



In-Silico Lead Druggable Compounds Identification against SARS COVID-19 Main Protease Target from In-House, Chembridge and Zinc Databases by Structure-Based Virtual Screening, Molecular Docking and Molecular Dynamics Simulations
 , , ,
, , ,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Protein Preparation
2.2. Preparation of Databases
2.3. Structure-Based Virtual Screening
2.4. Molecular Docking
2.5. Molecular Dynamics Simulation (MDS)
2.6. Assessment of Binding Free Energy
3. Results and Discussion
3.1. Structure-Based Virtual Screening
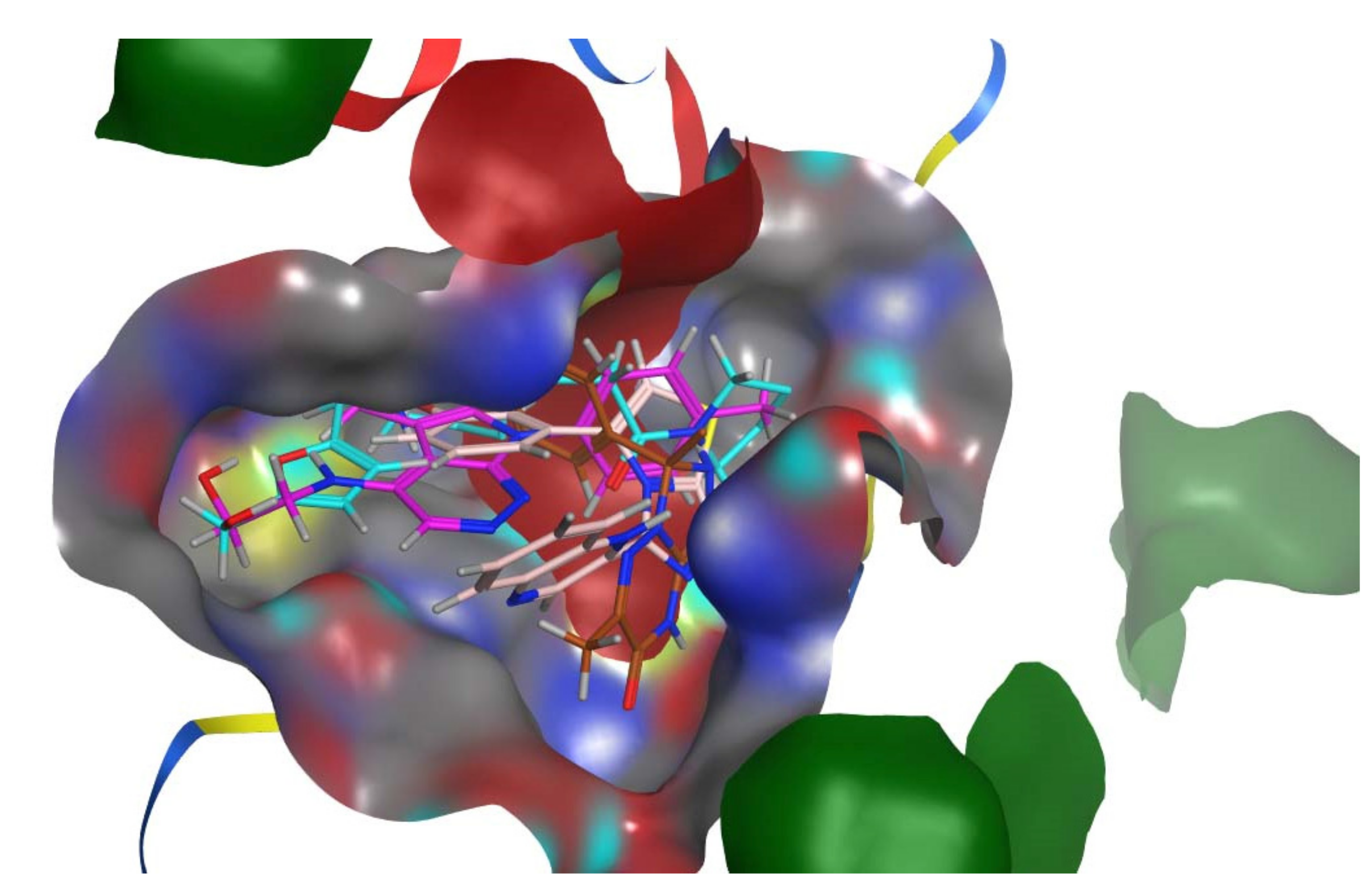
3.2. Molecular Docking
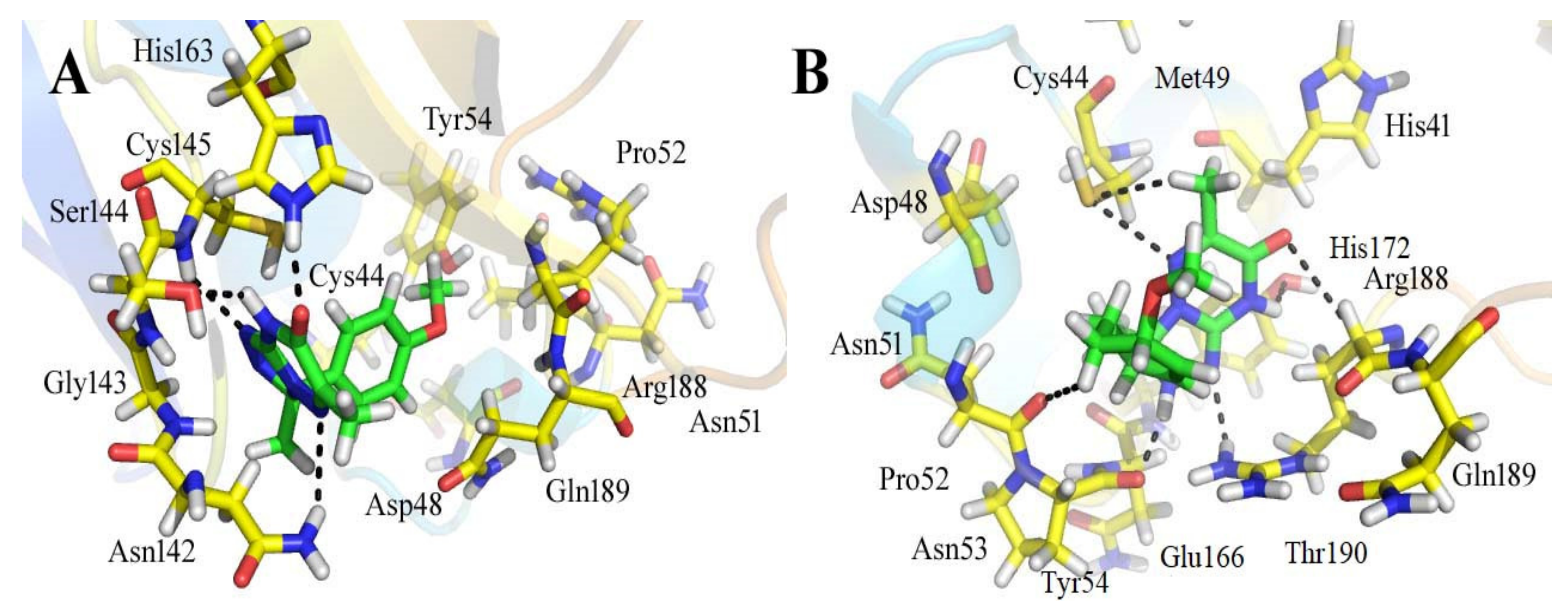
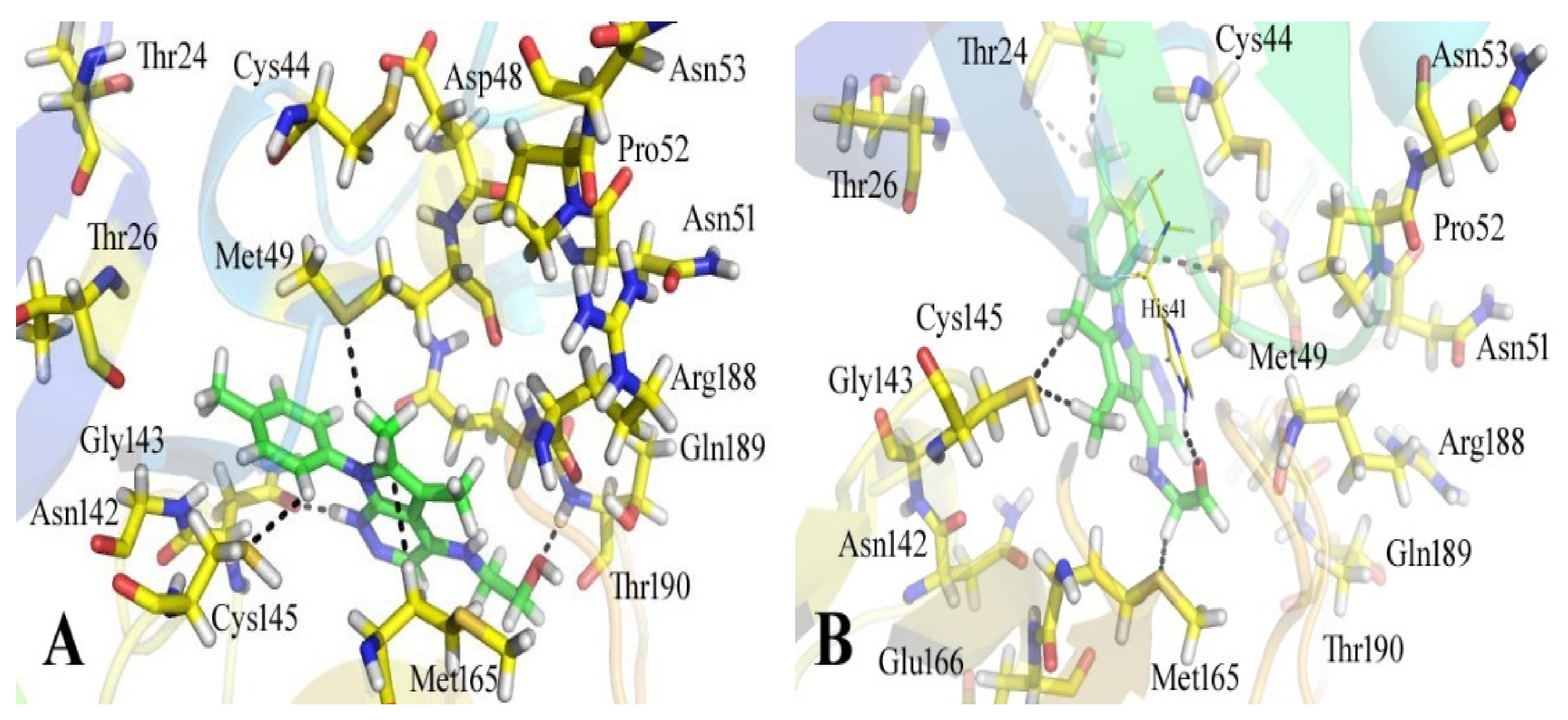
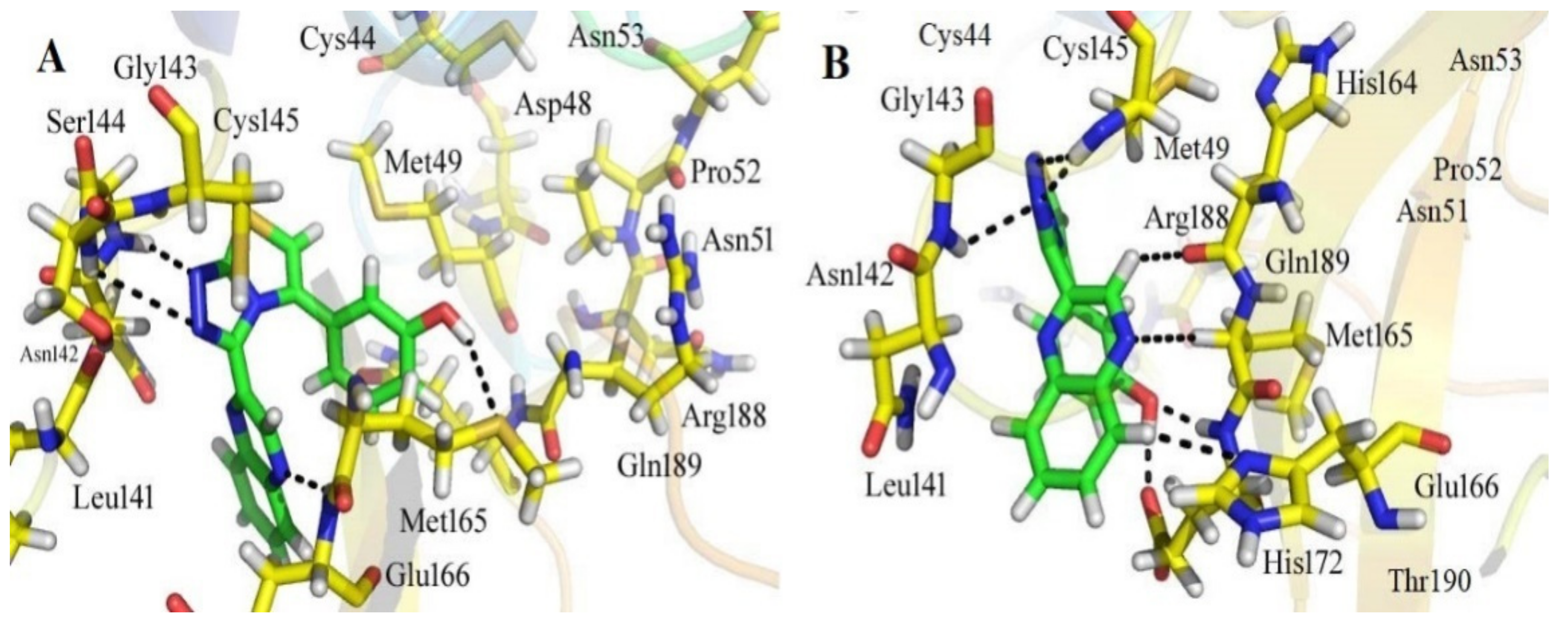
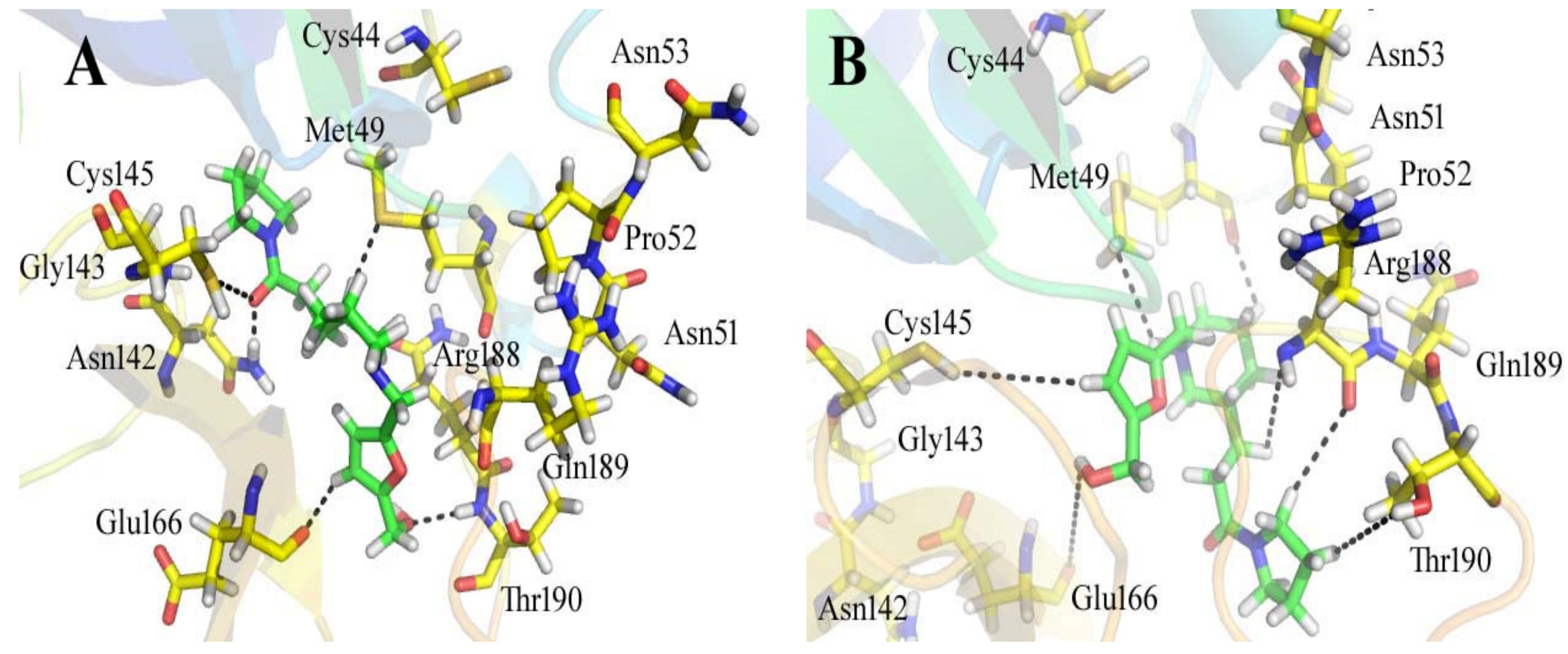
3.3. Analyses of the Binding Interactions of Finally Selected Drug-like Compounds
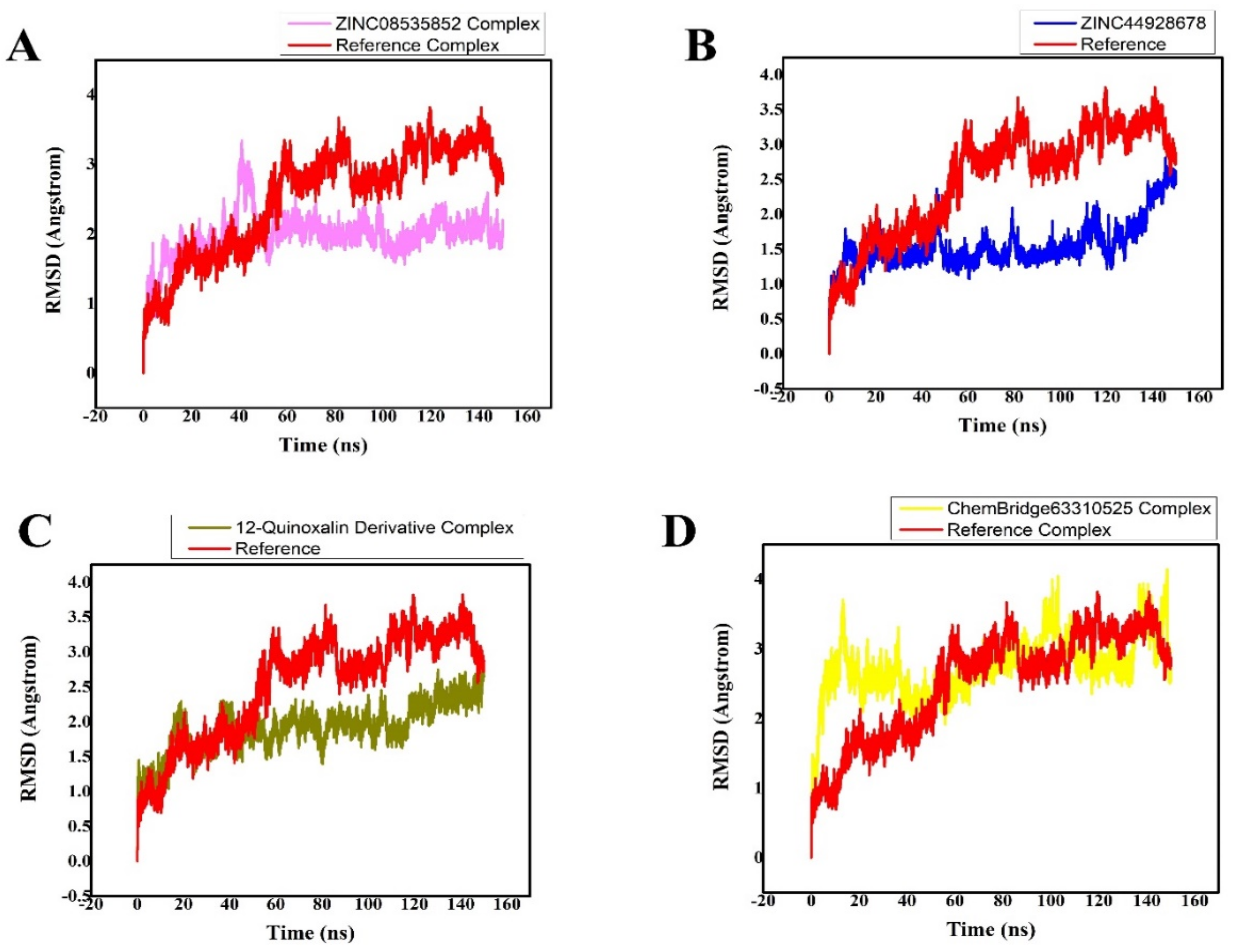
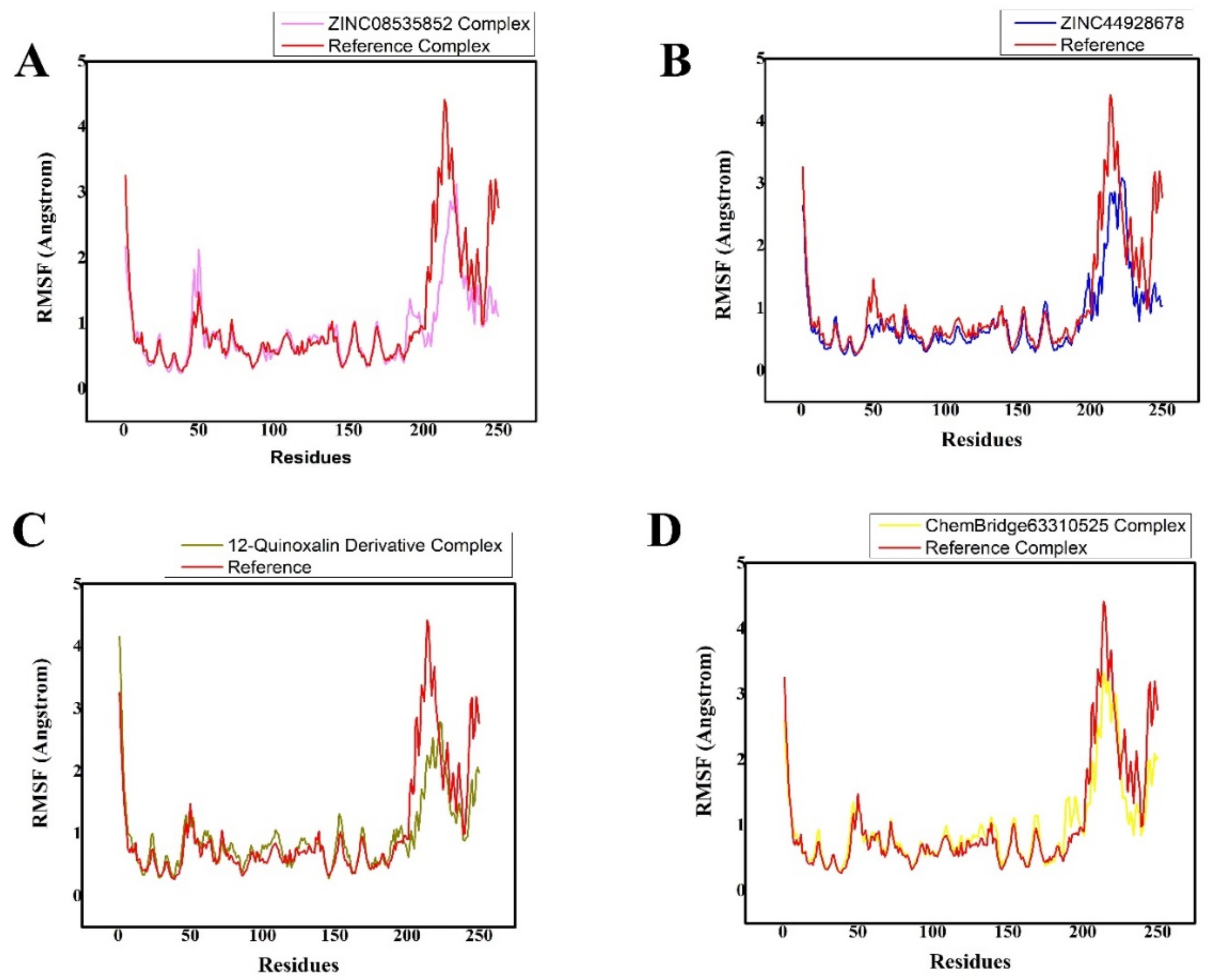
3.4. MD Simulation Analysis of the Final Lead Hit/Mpro Complexes
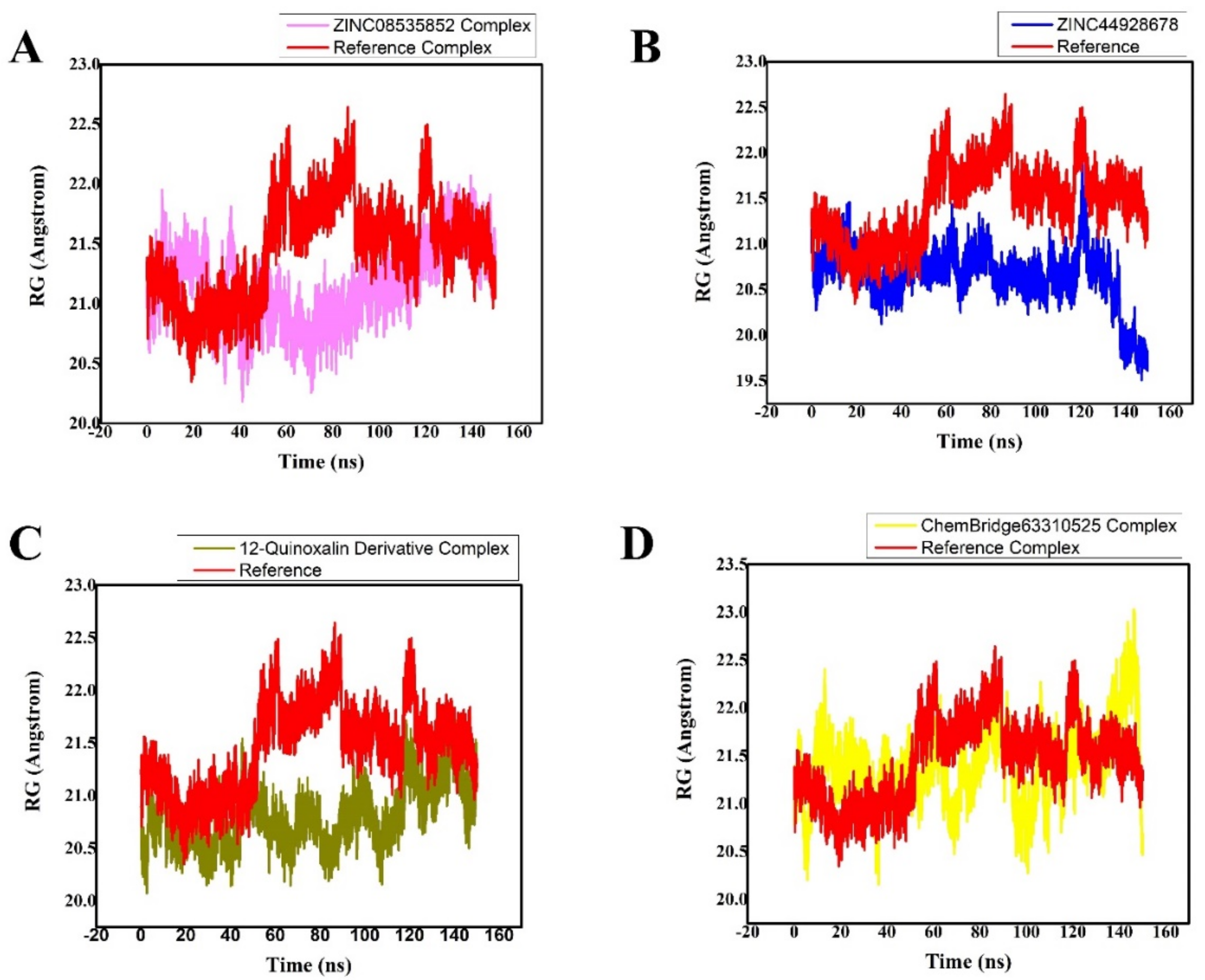
3.5. Radius of Gyration (Rg)
3.6. Molecular Mechanics with Generalized Born and Surface Area Solvation (MMGBSA)
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Team, E. The epidemiological characteristics of an outbreak of 2019 novel coronavirus diseases (COVID-19)—China, 2020. China CDC Wkly. 2020, 2, 113. [Google Scholar]
- Gorbalenya, A.E.; Baker, S.C.; Baric, R.; Groot, R.J.d.; Drosten, C.; Gulyaeva, A.A.; Haagmans, B.L.; Lauber, C.; Leontovich, A.M.; Neuman, B.W. Severe acute respiratory syndrome-related coronavirus: The species and its viruses–a statement of the Coronavirus Study Group. BioRxiv 2020, 1–15. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Tenchov, R.; Smoot, J.; Liu, C.; Watkins, S.; Zhou, Q. A comprehensive review of the global efforts on COVID-19 vaccine development. ACS Cent. Sci. 2021, 7, 512–533. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Li, F.; Shi, Z.-L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef]
- Jalal, K.; Khan, K.; Basharat, Z.; Abbas, M.N.; Uddin, R.; Ali, F.; Khan, S.A. Reverse vaccinology approach for multi-epitope centered vaccine design against delta variant of the SARS-CoV-2. Environ. Sci. Pollut. Res. 2022, 29, 60035–60053. [Google Scholar] [CrossRef]
- Li, Q.; Wu, J.; Nie, J.; Zhang, L.; Hao, H.; Liu, S.; Zhao, C.; Zhang, Q.; Liu, H.; Nie, L. The impact of mutations in SARS-CoV-2 spike on viral infectivity and antigenicity. Cell 2020, 182, 1284–1294.e9. [Google Scholar] [CrossRef]
- Plante, J.A.; Liu, Y.; Liu, J.; Xia, H.; Johnson, B.A.; Lokugamage, K.G.; Zhang, X.; Muruato, A.E.; Zou, J.; Fontes-Garfias, C.R. Spike mutation D614G alters SARS-CoV-2 fitness. Nature 2021, 592, 116–121. [Google Scholar] [CrossRef]
- Ge, X.-Y.; Li, J.-L.; Yang, X.-L.; Chmura, A.A.; Zhu, G.; Epstein, J.H.; Mazet, J.K.; Hu, B.; Zhang, W.; Peng, C. Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature 2013, 503, 535–538. [Google Scholar] [CrossRef]
- Liu, C.; Zhou, Q.; Li, Y.; Garner, L.V.; Watkins, S.P.; Carter, L.J.; Smoot, J.; Gregg, A.C.; Daniels, A.D.; Jervey, S. Research and Development on Therapeutic Agents and Vaccines for COVID-19 and Related Human Coronavirus Diseases. ACS Cent. Sci. 2020, 6, 315–331. [Google Scholar] [CrossRef] [PubMed]
- Zumla, A.; Chan, J.F.; Azhar, E.I.; Hui, D.S.; Yuen, K.-Y. Coronaviruses—Drug discovery and therapeutic options. Nat. Rev. Drug Discov. 2016, 15, 327–347. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Xi, K.; Grum-Tokars, V.; Xu, X.; Ratia, K.; Fu, W.; Houser, K.V.; Baker, S.C.; Johnson, M.E.; Mesecar, A.D. Structure-based design, synthesis, and biological evaluation of peptidomimetic SARS-CoV 3CLpro inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 5876–5880. [Google Scholar] [CrossRef]
- Wu, C.; Liu, Y.; Yang, Y.; Zhang, P.; Zhong, W.; Wang, Y.; Wang, Q.; Xu, Y.; Li, M.; Li, X. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B 2020, 10, 766–788. [Google Scholar] [CrossRef]
- Zhavoronkov, A. Medicinal chemists versus machines challenge: What will it take to adopt and advance artificial intelligence for drug discovery? J. Chem. Inf. Model. 2020, 60, 2657–2659. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, A.; Narwal, M.; Majowicz, S.A.; Varricchio, C.; Toner, S.A.; Ballatore, C.; Brancale, A.; Murakami, K.S.; Jose, J. Identification of SARS-CoV-2 inhibitors targeting Mpro and PLpro using in-cell-protease assay. Commun. Biol. 2022, 5, 169. [Google Scholar] [CrossRef]
- Shams ul Hassan, S.; Abbas, S.Q.; Hassan, M.; Jin, H.-Z. Computational Exploration of Anti-Cancer Potential of GUAIANE Dimers from Xylopia vielana by Targeting B-Raf Kinase Using Chemo-Informatics, Molecular Docking, and MD Simulation Studies. Anti-Cancer Agents Med. Chem. 2022, 22, 731–746. [Google Scholar] [CrossRef]
- Biswas, D.; Nandy, S.; Mukherjee, A.; Pandey, D.; Dey, A. Moringa oleifera Lam. and derived phytochemicals as promising antiviral agents: A review. S. Afr. J. Bot. 2020, 129, 272–282. [Google Scholar] [CrossRef]
- Keretsu, S.; Bhujbal, S.P.; Cho, S.J. Molecular modeling studies of pyrrolo [2,3-d] pyrimidin-4-amine derivatives as JAK1 inhibitors based on 3D-QSAR, molecular docking, molecular dynamics (MD) and MM-PBSA calculations. J. Biomol. Struct. Dyn. 2021, 39, 753–765. [Google Scholar] [CrossRef]
- Zhou, T.; Wu, Z.; Das, S.; Eslami, H.; Müller-Plathe, F. How Ethanolic Disinfectants Disintegrate Coronavirus Model Membranes: A Dissipative Particle Dynamics Simulation Study. J. Chem. Theory Comput. 2022, 18, 2597–2615. [Google Scholar] [CrossRef] [PubMed]
- Jakalian, A.; Jack, D.B.; Bayly, C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 2002, 23, 1623–1641. [Google Scholar] [CrossRef]
- Ghufran, M.; Khan, H.A.; Ullah, M.; Ghufran, S.; Ayaz, M.; Siddiq, M.; Hassan, S.S.u.; Bungau, S. In Silico Strategies for Designing of Peptide Inhibitors of Oncogenic K-Ras G12V Mutant: Inhibiting Cancer Growth and Proliferation. Cancers 2022, 14, 4884. [Google Scholar] [CrossRef]
- Ghufran, M.; Rehman, A.U.; Ayaz, M.; Ul-Haq, Z.; Uddin, R.; Azam, S.S.; Wadood, A. New lead compounds identification against KRas mediated cancers through pharmacophore-based virtual screening and in vitro assays. J. Biomol. Struct. Dyn. 2022, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Hassan, S.S.; Tiwari, S.; Guimarães, L.C.; Jamal, S.B.; Folador, E.; Sharma, N.B.; de Castro Soares, S.; Almeida, S.; Ali, A.; Islam, A. Proteome scale comparative modeling for conserved drug and vaccine targets identification in Corynebacterium pseudotuberculosis. BMC Genom. 2014, 15, S3. [Google Scholar] [CrossRef]
- Ghufran, M.; Rehman, A.U.; Shah, M.; Ayaz, M.; Ng, H.L.; Wadood, A. In-silico design of peptide inhibitors of K-Ras target in cancer disease. J. Biomol. Struct. Dyn. 2020, 38, 5488–5499. [Google Scholar] [CrossRef]
- Taj, S.; Ahmad, M.; Alshammari, A.; Alghamdi, A.; Ashfaq, U.A. Exploring the therapeutic potential of benzothiazine-pyrazole hybrid molecules against alpha-glucosidase: Pharmacological and molecular modelling based approach. Saudi J. Biol. Sci. 2022, 29, 1416–1421. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed]
- Zwanzig, R. Nonlinear generalized Langevin equations. J. Stat. Phys. 1973, 9, 215–220. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Gotz, A.W.; Williamson, M.J.; Xu, D.; Poole, D.; Le Grand, S.; Walker, R.C. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 1. Generalized born. J. Chem. Theory Comput. 2012, 8, 1542–1555. [Google Scholar] [CrossRef]
- Sun, H.; Li, Y.; Tian, S.; Xu, L.; Hou, T. Assessing the performance of MM/PBSA and MM/GBSA methods. 4. Accuracies of MM/PBSA and MM/GBSA methodologies evaluated by various simulation protocols using PDBbind data set. Phys. Chem. Chem. Phys. 2014, 16, 16719–16729. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.R., III; McGee, T.D., Jr.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA. py: An efficient program for end-state free energy calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]
- Hui, D.S.; Azhar, E.I.; Madani, T.A.; Ntoumi, F.; Kock, R.; Dar, O.; Ippolito, G.; Mchugh, T.D.; Memish, Z.A.; Drosten, C. The continuing 2019-nCoV epidemic threat of novel coronaviruses to global health—The latest 2019 novel coronavirus outbreak in Wuhan, China. Int. J. Infect. Dis. 2020, 91, 264–266. [Google Scholar] [CrossRef]
- Wang, F.; Chen, C.; Tan, W.; Yang, K.; Yang, H. Structure of main protease from human coronavirus NL63: Insights for wide spectrum anti-coronavirus drug design. Sci. Rep. 2016, 6, 22677. [Google Scholar] [CrossRef] [PubMed]
- Maia, E.H.B.; Assis, L.C.; De Oliveira, T.A.; Da Silva, A.M.; Taranto, A.G. Structure-based virtual screening: From classical to artificial intelligence. Front. Chem. 2020, 8, 343. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Lipinski, C.A. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods 2000, 44, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Mahgoub, R.E.; Mohamed, F.E.; Alzyoud, L.; Ali, B.R.; Ferreira, J.; Rabeh, W.M.; AlNeyadi, S.S.; Atatreh, N.; Ghattas, M.A. The Discovery of Small Allosteric and Active Site Inhibitors of the SARS-CoV-2 Main Protease via Structure-Based Virtual Screening and Biological Evaluation. Molecules 2022, 27, 6710. [Google Scholar] [CrossRef]
- Kumar, P.; Choonara, Y.E.; Pillay, V. In silico affinity profiling of neuroactive polyphenols for post-traumatic calpain inactivation: A molecular docking and atomistic simulation sensitivity analysis. Molecules 2014, 20, 135–168. [Google Scholar] [CrossRef]
- Li, W.-Y.; Duan, Y.-Q.; Ma, Y.-C.; Lu, X.-H.; Ma, Y.; Wang, R.-L. Exploring the cause of the inhibitor 4AX attaching to binding site disrupting protein tyrosine phosphatase 4A1 trimerization by molecular dynamic simulation. J. Biomol. Struct. Dyn. 2019, 37, 4840–4851. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. NO | Compound Names | Structures | Docking Scores |
|---|---|---|---|
| 1 | ZINC08535852 |  | −41.3801 |
| 2 | ZINC44928678 |  | −41.0291 |
| 3 | ZINC72171104 |  | −39.5487 |
| 4 | 12-quinoxaline derivative |  | −38.7102 |
| 5 | ChemBridge63310525 |  | −38.0478 |
| 6 | 18-quinoxaline derivative |  | −37.5300 |
| 7 | ChemBridge53208972 |  | −35.4302 |
| 8 | 25-quinoxaline derivative |  | −34.3177 |
| Reference | N3 |  | −29.5841 |
| Compounds IDs | Ligand | Receptor | Interaction | Distance | E (kcal/mol) | ||
|---|---|---|---|---|---|---|---|
| ZINC08535852 | C1 1 | SG | CYS | 44 | H-donor | 3.91 | −0.2 |
| N1 3 | SG | CYS | 44 | H-donor | 3.34 | −0.6 | |
| N4 7 | O | PRO | 52 | H-donor | 3.09 | −1.1 | |
| C6 9 | O | ASN | 51 | H-donor | 3.22 | −0.1 | |
| NH 11 | O | Tyr | 54 | H-donor | 3.69 | −0.1 | |
| N3 6 | NH1 | ARG | 188 | H-acceptor | 3.01 | −0.7 | |
| O2 19 | CA | ARG | 188 | H-acceptor | 3.85 | −0.1 | |
| ZINC44928678 | C1 1 | OG1 | THR | 24 | H-donor | 3.65 | −0.1 |
| C1 1 | O | THR | 24 | H-donor | 3.65 | −0.1 | |
| C6 6 | SD | MET | 49 | H-donor | 3.82 | −0.2 | |
| C14 18 | SD | MET | 165 | H-donor | 3.58 | −0.1 | |
| C16 21 | SG | CYS | 145 | H-donor | 3.88 | −0.1 | |
| C17 22 | SG | CYS | 145 | H-donor | 4.1 | −0.3 | |
| O1 20 | NE2 | HIS | 41 | H-acceptor | 2.98 | −0.9 | |
| ZINC72171104 | N2 8 | SG | CYS | 145 | H-donor | 3.33 | −2.2 |
| N5 20 | O | THR | 190 | H-donor | 2.83 | −4.7 | |
| 5-ring | CA | ASN | 142 | pi-H | 4.1 | −0.4 | |
| 5-ring | CA | MET | 165 | pi-H | 3.51 | −0.3 | |
| 6-ring | CB | MET | 165 | pi-H | 3.59 | −0.5 | |
| 6-ring | CD | PRO | 168 | pi-H | 4.88 | −0.3 | |
| 5-ring | CD | PRO | 168 | pi-H | 4.43 | −0.3 | |
| 5-ring | CA | GLN | 189 | pi-H | 3.64 | −1 | |
| 6-ring | CG | GLN | 189 | pi-H | 4.21 | −0.7 | |
| 12-quinoxaline derivative | C3 3 | ND1 | HIE | 172 | H-donor | 3.86 | −0.2 |
| C24 24 | O | HIE | 164 | H-donor | 3.43 | −0.3 | |
| O25 25 | OE1 | GLU | 166 | H-donor | 2.53 | −4.7 | |
| O25 25 | NH | GLU | 166 | H-acceptor | 2.4 | −4.8 | |
| N12 12 | N | CYS | 145 | H-acceptor | 3.12 | −0.8 | |
| N11 11 | N | CYS | 145 | H-acceptor | 3.12 | −0.8 | |
| N4 4 | CA | MET | 165 | H-acceptor | 3.25 | −0.1 | |
| O25 25 | N | GLU | 166 | H-acceptor | 2.86 | −1.3 | |
| 18-quinoxaline derivative | C3 3 | SG | CYS | 44 | H-donor | 2.97 | −0.2 |
| O27 27 | NH | ASN | 142 | H-donor | 2.8 | −0.6 | |
| O27 27 | O | GLY | 143 | H-acceptor | 2.5 | −0.9 | |
| C24 24 | NH | GLY | 143 | H-acceptor | 1.6 | −1.8 | |
| C26 28 | SG | CYS | 145 | H-donor | 2.39 | −0.1 | |
| 6-ring | CG | MET | 49 | pi-H | 4.73 | −0.1 | |
| 6-ring | CG | MET | 49 | pi-H | 3.86 | −1 | |
| C3 3 | CA | MET | 49 | pi-H | 4.23 | −0.8 | |
| 25-quinoxaline derivative | C3 3 | SD | MET | 49 | H-donor | 2.84 | −0.2 |
| O26 26 | O | GLU | 166 | H-donor | 2.7 | −2.3 | |
| O25 25 | NH | GLU | 166 | H-donor | 2.8 | −2.1 | |
| N13 13 | CB | THR | 190 | H-acceptor | 2.58 | −0.2 | |
| N7 7 | CA | GLN | 189 | H-donor | 3.36 | −0.3 | |
| N10 10 | NH | ARG | 188 | H-acceptor | 3.31 | −0.1 | |
| 5-ring | N | THR | 190 | pi-H | 3.67 | −0.3 | |
| ChemBridge63310525 | C9 9 | O | ARG | 188 | H-donor | 2.93 | −0.5 |
| C11 11 | NH | ARG | 188 | H-donor | 2.5 | −0.8 | |
| C8 8 | SD | CYS | 145 | H-donor | 3.59 | −0.1 | |
| O19 19 | O | GLU | 166 | H-donor | 3.44 | −0.4 | |
| C14 14 | O | MET | 49 | H-donor | 3.13 | −0.2 | |
| C22 22 | OH | THR | 190 | H-acceptor | 2.53 | −0.1 | |
| C20 14 | C | MET | 49 | H-donor | 3.13 | −0.2 | |
| ChemBridge53208972 | C15 22 | O | HIE | 41 | H-donor | 3.75 | −0.1 |
| C18 28 | O | HIP | 164 | H-donor | 3.3 | −0.2 | |
| C19 29 | SD | MET | 49 | H-donor | 4.48 | −0.1 | |
| C18 28 | 5-ring | HIE | 41 | H-pi | 3.58 | −0.1 | |
| 6-ring | CA | ARG | 188 | pi-H | 4.02 | −0.4 | |
| 6-ring | CD | ARG | 188 | pi-H | 4.42 | −0.1 | |
| 5-ring | CD | ARG | 188 | pi-H | 4.52 | −0.1 | |
| 6-ring | N | GLN | 189 | pi-H | 4.76 | −0.3 | |
| N3 (reference ligand) | N 13 | O | THR | 190 | H-donor | 2.85 | −2.6 |
| N 23 | O | GLU | 166 | H-donor | 2.83 | −4.8 | |
| N 39 | OE1 | GLN | 189 | H-donor | 2.93 | −3.3 | |
| O 85 | N | GLY | 143 | H-acceptor | 2.80 | −1.0 | |
| CD1 50 | 5-ring | HIS | 41 | H-pi | 4.08 | −0.5 | |
| Compound Names | RMSD (Å) | Binding Free Energies (Kcal/mol) |
|---|---|---|
| ZINC08535852 (ZINC database) | 1.8 0.005 | −39.5546 0.3671 |
| ZINC44928678 (ZINC database) | 2.5 0.005 | −35.8398 0.1901 |
| 12-quinoxaline derivative in-house database | 2.4 0.005 | −37.8210 0.5091 |
| ChemBridge63310525 (ChemBridge database) | 2.80.005 | −33.2041 0.2102 |
| Reference (N3) | 2.80.0126 | −20.7812 0.4214 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghufran, M.; Ullah, M.; Khan, H.A.; Ghufran, S.; Ayaz, M.; Siddiq, M.; Abbas, S.Q.; Hassan, S.S.u.; Bungau, S. In-Silico Lead Druggable Compounds Identification against SARS COVID-19 Main Protease Target from In-House, Chembridge and Zinc Databases by Structure-Based Virtual Screening, Molecular Docking and Molecular Dynamics Simulations. Bioengineering 2023, 10, 100. https://doi.org/10.3390/bioengineering10010100
Ghufran M, Ullah M, Khan HA, Ghufran S, Ayaz M, Siddiq M, Abbas SQ, Hassan SSu, Bungau S. In-Silico Lead Druggable Compounds Identification against SARS COVID-19 Main Protease Target from In-House, Chembridge and Zinc Databases by Structure-Based Virtual Screening, Molecular Docking and Molecular Dynamics Simulations. Bioengineering. 2023; 10(1):100. https://doi.org/10.3390/bioengineering10010100
Chicago/Turabian StyleGhufran, Mehreen, Mehran Ullah, Haider Ali Khan, Sabreen Ghufran, Muhammad Ayaz, Muhammad Siddiq, Syed Qamar Abbas, Syed Shams ul Hassan, and Simona Bungau. 2023. "In-Silico Lead Druggable Compounds Identification against SARS COVID-19 Main Protease Target from In-House, Chembridge and Zinc Databases by Structure-Based Virtual Screening, Molecular Docking and Molecular Dynamics Simulations" Bioengineering 10, no. 1: 100. https://doi.org/10.3390/bioengineering10010100
APA StyleGhufran, M., Ullah, M., Khan, H. A., Ghufran, S., Ayaz, M., Siddiq, M., Abbas, S. Q., Hassan, S. S. u., & Bungau, S. (2023). In-Silico Lead Druggable Compounds Identification against SARS COVID-19 Main Protease Target from In-House, Chembridge and Zinc Databases by Structure-Based Virtual Screening, Molecular Docking and Molecular Dynamics Simulations. Bioengineering, 10(1), 100. https://doi.org/10.3390/bioengineering10010100