wang, J.; Feng, J.; Xu, M.; Chen, Y.; Song, Q.; Jiang, G.; Gu, X.; et al. Sequential Targeting TGF-β Signaling and KRAS Mutation Increases Therapeutic Efficacy in Pancreatic Cancer. Small 2019, 15, e1900631. [Google Scholar] [CrossRef]
- Noel, P.; Von Hoff, D.D.; Saluja, A.K.; Velagapudi, M.; Borazanci, E.; Han, H. Triptolide and Its Derivatives as Cancer Therapies. Trends Pharmacol. Sci. 2019, 40, 327–341. [Google Scholar] [CrossRef]
- Zhang, B.; Jiang, T.; Shen, S.; She, X.; Tuo, Y.; Hu, Y.; Pang, Z.; Jiang, X. Cyclopamine disrupts tumor extracellular matrix and improves the distribution and efficacy of nanotherapeutics in pancreatic cancer. Biomaterials 2016, 103, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Wang, H.; Hsiao, C.-H.; Chow, D.S.L.; Koay, E.J.; Kang, Y.; Wen, X.; Huang, Q.; Ma, Y.; Bankson, J.A.; et al. Simultaneous inhibition of hedgehog signaling and tumor proliferation remodels stroma and enhances pancreatic cancer therapy. Biomaterials 2018, 159, 215–228. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Wu, C.; Abbruzzese, J.; Hwang, R.F.; Li, C. Cyclopamine-Loaded Core-Cross-Linked Polymeric Micelles Enhance Radiation Response in Pancreatic Cancer and Pancreatic Stellate Cells. Mol. Pharm. 2015, 12, 2093–2100. [Google Scholar] [CrossRef] [PubMed]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of Hedgehog Signaling Enhances Delivery of Chemotherapy in a Mouse Model of Pancreatic Cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Qu, C.; Xie, F.; Chen, L.; Liu, L.; Liang, X.; Wu, X.; Wang, P.; Meng, Z. Curcumin suppresses epithelial-to-mesenchymal transition and metastasis of pancreatic cancer cells by inhibiting cancer-associated fibroblasts. Am. J. Cancer Res. 2017, 7, 125–133. [Google Scholar] [CrossRef]
- Ji, T.; Lang, J.; Wang, J.; Cai, R.; Zhang, Y.; Qi, F.; Zhang, L.; Zhao, X.; Wu, W.; Hao, J.; et al. Designing Liposomes To Suppress Extracellular Matrix Expression To Enhance Drug Penetration and Pancreatic Tumor Therapy. ACS Nano 2017, 11, 8668–8678. [Google Scholar] [CrossRef]
- Mei, K.-C.; Liao, Y.-P.; Jiang, J.; Chiang, M.; Khazaieli, M.; Liu, X.; Wang, X.; Liu, Q.; Chang, C.H.; Zhang, X.; et al. Liposomal Delivery of Mitoxantrone and a Cholesteryl Indoximod Prodrug Provides Effective Chemo-immunotherapy in Multiple Solid Tumors. ACS Nano 2020, 14, 13343–13366. [Google Scholar] [CrossRef]
- Vennin, C.; Murphy, K.J.; Morton, J.P.; Cox, T.R.; Pajic, M.; Timpson, P. Reshaping the Tumor Stroma for Treatment of Pancreatic Cancer. Gastroenterology 2018, 154, 820–838. [Google Scholar] [CrossRef]
- Ho, W.J.; Jaffee, E.M.; Zheng, L. The tumour microenvironment in pancreatic cancer—Clinical challenges and opportunities. Nat. Rev. Clin. Oncol. 2020, 17, 527–540. [Google Scholar] [CrossRef]
- Ligorio, M.; Sil, S.; Malagon-Lopez, J.; Nieman, L.T.; Misale, S.; Di Pilato, M.; Ebright, R.Y.; Karabacak, M.N.; Kulkarni, A.S.; Liu, A.; et al. Stromal Microenvironment Shapes the Intratumoral Architecture of Pancreatic Cancer. Cell 2019, 178, 160–175.e27. [Google Scholar] [CrossRef]
- Calon, A.; Tauriello, D.V.; Batlle, E. TGF-beta in CAF-mediated tumor growth and metastasis. Semin. Cancer Biol. 2014, 25, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Al-toub, M.; Almusa, A.; Almajed, M.; Al-Nbaheen, M.; Kassem, M.; Aldahmash, A.; Alajez, N.M. Pleiotropic effects of cancer cells’ secreted factors on human stromal (mesenchymal) stem cells. Stem Cell Res. Ther. 2013, 4, 114. [Google Scholar] [CrossRef] [PubMed]
- De Boeck, A.; Hendrix, A.; Maynard, D.; Van Bockstal, M.; Daniëls, A.; Pauwels, P.; Gespach, C.; Bracke, M.; De Wever, O. Differential secretome analysis of cancer-associated fibroblasts and bone marrow-derived precursors to identify microenvironmental regulators of colon cancer progression. Proteomics 2013, 13, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Qian, Y.; Ye, Z.; Chen, H.; Xie, H.; Zhou, L.; Shen, Y.; Zheng, S. Cancer-associated fibroblasts promote M2 polarization of macrophages in pancreatic ductal adenocarcinoma. Cancer Med. 2017, 6, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N. Pro-resolving lipid mediators are leads for resolution physiology. Nature 2014, 510, 92–101. [Google Scholar] [CrossRef]
- Zong, L.; Li, J.; Chen, X.; Chen, K.; Li, W.; Li, X.; Zhang, L.; Duan, W.; Lei, J.; Xu, Q.; et al. Lipoxin A4 Attenuates Cell Invasion by Inhibiting ROS/ERK/MMP Pathway in Pancreatic Cancer. Oxidative Med. Cell. Longev. 2016, 2016, 6815727. [Google Scholar] [CrossRef]
- Blogowski, W.; Dolegowska, K.; Deskur, A.; Dolegowska, B.; Starzynska, T. Lipoxins and Resolvins in Patients With Pancreatic Cancer: A Preliminary Report. Front. Oncol. 2022, 11, 757073. [Google Scholar] [CrossRef]
- Serhan, C.N.; Chiang, N.; Van Dyke, T.E. Resolving inflammation: Dual anti-inflammatory and pro-resolution lipid mediators. Nat. Rev. Immunol. 2008, 8, 349–361. [Google Scholar] [CrossRef]
- Roach, K.M.; Feghali-Bostwick, C.A.; Amrani, Y.; Bradding, P. Lipoxin A4 Attenuates Constitutive and TGF-β1-Dependent Profibrotic Activity in Human Lung Myofibroblasts. J. Immunol. 2015, 195, 2852–2860. [Google Scholar] [CrossRef]
- Brennan, E.P.; Nolan, K.A.; Börgeson, E.; Gough, O.S.; McEvoy, C.M.; Docherty, N.G.; Higgins, D.F.; Murphy, M.; Sadlier, D.M.; Ali-Shah, S.T.; et al. Lipoxins attenuate renal fibrosis by inducing let-7c and suppressing TGFβR1. J. Am. Soc. Nephrol. 2013, 24, 627–637. [Google Scholar] [CrossRef]
- Zong, L.; Chen, K.; Jiang, Z.; Chen, X.; Sun, L.; Ma, J.; Zhou, C.; Xu, Q.; Duan, W.; Han, L.; et al. Lipoxin A4 reverses mesenchymal phenotypes to attenuate invasion and metastasis via the inhibition of autocrine TGF-β1 signaling in pancreatic cancer. J. Exp. Clin. Cancer Res. 2017, 36, 181. [Google Scholar] [CrossRef] [PubMed]
- Maddox, J.F.; Hachicha, M.; Takano, T.; Petasis, N.A.; Fokin, V.V.; Serhan, C.N. Lipoxin A4 Stable Analogs Are Potent Mimetics That Stimulate Human Monocytes and THP-1 Cells via a G-protein-linked Lipoxin A4 Receptor*. J. Biol. Chem. 1997, 272, 6972–6978. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Hu, S.; Shangguan, J.; Eresen, A.; Li, Y.; Ma, Q.; Yaghmai, V.; Benson Iii, A.B.; Zhang, Z. Dinaciclib prolongs survival in the LSL-Kras(G12D/+); LSL-Trp53(R172H/+); Pdx-1-Cre (KPC) transgenic murine models of pancreatic ductal adenocarcinoma. Am. J. Transl. Res. 2020, 12, 1031–1043. [Google Scholar] [PubMed]
- Simões, R.L.; De-Brito, N.M.; Cunha-Costa, H.; Morandi, V.; Fierro, I.M.; Roitt, I.M.; Barja-Fidalgo, C. Lipoxin A(4) selectively programs the profile of M2 tumor-associated macrophages which favour control of tumor progression. Int. J. Cancer 2017, 140, 346–357. [Google Scholar] [CrossRef]
- Coppola, D.; Nebozhyn, M.; Khalil, F.; Dai, H.; Yeatman, T.; Loboda, A.; Mulé, J.J. Unique ectopic lymph node-like structures present in human primary colorectal carcinoma are identified by immune gene array profiling. Am. J. Pathol. 2011, 179, 37–45. [Google Scholar] [CrossRef]
- Dieu-Nosjean, M.C.; Antoine, M.; Danel, C.; Heudes, D.; Wislez, M.; Poulot, V.; Rabbe, N.; Laurans, L.; Tartour, E.; de Chaisemartin, L.; et al. Long-term survival for patients with non-small-cell lung cancer with intratumoral lymphoid structures. J. Clin. Oncol. 2008, 26, 4410–4417. [Google Scholar] [CrossRef] [PubMed]
- Messina, J.L.; Fenstermacher, D.A.; Eschrich, S.; Qu, X.; Berglund, A.E.; Lloyd, M.C.; Schell, M.J.; Sondak, V.K.; Weber, J.S.; Mulé, J.J. 12-Chemokine gene signature identifies lymph node-like structures in melanoma: Potential for patient selection for immunotherapy? Sci. Rep. 2012, 2, 765. [Google Scholar] [CrossRef] [PubMed]
- Pitzalis, C.; Jones, G.W.; Bombardieri, M.; Jones, S.A. Ectopic lymphoid-like structures in infection, cancer and autoimmunity. Nat. Rev. Immunol. 2014, 14, 447–462. [Google Scholar] [CrossRef]
- Cox, A.D.; Fesik, S.W.; Kimmelman, A.C.; Luo, J.; Der, C.J. Drugging the undruggable RAS: Mission possible? Nat. Rev. Drug Discov. 2014, 13, 828–851. [Google Scholar] [CrossRef]
- Chan-Seng-Yue, M.; Kim, J.C.; Wilson, G.W.; Ng, K.; Figueroa, E.F.; O’Kane, G.M.; Connor, A.A.; Denroche, R.E.; Grant, R.C.; McLeod, J.; et al. Transcription phenotypes of pancreatic cancer are driven by genomic events during tumor evolution. Nat. Genet. 2020, 52, 231–240. [Google Scholar] [CrossRef]
- Mullard, A. Cracking KRAS. Nat. Rev. Drug Discov. 2019, 18, 887–891. [Google Scholar] [CrossRef] [PubMed]
- Oldfield, L.E.; Connor, A.A.; Gallinger, S. Molecular Events in the Natural History of Pancreatic Cancer. Trends Cancer 2017, 3, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.J.; Yu, Z.; Griffith, K.; Hanada, K.; Restifo, N.P.; Yang, J.C. Identification of T-cell Receptors Targeting KRAS-Mutated Human Tumors. Cancer Immunol. Res. 2016, 4, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Dillard, P.; Casey, N.; Pollmann, S.; Vernhoff, P.; Gaudernack, G.; Kvalheim, G.; Wälchli, S.; Inderberg, E.M. Targeting KRAS mutations with HLA class II-restricted TCRs for the treatment of solid tumors. Oncoimmunology 2021, 10, 1936757. [Google Scholar] [CrossRef] [PubMed]
- Carbone, D.P.; Ciernik, I.F.; Kelley, M.J.; Smith, M.C.; Nadaf, S.; Kavanaugh, D.; Maher, V.E.; Stipanov, M.; Contois, D.; Johnson, B.E.; et al. Immunization With Mutant p53- and K-ras–Derived Peptides in Cancer Patients: Immune Response and Clinical Outcome. J. Clin. Oncol. 2005, 23, 5099–5107. [Google Scholar] [CrossRef]
- Gjertsen, M.K.; Buanes, T.; Rosseland, A.R.; Bakka, A.; Gladhaug, I.; Søreide, O.; Eriksen, J.A.; Møller, M.; Baksaas, I.; Lothe, R.A.; et al. Intradermal ras peptide vaccination with granulocyte-macrophage colony-stimulating factor as adjuvant: Clinical and immunological responses in patients with pancreatic adenocarcinoma. Int. J. Cancer 2001, 92, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Domblides, C.; Rochefort, J.; Riffard, C.; Panouillot, M.; Lescaille, G.; Teillaud, J.-L.; Mateo, V.; Dieu-Nosjean, M.-C. Tumor-Associated Tertiary Lymphoid Structures: From Basic and Clinical Knowledge to Therapeutic Manipulation. Front. Immunol. 2021, 12, 698604. [Google Scholar] [CrossRef]
- Lutz, E.R.; Wu, A.A.; Bigelow, E.; Sharma, R.; Mo, G.; Soares, K.; Solt, S.; Dorman, A.; Wamwea, A.; Yager, A.; et al. Immunotherapy converts nonimmunogenic pancreatic tumors into immunogenic foci of immune regulation. Cancer Immunol. Res. 2014, 2, 616–631. [Google Scholar] [CrossRef]
- Zheng, L.; Ding, D.; Edil, B.H.; Judkins, C.; Durham, J.N.; Thomas, D.L., 2nd; Bever, K.M.; Mo, G.; Solt, S.E.; Hoare, J.A.; et al. Vaccine-Induced Intratumoral Lymphoid Aggregates Correlate with Survival Following Treatment with a Neoadjuvant and Adjuvant Vaccine in Patients with Resectable Pancreatic Adenocarcinoma. Clin. Cancer Res. 2021, 27, 1278–1286. [Google Scholar] [CrossRef]
- Moynihan, K.D.; Holden, R.L.; Mehta, N.K.; Wang, C.; Karver, M.R.; Dinter, J.; Liang, S.; Abraham, W.; Melo, M.B.; Zhang, A.Q.; et al. Enhancement of Peptide Vaccine Immunogenicity by Increasing Lymphatic Drainage and Boosting Serum Stability. Cancer Immunol. Res. 2018, 6, 1025–1038. [Google Scholar] [CrossRef]
- Irvine, D.J.; Aung, A.; Silva, M. Controlling timing and location in vaccines. Adv. Drug Deliv. Rev. 2020, 158, 91–115. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.J.; Suh, H.; Bershteyn, A.; Stephan, M.T.; Liu, H.; Huang, B.; Sohail, M.; Luo, S.; Um, S.H.; Khant, H.; et al. Interbilayer-crosslinked multilamellar vesicles as synthetic vaccines for potent humoral and cellular immune responses. Nat. Mater. 2011, 10, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.I.; Barrios, K.; Lee, Y.R.; Linowski, A.K.; Celis, E. BiVax: A peptide/poly-IC subunit vaccine that mimics an acute infection elicits vast and effective anti-tumor CD8 T-cell responses. Cancer Immunol. Immunother. 2013, 62, 787–799. [Google Scholar] [CrossRef] [PubMed]
- Reddy, S.T.; van der Vlies, A.J.; Simeoni, E.; Angeli, V.; Randolph, G.J.; O’Neil, C.P.; Lee, L.K.; Swartz, M.A.; Hubbell, J.A. Exploiting lymphatic transport and complement activation in nanoparticle vaccines. Nat. Biotechnol. 2007, 25, 1159–1164. [Google Scholar] [CrossRef]
- Bershteyn, A.; Hanson, M.C.; Crespo, M.P.; Moon, J.J.; Li, A.V.; Suh, H.; Irvine, D.J. Robust IgG responses to nanograms of antigen using a biomimetic lipid-coated particle vaccine. J. Control. Release 2012, 157, 354–365. [Google Scholar] [CrossRef]
- Mehta, N.K.; Pradhan, R.V.; Soleimany, A.P.; Moynihan, K.D.; Rothschilds, A.M.; Momin, N.; Rakhra, K.; Mata-Fink, J.; Bhatia, S.N.; Wittrup, K.D.; et al. Pharmacokinetic tuning of protein-antigen fusions enhances the immunogenicity of T-cell vaccines. Nat. Biomed. Eng. 2020, 4, 636–648. [Google Scholar] [CrossRef]
- Zhang, Q.; Wu, S. Tertiary lymphoid structures are critical for cancer prognosis and therapeutic response. Front. Immunol. 2022, 13, 1063711. [Google Scholar] [CrossRef]
- Delvecchio, F.R.; Fincham, R.E.A.; Spear, S.; Clear, A.; Roy-Luzarraga, M.; Balkwill, F.R.; Gribben, J.G.; Bombardieri, M.; Hodivala-Dilke, K.; Capasso, M.; et al. Pancreatic Cancer Chemotherapy Is Potentiated by Induction of Tertiary Lymphoid Structures in Mice. Cell Mol. Gastroenterol. Hepatol. 2021, 12, 1543–1565. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Thommen, D.S. Tertiary lymphoid structures in cancer. Science 2022, 375, eabf9419. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Active Agent | Pre-Clinical Model | Mechanism | References |
|---|---|---|---|---|
| ECM | PEGPH20 | Pancreatic tumor | Depletion of hyaluronic acid | [31,32] |
| ECM | Pamrevlumab (FG-3019) | Pancreatic tumor | Reduction in Connective Tissue Growth Factor (CTGF) expression | [33,34] |
| ECM | Simtuzumab (GS-6624) | Pancreatic tumor | Lysyl oxidase inhibitor | [35] |
| ECM | Losartan | Pancreatic tumor | Angiotensin inhibition reduces stromal collagen and hyaluronan production | [36] |
| CAF-cancer cell crosstalk | Resveratrol | Pancreatic tumor | Stromal remodeling by reducing the number of CAFs and leukocytes | [37] |
| ECM | Captopril | Pancreatic tumor | TGF-β pathway inhibition | [38] |
| ECM | Lipoxin A4 | Pancreatic tumor | TGF-β pathway inhibition | [39] |
| CAF-cancer cell crosstalk | Fraxinellone | Pancreatic tumor | TGF-β pathway inhibition | [40] |
| CAF-cancer cell crosstalk | Triptolide | Pancreatic tumor | TGF-β pathway inhibition | [41] |
| ECM | Cyclopamine | Pancreatic tumor | Hedgehog pathway inhibition | [42,43,44] |
| CAF activation | Vismodegib (GDC-0449) | Pancreatic tumor | Hedgehog pathway inhibition | [45] |
| CAF-cancer cell crosstalk | Curcumin | Pancreatic tumor | EMT inhibition | [46] |
| ECM | Pirfenidone | Pancreatic tumor | MMP-2 reduction | [47] |
| Liposomes (0.2 mol% LXA4) | Size (PDI) | Zeta Potential | Loading Capacity | Loading Efficiency |
|---|---|---|---|---|
| (Units) | (nm) | (mV) | (%) | (%) |
| Pre-loading | 88.2 (0.15) | −3.06 ± 2.1 | -- | -- |
| Post loading | 76.2 (0.09) | −2.53 ± 3.87 | 28.95 | 82.09 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chattopadhyay, S.; Liao, Y.-P.; Wang, X.; Nel, A.E. Use of Stromal Intervention and Exogenous Neoantigen Vaccination to Boost Pancreatic Cancer Chemo-Immunotherapy by Nanocarriers. Bioengineering 2023, 10, 1205. https://doi.org/10.3390/bioengineering10101205
Chattopadhyay S, Liao Y-P, Wang X, Nel AE. Use of Stromal Intervention and Exogenous Neoantigen Vaccination to Boost Pancreatic Cancer Chemo-Immunotherapy by Nanocarriers. Bioengineering. 2023; 10(10):1205. https://doi.org/10.3390/bioengineering10101205
Chicago/Turabian StyleChattopadhyay, Saborni, Yu-Pei Liao, Xiang Wang, and André E. Nel. 2023. "Use of Stromal Intervention and Exogenous Neoantigen Vaccination to Boost Pancreatic Cancer Chemo-Immunotherapy by Nanocarriers" Bioengineering 10, no. 10: 1205. https://doi.org/10.3390/bioengineering10101205
APA StyleChattopadhyay, S., Liao, Y.-P., Wang, X., & Nel, A. E. (2023). Use of Stromal Intervention and Exogenous Neoantigen Vaccination to Boost Pancreatic Cancer Chemo-Immunotherapy by Nanocarriers. Bioengineering, 10(10), 1205. https://doi.org/10.3390/bioengineering10101205