
Laboratory Experiments to Evaluate the Effectiveness of Persulfate to Oxidize BTEX in Saline Environment and at Elevated Temperature Using Stable Isotopes


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Experimental Procedure
2.3. Isotope Analyses
3. Results and Discussion
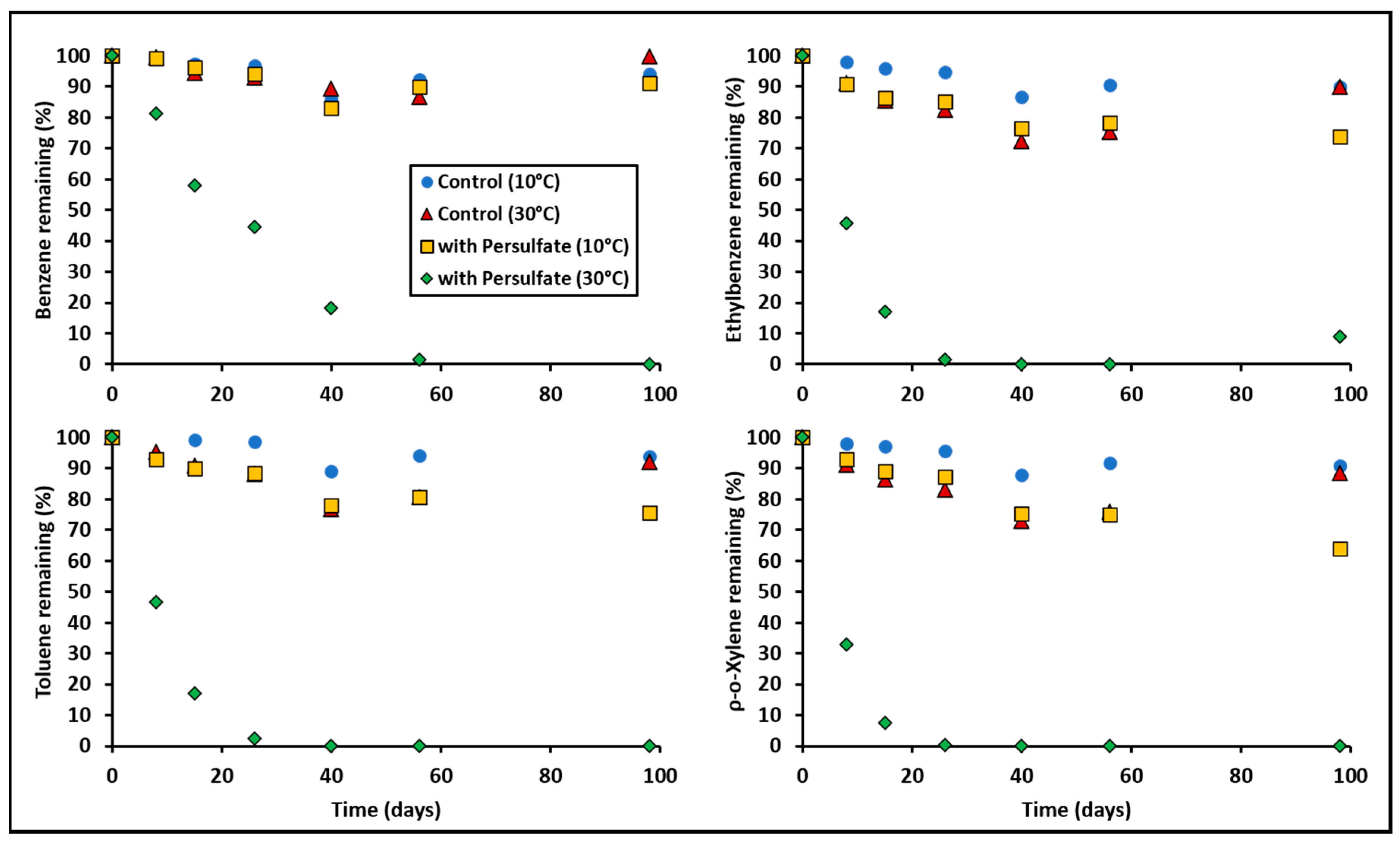
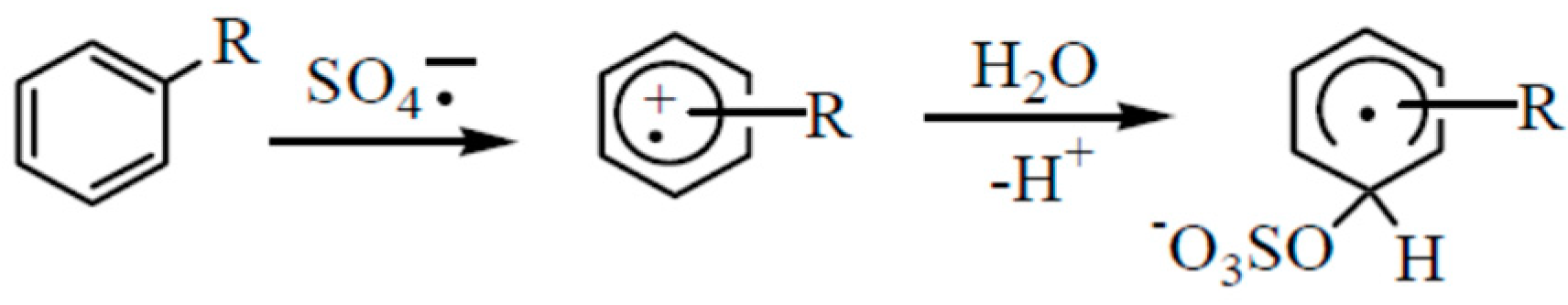
3.1. BTEX Oxidation by Persulfate
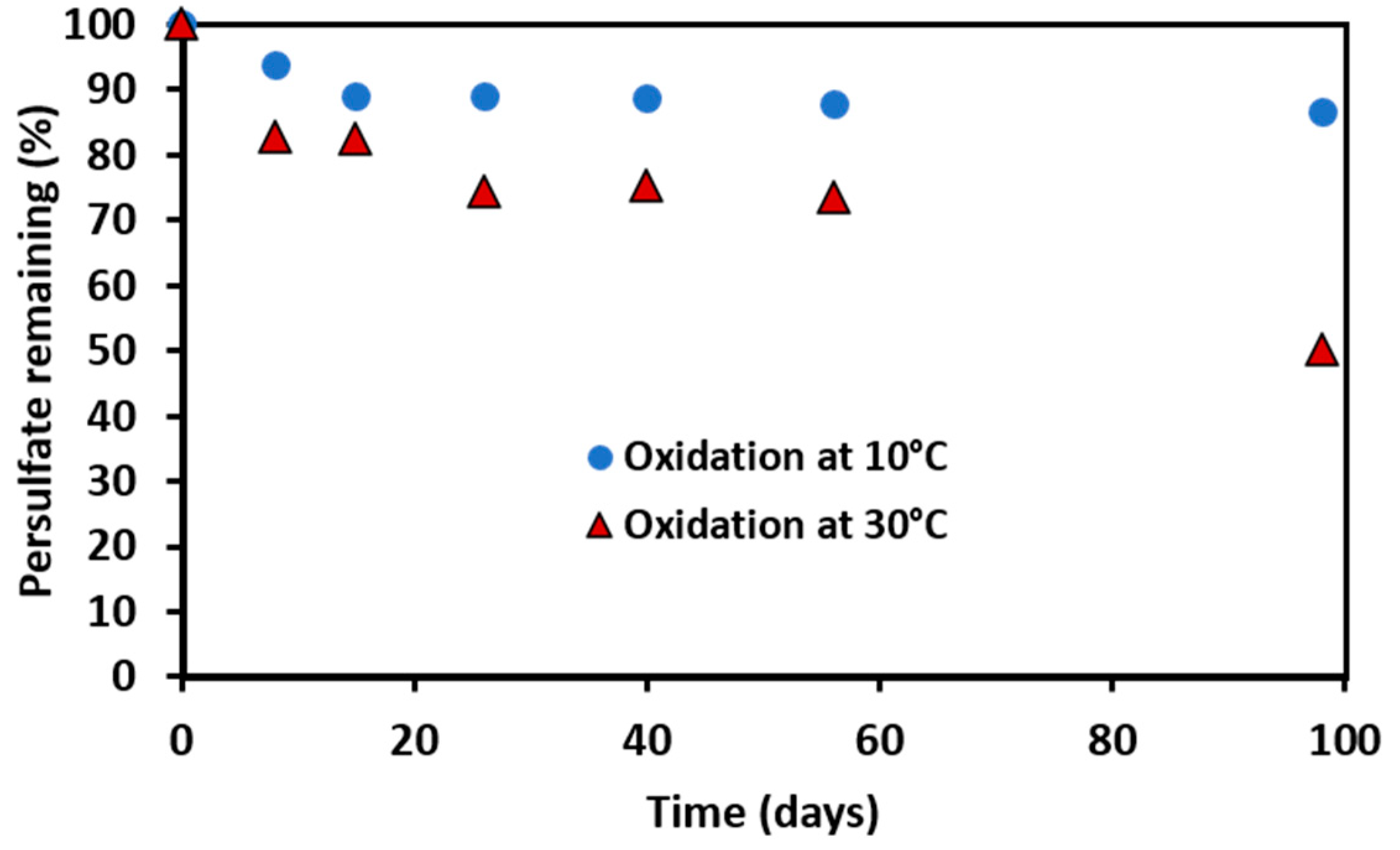
3.2. Persistence of Persulfate
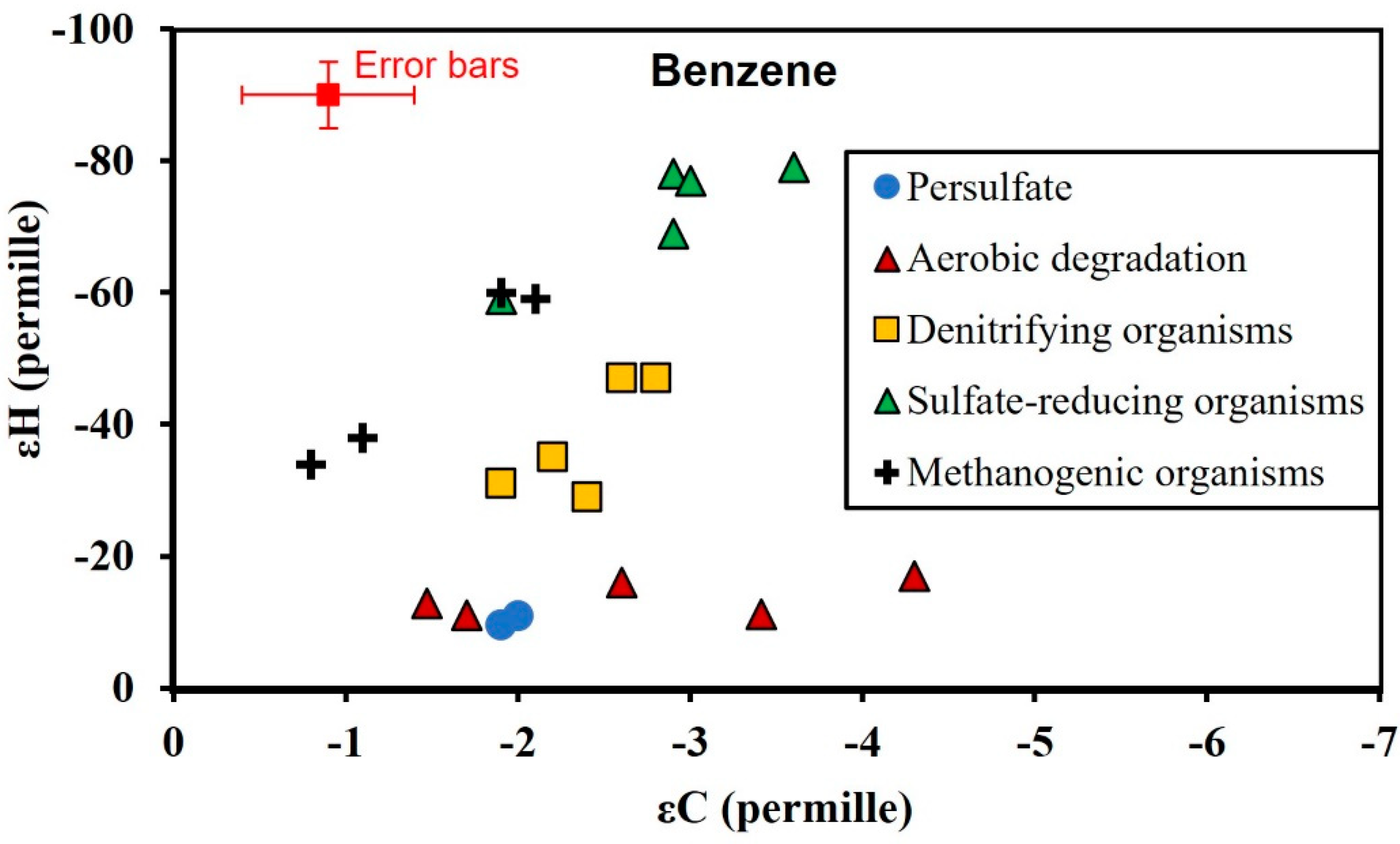
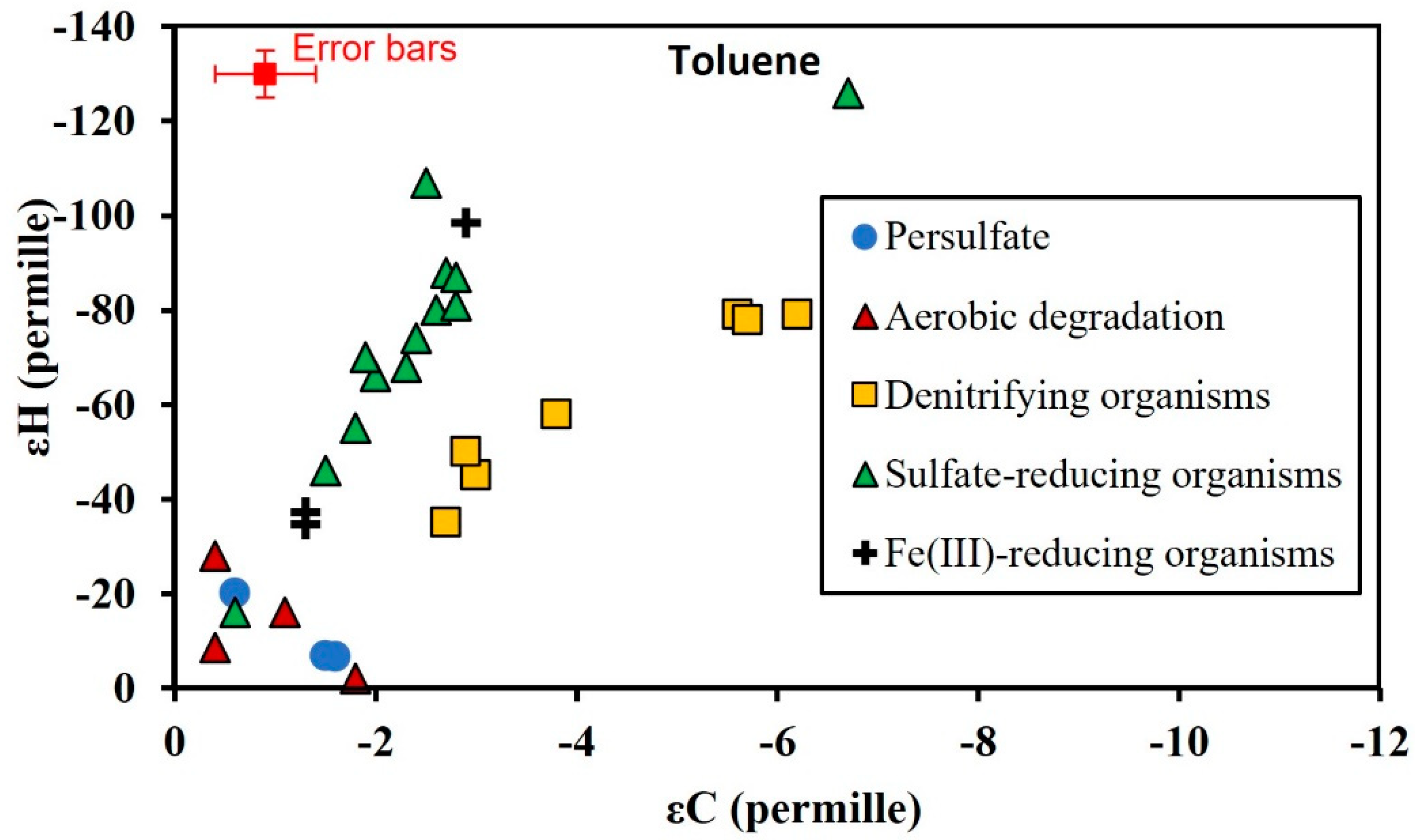
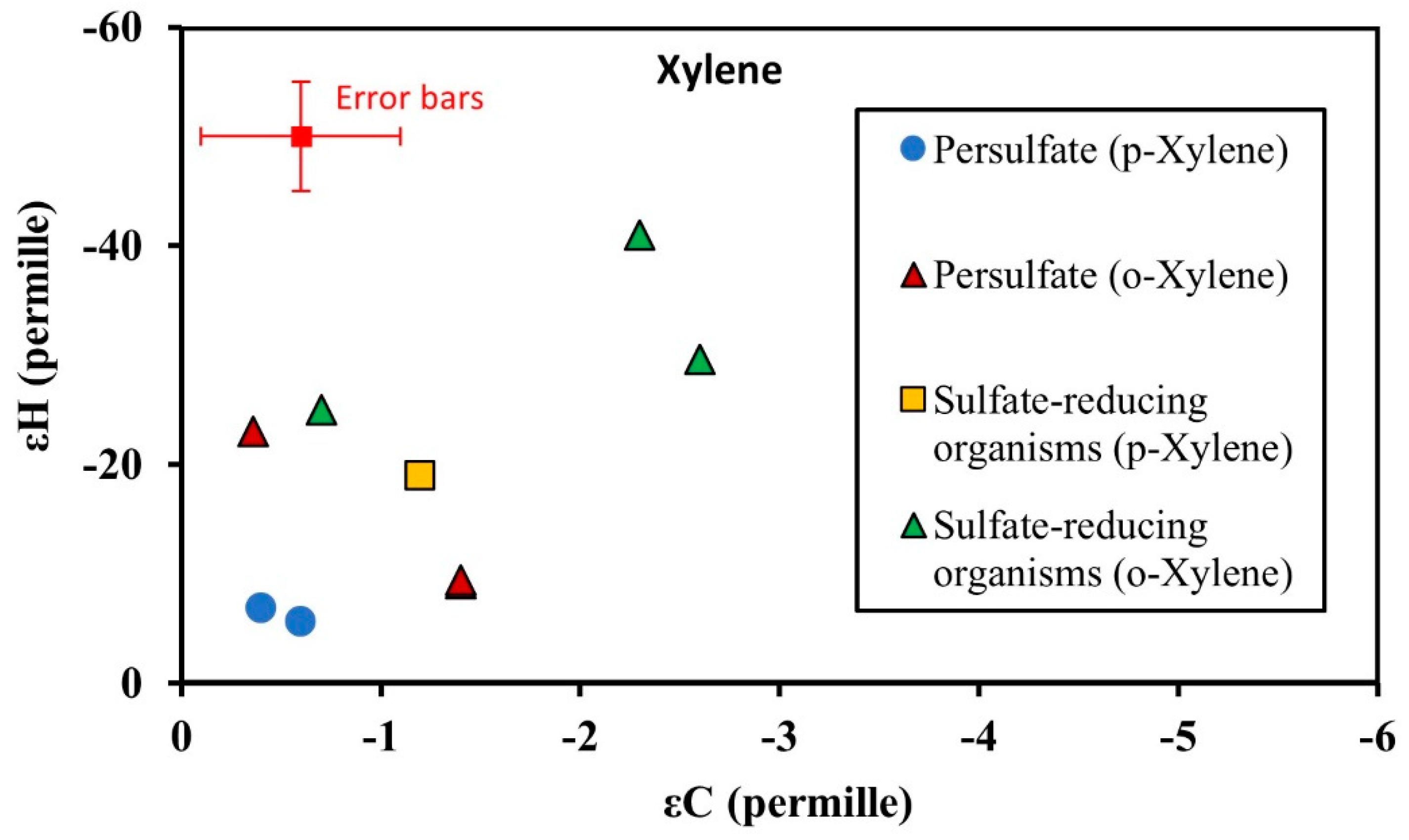
3.3. Isotopic Enrichment Factors for Carbon and Hydrogen during BTEX Oxidation
3.4. Two-Dimensional Isotope Fractionation Analyses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Schwartz, F.W. Physical and Chemical Hydrogeology; John Wiley & Sons Inc.: Hoboken, NJ, USA, 1998. [Google Scholar]
- Huling, S.; Pivetz, B. In Situ Chemical Oxidation; Publication EPA/600/R-06/072; U.S. EPA: Washington, DC, USA, 2006.
- Tsitonaki, A.; Petri, B.; Crimi, M.; Mosbk, H.; Siegrist, R.L.; Bjerg, P.L. In situ chemical oxidation of contaminated soil and groundwater using persulfate: A review. Crit. Rev. Environ. Sci. Technol. 2010, 40, 55–91. [Google Scholar] [CrossRef]
- Liang, C.; Huang, C.; Chen, Y. Potential for activated persulfate degradation of BTEX contamination. Water Res. 2008, 42, 4091–4100. [Google Scholar] [CrossRef]
- Sra, K.S. Persulfate Persistence and Treatability of Gasoline Compounds. Ph.D. Thesis, University of Waterloo, Waterloo, ON, Canada, 2010. [Google Scholar]
- Sra, K.S.; Thomson, N.R.; Barker, J.F. Persulfate treatment of dissolved gasoline compounds. J. Hazard. Toxic Radioact. Waste 2012, 17, 9–15. [Google Scholar] [CrossRef]
- Sra, K.S.; Thomson, N.R.; Barker, J.F. Persulfate injection into a gasoline source zone. J. Contam. Hydrol. 2013, 150, 35–44. [Google Scholar] [CrossRef]
- Watts, R.J.; Teel, A.L. Treatment of contaminated soils and groundwater using ISCO. Pract. Period. Hazard. Toxic Radioact. Waste Manag. 2006, 10, 2–9. [Google Scholar] [CrossRef]
- Crimi, M.L.; Taylor, J. Experimental evaluation of catalyzed hydrogen peroxide and sodium persulfate for destruction of BTEX contaminants. Soil Sediment Contam. 2007, 16, 29–45. [Google Scholar] [CrossRef]
- Petri, B.G.; Watts, R.J.; Tsitonaki, A.; Crimi, M.; Thomson, N.R.; Teel, A.L. Fundamentals of ISCO Using Persulfate. In In Situ Chemical Oxidation for Groundwater Remediation; Springer: New York, NY, USA, 2011; pp. 147–191. [Google Scholar]
- Solano, F.M.; Marchesi, M.; Thomson, N.R.; Bouchard, D.; Aravena, R. Carbon and hydrogen isotope fractionation of benzene, toluene, and o-Xylene during chemical oxidation by persulfate. Groundw. Monit. Remediat. 2018, 38, 62–72. [Google Scholar] [CrossRef] [Green Version]
- Price, S.L.; Kasevich, R.S.; Johnson, M.A.; Wiberg, D.; Marley, M.C. Radio frequency heating for soil remediation. J. Air Waste Manag. Assoc. 1999, 49, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Buettner, H.M.; Daily, W.D. Cleaning contaminated soil using electrical heating and air stripping. J. Environ. Eng. 1995, 121, 580–589. [Google Scholar] [CrossRef]
- Rosansky, S.; Dindal, A. Cost and Performance Report for Persulfate Treatability Studies; NAVFAC Engineering Service Center: Port Hueneme, CA, USA, 2010. [Google Scholar]
- Thomson, N.R.; Hood, E.D.; Farquhar, G.J. Permanganate treatment of an emplaced DNAPL source. Groundw. Monit. Remediat. 2007, 27, 74–85. [Google Scholar] [CrossRef]
- Thomson, N.; Fraser, M.; Lamarche, C.; Barker, J.; Forsey, S. Rebound of a coal tar creosote plume following partial source zone treatment with permanganate. J. Contam. Hydrol. 2008, 102, 154–171. [Google Scholar] [CrossRef] [PubMed]
- Morasch, B.; Richnow, H.H.; Schink, B.; Vieth, A.; Meckenstock, R.U. Carbon and hydrogen stable isotope fractionation during aerobic bacterial degradation of aromatic hydrocarbons. Appl. Environ. Microbiol. 2002, 68, 5191–5194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunkeler, D.; Aravena, R.; Parker, B.; Cherry, J.; Diao, X. Monitoring oxidation of chlorinated ethenes by permanganate in groundwater using stable isotopes: Laboratory and field studies. Environ. Sci. Technol. 2003, 37, 798–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mancini, S.A.; Ulrich, A.C.; Lacrampe-Couloume, G.; Sleep, B.; Edwards, E.A.; Lollar, B.S. Carbon and hydrogen isotopic fractionation during anaerobic biodegradation of benzene. Appl. Environ. Microbiol. 2003, 69, 191–198. [Google Scholar] [CrossRef] [Green Version]
- Morasch, B.; Richnow, H.H.; Vieth, A.; Schink, B.; Meckenstock, R.U. Stable isotope fractionation caused by glycyl radical enzymes during bacterial degradation of aromatic compounds. Appl. Environ. Microbiol. 2004, 70, 2935–2940. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, T.C.; Zwank, L.; Elsner, M.; Berg, M.; Meckenstock, R.U.; Haderlein, S.B. Compound-specific stable isotope analysis of organic contaminants in natural environments: A critical review of the state of the art, prospects, and future challenges. Anal. Bioanal. Chem. 2004, 378, 283–300. [Google Scholar]
- Fischer, A.; Herklotz, I.; Herrmann, S.; Thullner, M.; Weelink, S.A.B.; Stams, A.J.M.; Vogt, C. Combined carbon and hydrogen isotope fractionation investigations for elucidating benzene biodegradation pathways. Environ. Sci. Technol. 2008, 42, 4356–4363. [Google Scholar] [CrossRef]
- Mancini, S.A.; Devine, C.E.; Elsner, M.; Nandi, M.E.; Ulrich, A.C.; Edwards, E.A.; Lollar, B.S. Isotopic evidence suggests different initial reaction mechanisms for anaerobic benzene biodegradation. Environ. Sci. Technol. 2008, 42, 8290–8296. [Google Scholar] [CrossRef]
- Fischer, A.; Gehre, M.; Breitfeld, J.; Richnow, H.; Vogt, C. Carbon and hydrogen isotope fractionation of benzene during biodegradation under sulfate-reducing conditions: A laboratory to field site approach. Rapid Commun. Mass Spectrom. 2009, 23, 2439–2447. [Google Scholar] [CrossRef]
- Saeed, W. The Effectiveness of Persulfate in the Oxidation of Petroleum Contaminants in Saline Environment at Elevated Groundwater Temperature. Master’s Thesis, University of Waterloo, Waterloo, ON, Canada, 2011. [Google Scholar]
- Dessort, D.; Hassan, H. Monitoring of biodegradation of BTEX in water using isotopes ratios. In Proceedings of the Qatar Foundation Annual Research Forum, Doha, Qatar, 21–23 October 2012. [Google Scholar]
- Marchesi, M.; Thomson, N.R.; Aravena, R.; Sra, K.S.; Otero, N.; Soler, A. Carbon isotope fractionation of 1, 1, 1-trichloroethane during base-catalyzed persulfate treatment. J. Hazard. Mater. 2013, 260, 61–66. [Google Scholar] [CrossRef]
- Wijker, R.S.; Adamczyk, P.; Bolotin, J.; Paneth, P.; Hofstetter, T.B. Isotopic analysis of oxidative pollutant degradation pathways exhibiting large H isotope fractionation. Environ. Sci. Technol. 2013, 47, 13459–13468. [Google Scholar] [CrossRef]
- Hunkeler, D.; Morasch, B. Isotope fractionation during transformation processes. In Environmental Isotopes in Biodegradation and Bioremediation, 1st ed.; Aelion, C., Höhener, P., Hunkeler, D., Aravena, R., Eds.; Taylor & Francis Group: Boca Raton, FL, USA, 2010. [Google Scholar]
- Elsner, M.; Zwank, L.; Hunkeler, D.; Schwarzenbach, R.P. A new concept linking observable stable isotope fractionation to transformation pathways of organic pollutants. Environ. Sci. Technol. 2005, 39, 6896–6916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zwank, L.; Berg, M.; Elsner, M.; Schmidt, T.C.; Schwarzenbach, R.P.; Haderlein, S.B. New evaluation scheme for two-dimensional isotope analysis to decipher biodegradation processes: Application to groundwater contamination by MTBE. Environ. Sci. Technol. 2005, 39, 1018–1029. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, S.; Vogt, C.; Fischer, A.; Kuppardt, A.; Richnow, H. Characterization of anaerobic xylene biodegradation by two-dimensional isotope fractionation analysis. Environ. Microbiol. Rep. 2009, 1, 535–544. [Google Scholar] [CrossRef]
- Puls, R.W.; Barcelona, M.J. Low-Flow (Minimal Drawdown) Groundwater Sampling Procedures; EPA Groundwater Issue EPA/540/S-95/504; US Environmental Protection Agency, Office of Solid Waste and Emergency Response: Washington, DC, USA, 1996.
- Freitas, J.G.; Barker, J.F. Sampling VOCs with porous suction samplers in the presence of ethanol: How much are we losing? Ground Water Monit. Remediat. 2008, 28, 83–92. [Google Scholar] [CrossRef]
- Huang, K.C.; Couttenye, R.A.; Hoag, G.E. Kinetics of heat-assisted persulfate oxidation of methyl tert-butyl ether (MTBE). Chemosphere 2002, 49, 413–420. [Google Scholar] [CrossRef]
- Block, P.A.; Brown, R.A.; Robinson, D. Novel activation technologies for sodium persulfate in situ chemical oxidation. In Proceedings of the Fourth International Conference on the Remediation of Chlorinated and Recalcitrant Compounds, Monterey, CA, USA, 24–27 May 2004; pp. 24–27. [Google Scholar]
- Peyton, G.R. The free-radical chemistry of persulfate-based total organic carbon analyzers. Mar. Chem. 1993, 41, 91–103. [Google Scholar] [CrossRef]
- Huang, K.; Zhao, Z.; Hoag, G.E.; Dahmani, A.; Block, P.A. Degradation of volatile organic compounds with thermally activated persulfate oxidation. Chemosphere 2005, 61, 551–560. [Google Scholar] [CrossRef]
- Ahad, J.M.E.; Slater, G.F. Carbon isotope effects associated with Fenton-like degradation of toluene: Potential for differentiation of abiotic and biotic degradation. Sci. Total. Environ. 2008, 401, 194–198. [Google Scholar] [CrossRef]
- Forsey, S. In Situ Chemical Oxidation of Creosote/Coal Tar Residuals: Experimental and Numerical Investigation. Ph.D. Thesis, University of Waterloo, Waterloo, ON, Canada, 2004. [Google Scholar]
- Ahad, J.M.E.; Lollar, B.S.; Edwards, E.A.; Slater, G.F.; Sleep, B.E. Carbon isotope fractionation during anaerobic biodegradation of toluene: Implications for intrinsic bioremediation. Environ. Sci. Technol. 2000, 34, 892–896. [Google Scholar] [CrossRef]
- Vogt, C.; Cyrus, E.; Herklotz, I.; Schlosser, D.; Bahr, A.; Herrmann, S.; Fischer, A. Evaluation of toluene degradation pathways by two-dimensional stable isotope fractionation. Environ. Sci. Technol. 2008, 42, 7793–7800. [Google Scholar] [CrossRef] [PubMed]
- Hunkeler, D.; Andersen, N.; Aravena, R.; Bernasconi, S.M.; Butler, B.J. Hydrogen and carbon isotope fractionation during aerobic biodegradation of benzene. Environ. Sci. Technol. 2001, 35, 3462–3467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tobler, N.B.; Hofstetter, T.B.; Schwarzenbach, R.P. Carbon and hydrogen isotope fractionation during anaerobic toluene oxidation by Geobacter metallireducens with different Fe(III) phases as terminal electron acceptors. Environ. Sci. Technol. 2008, 42, 7786–7792. [Google Scholar] [CrossRef]
- Steinbach, A.; Seifert, R.; Annweiler, E.; Michaelis, W. Hydrogen and carbon isotope fractionation during anaerobic biodegradation of aromatic hydrocarbons—A field study. Environ. Sci. Technol. 2004, 38, 609–616. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ca2+ | 31 mg/L | Alkalinity | 1020 mg/L | Br- | 5.6 mg/L |
| Mg2+ | 1940 mg/L | SO42− | <20 mg/L | TDS | 5330 mg/L |
| Na+ | 33.1 mg/L | Cl- | 3030 mg/L | pH | 7.7 |
| EC | 10400 µmhos/cm | Benzene | 22.0 mg/L | Ethylbenzene | 3.6 mg/L |
| ρ,m-xylene | 9.8 mg/L | Toluene | 41.1 mg/L | o-xylene | 6.1 mg/L |
| Compounds | Temperature (°C) | kobs [102 h−1] | (r2) |
|---|---|---|---|
| Benzene | 10 | 4.6 | 0.96 |
| Benzene | 30 | 236.4 | 0.96 |
| Toluene | 10 | 6.5 | 0.96 |
| Toluene | 30 | 226.6 | 0.99 |
| Ethylbenzene | 10 | 6.72 | 0.94 |
| Ethylbenzene | 30 | 312.7 | 0.99 |
| ρ,m-Xylene | 10 | 12.24 | 0.99 |
| ρ,m-Xylene | 30 | 377.0 | 0.99 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saeed, W.; Shouakar-Stash, O.; Barker, J.; Thomson, N.; McGregor, R. Laboratory Experiments to Evaluate the Effectiveness of Persulfate to Oxidize BTEX in Saline Environment and at Elevated Temperature Using Stable Isotopes. Hydrology 2021, 8, 139. https://doi.org/10.3390/hydrology8030139
Saeed W, Shouakar-Stash O, Barker J, Thomson N, McGregor R. Laboratory Experiments to Evaluate the Effectiveness of Persulfate to Oxidize BTEX in Saline Environment and at Elevated Temperature Using Stable Isotopes. Hydrology. 2021; 8(3):139. https://doi.org/10.3390/hydrology8030139
Chicago/Turabian StyleSaeed, Waleed, Orfan Shouakar-Stash, Jim Barker, Neil Thomson, and Rick McGregor. 2021. "Laboratory Experiments to Evaluate the Effectiveness of Persulfate to Oxidize BTEX in Saline Environment and at Elevated Temperature Using Stable Isotopes" Hydrology 8, no. 3: 139. https://doi.org/10.3390/hydrology8030139
APA StyleSaeed, W., Shouakar-Stash, O., Barker, J., Thomson, N., & McGregor, R. (2021). Laboratory Experiments to Evaluate the Effectiveness of Persulfate to Oxidize BTEX in Saline Environment and at Elevated Temperature Using Stable Isotopes. Hydrology, 8(3), 139. https://doi.org/10.3390/hydrology8030139