




Lignin Valorization for Added-Value Chemicals: Kraft Lignin versus Lignin Fractions

 ,
,  , and
, and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Isolation of Kraft Lignin
2.2. Lignin Solubility
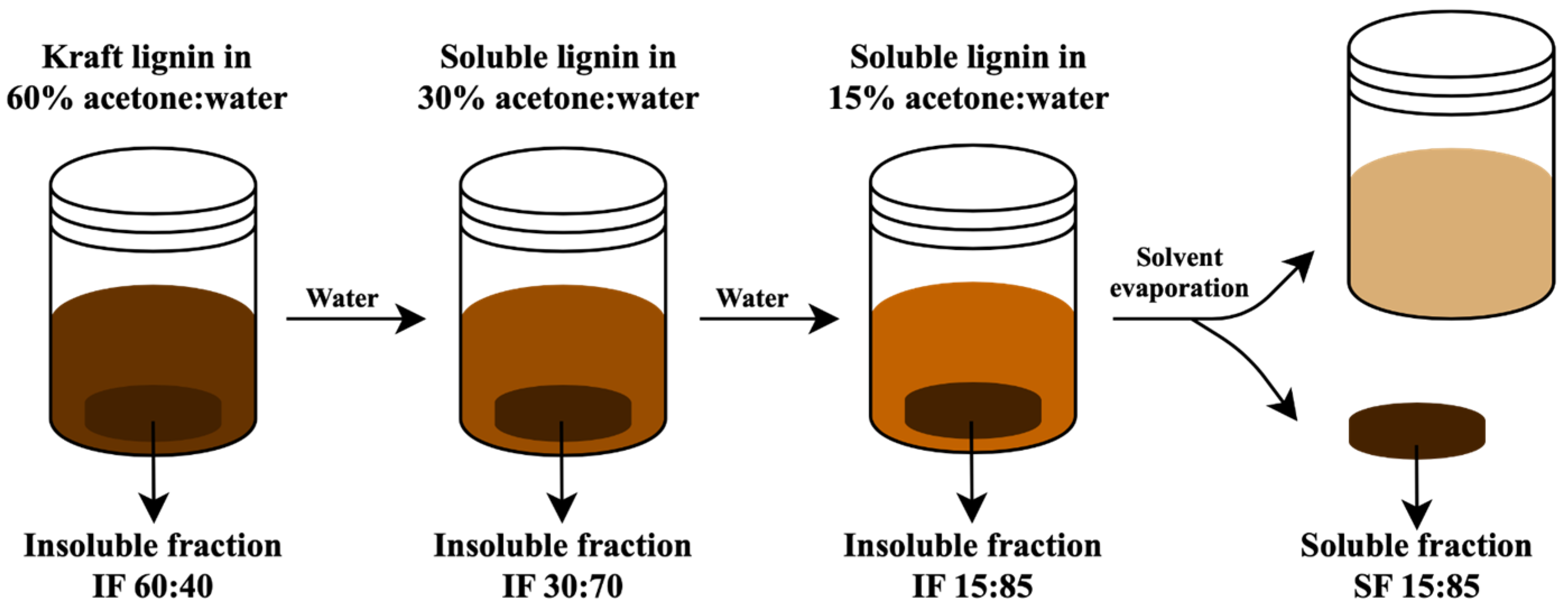
2.3. Fractionation Process of Kraft Lignin
2.4. Composition and Structural Characterization of Kraft Lignin and Lignin Fractions
2.4.1. Inorganics Content Determination
2.4.2. Gel Permeation Chromatography (GPC)
2.4.3. Nitrobenzene Oxidation (NO)
2.4.4. 13C NMR
2.4.5. 31P NMR
2.4.6. Oxidative Depolymerization
3. Results and Discussion
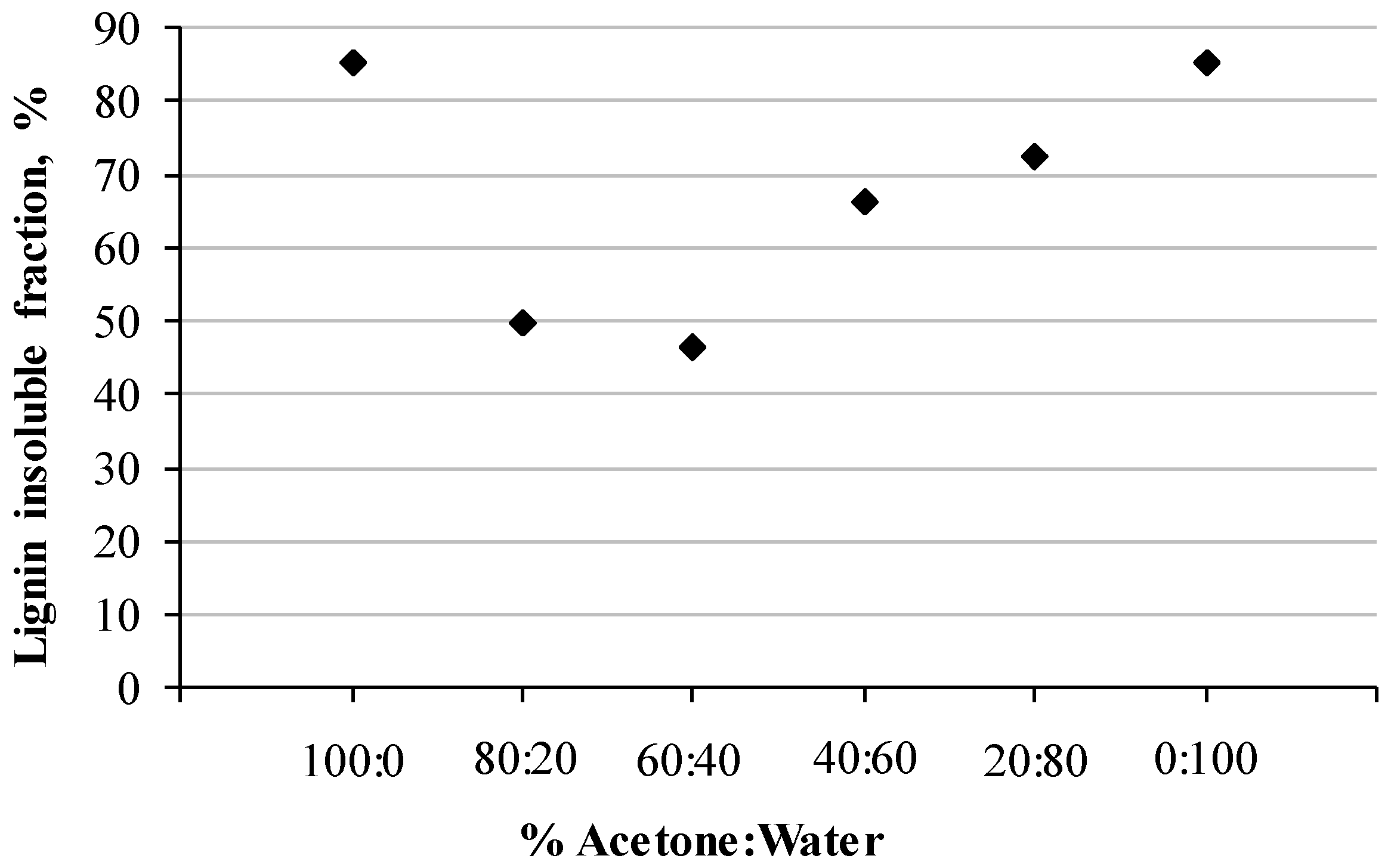
3.1. Kraft Lignin Solubility in Acetone Aqueous Solutions
3.2. Fractionation Yields of Kraft Lignin Fractions
3.3. Inorganics Content of Kraft Lignin and Lignin Fractions
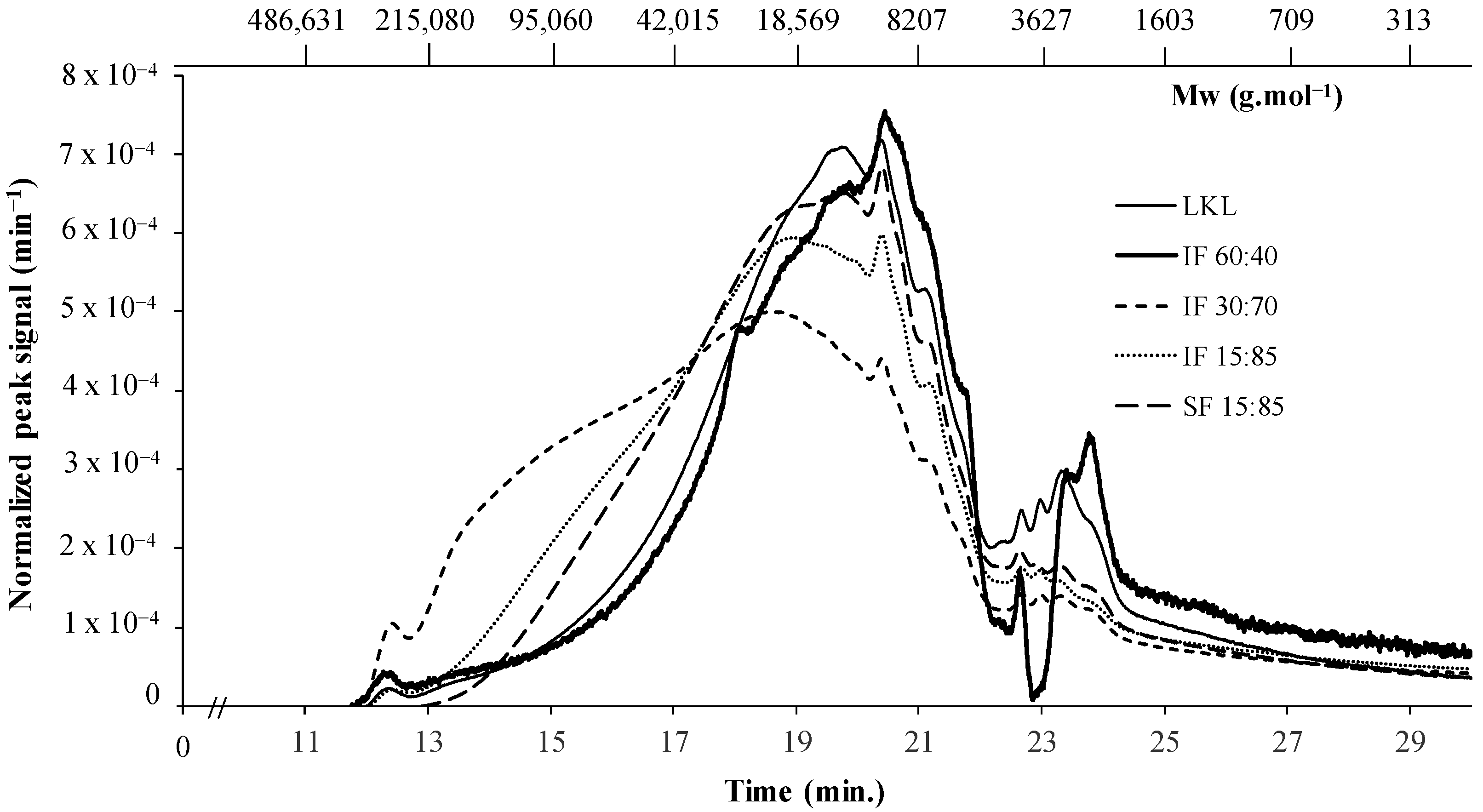
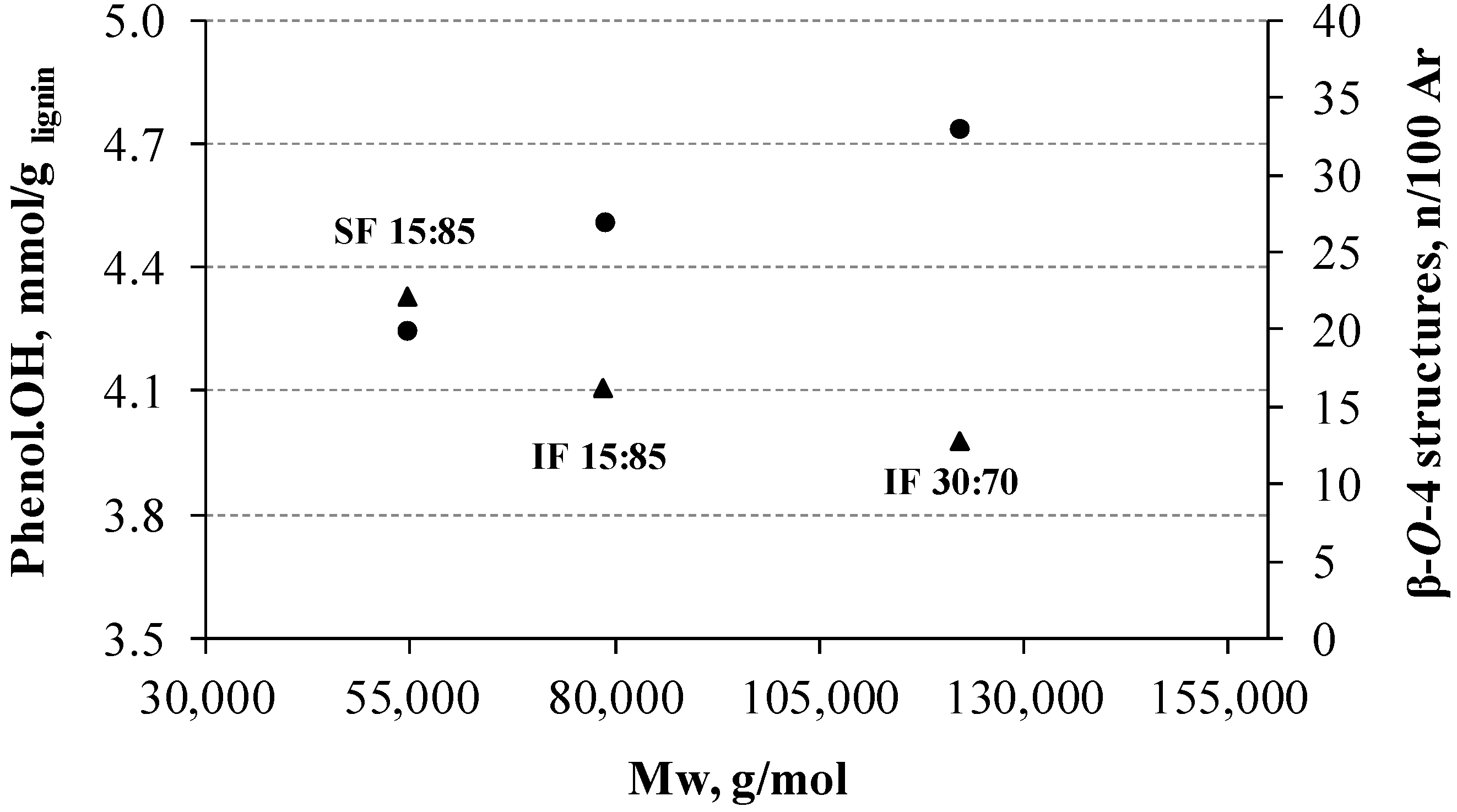
3.4. Molecular Weight Distribution of Lignins
3.5. Nitrobenzene Oxidation
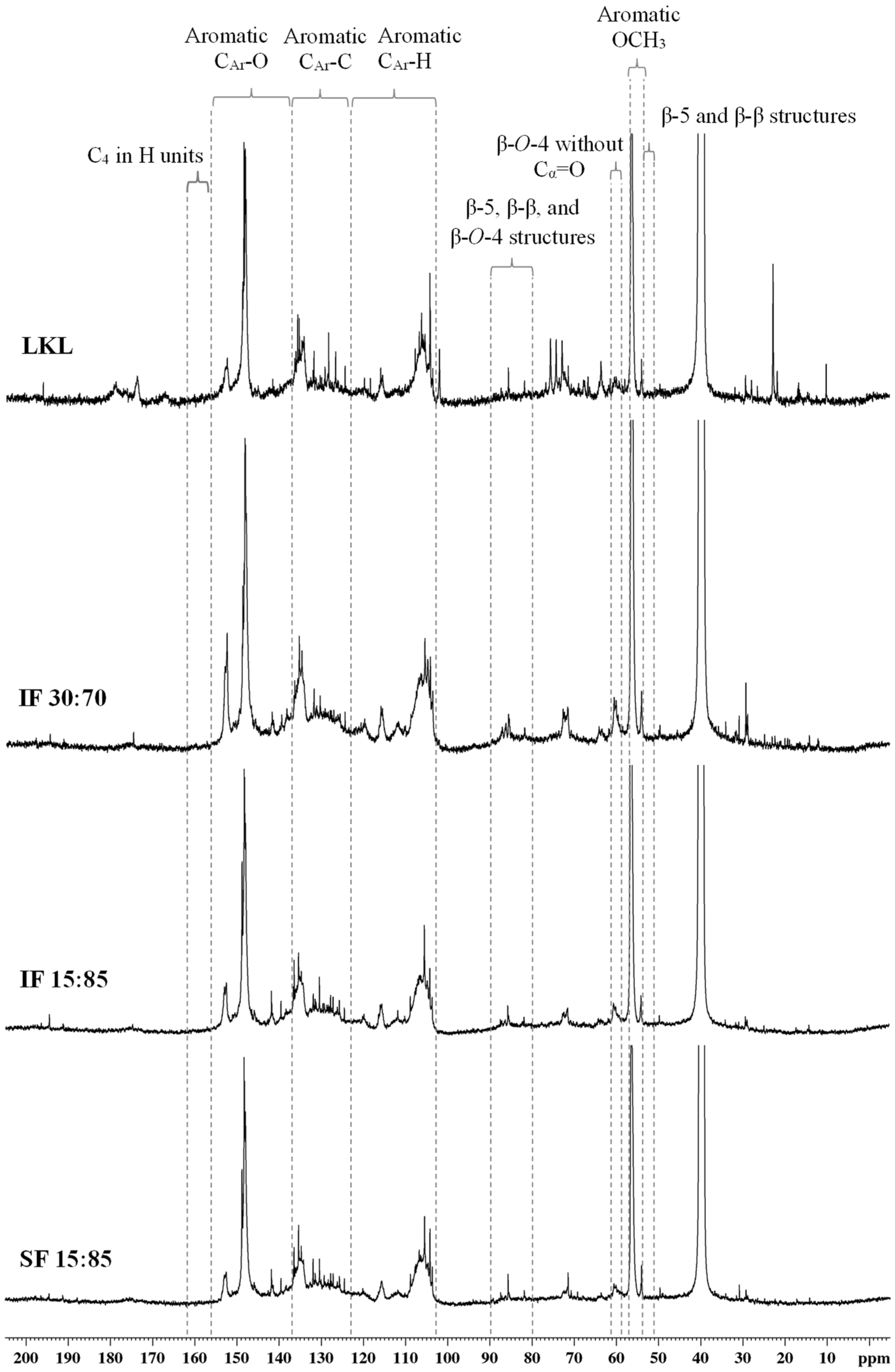
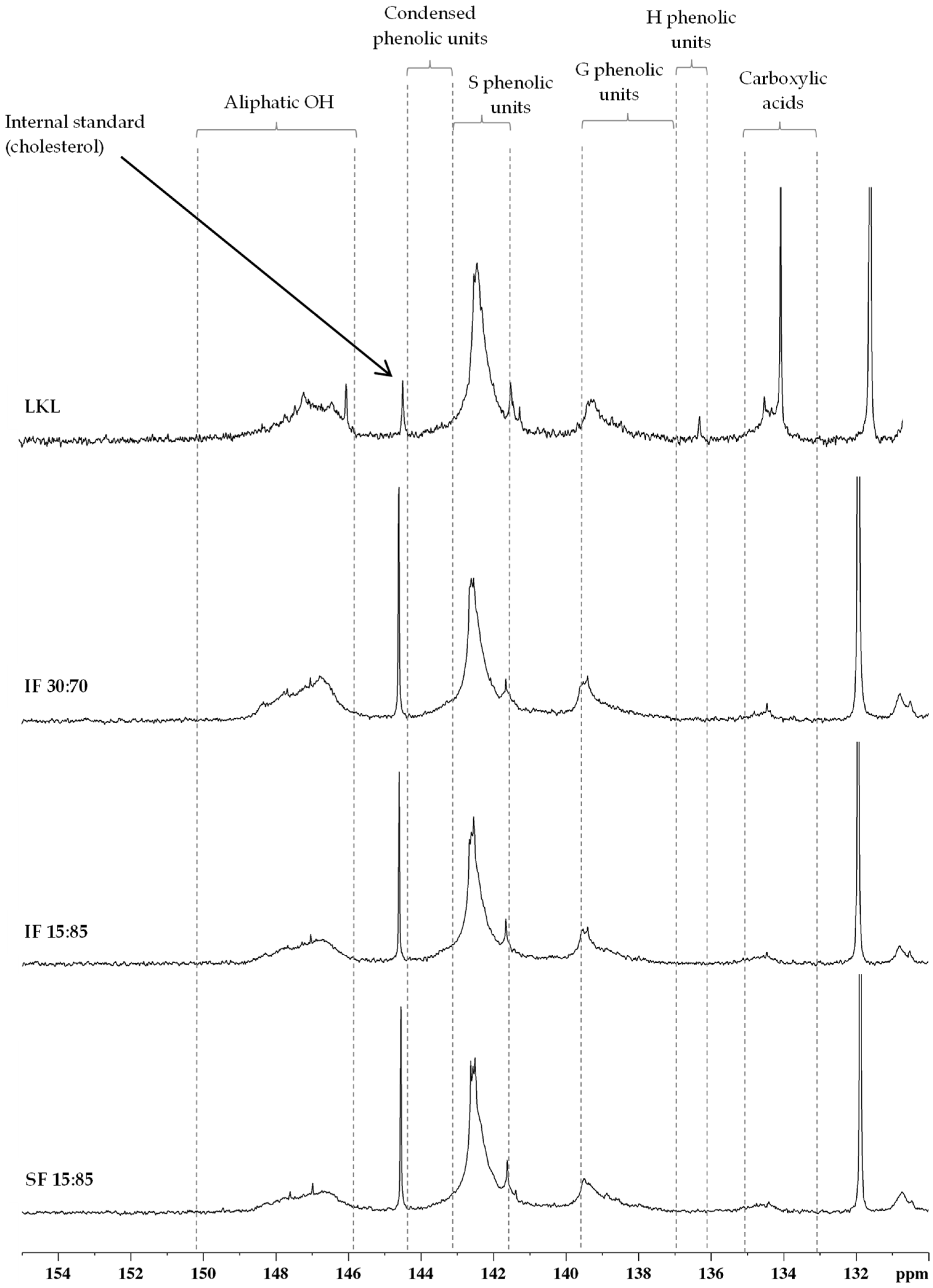
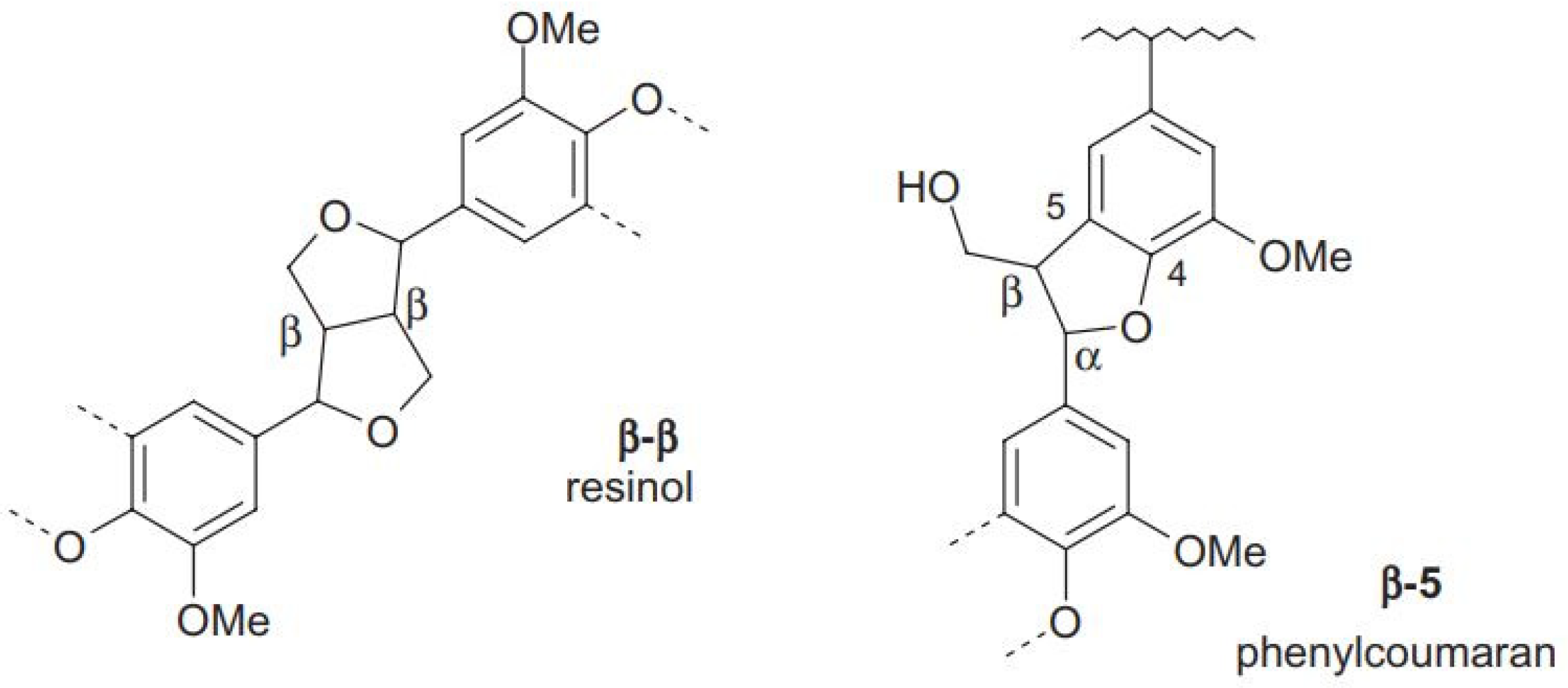
3.6. 13C NMR
3.7. 31P NMR
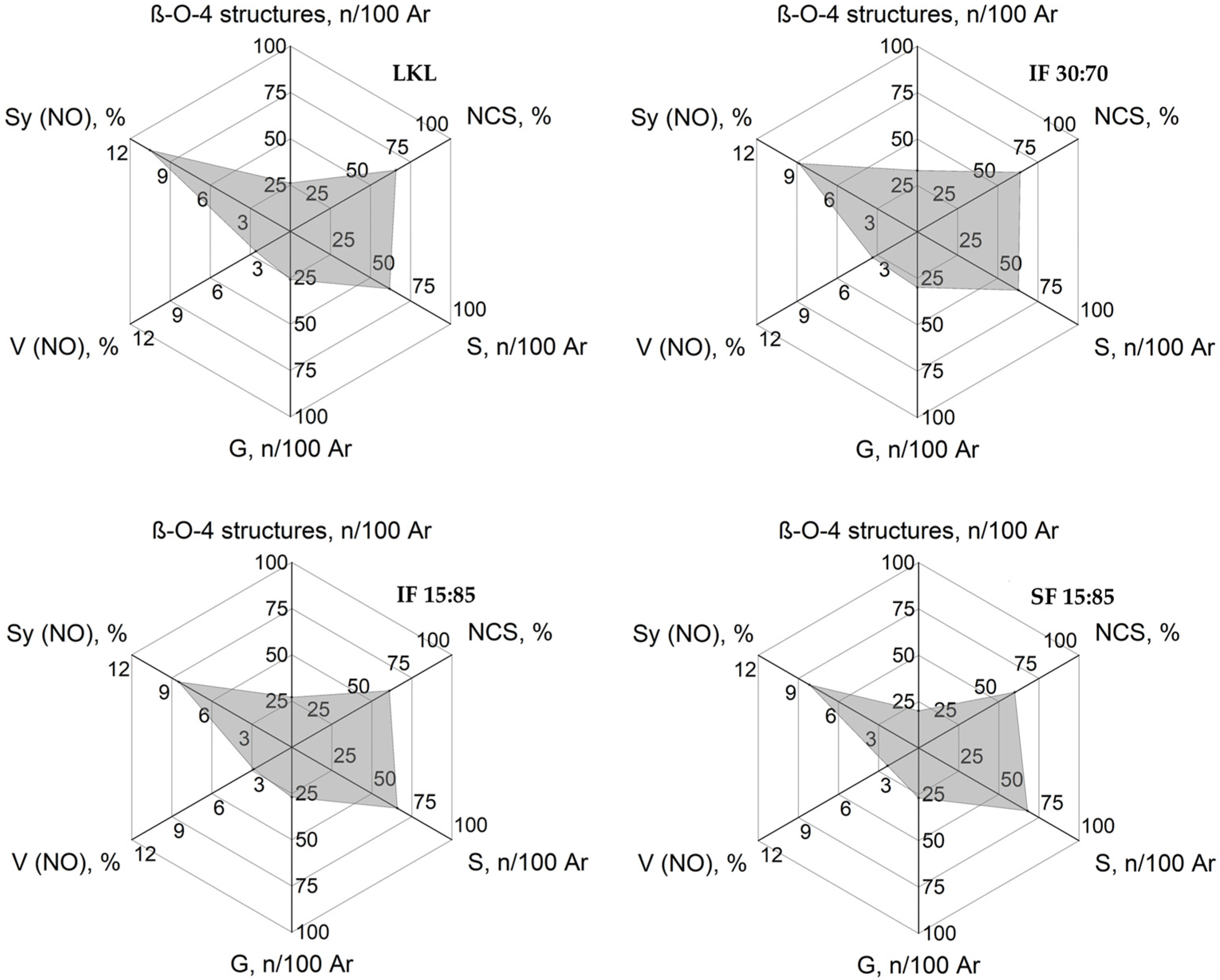
3.8. Radar Classification of Kraft Lignin and Lignin Fractions
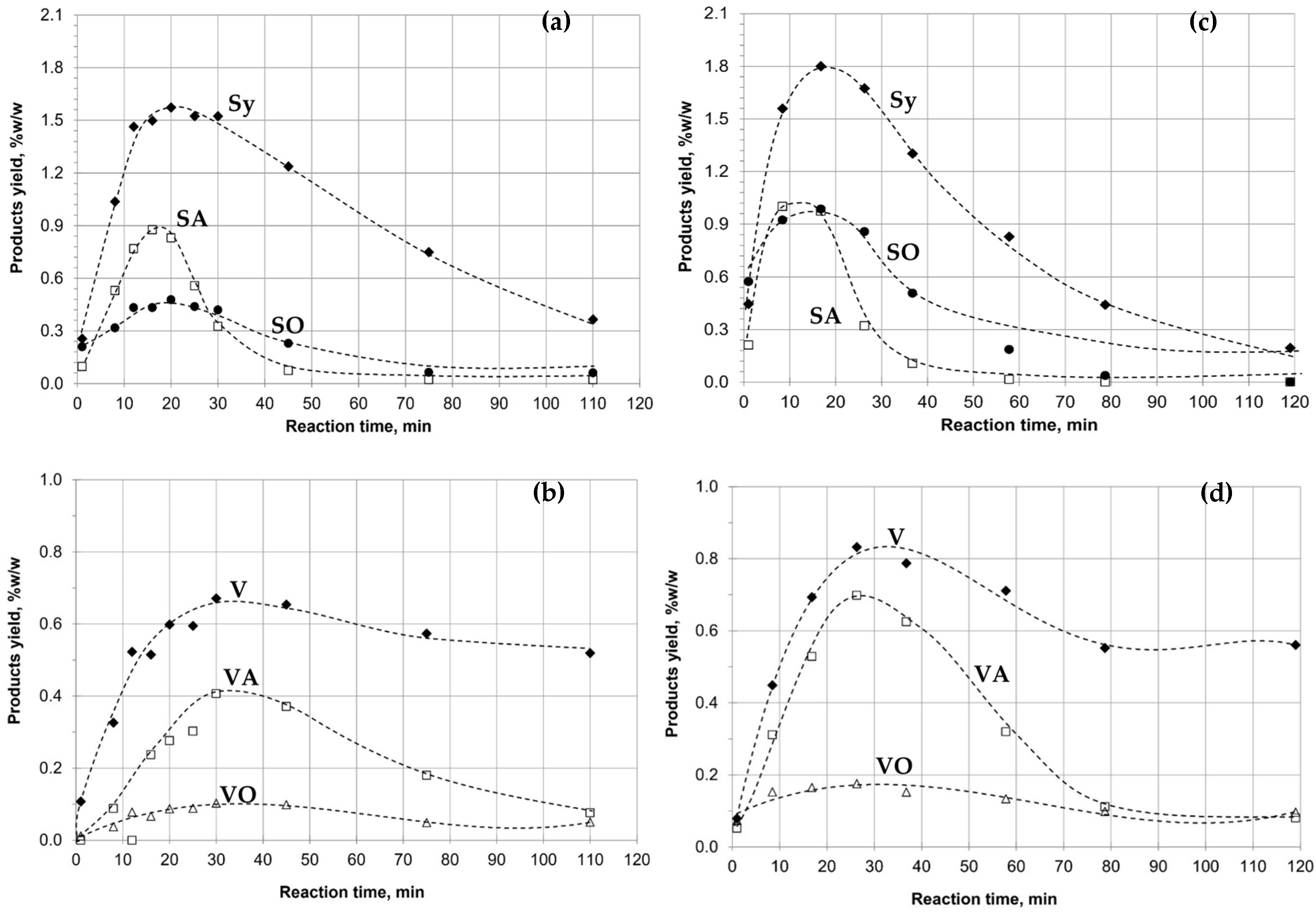
3.9. Oxidative Depolymerization of Kraft Lignin and Fraction IF 30:70
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Rodrigues, A.E.; Pinto, P.C.R.; Barreiro, M.F.; Costa, C.A.E.; Mota, M.I.F.; Fernandes, I. An Integrated Approach for Added-Value Products from Lignocellulosic Biorefineries: Vanillin, Syringaldehyde, Polyphenols and Polyurethane; Springer International Publishing: Berlin/Heidelberg, Germany, 2018. [Google Scholar]
- Yan, J.; Tan, E.C.D.; Katahira, R.; Pray, T.R.; Sun, N. Fractionation of lignin streams using tangential flow filtration. Ind. Eng. Chem. Res. 2022, 61, 4407–4417. [Google Scholar] [CrossRef]
- Azadi, P.; Inderwildi, O.R.; Farnood, R.; King, D.A. Liquid fuels, hydrogen and chemicals from lignin: A critical review. Renew. Sustain. Energy Rev. 2013, 21, 506–523. [Google Scholar] [CrossRef]
- Heitner, C.; Dimmel, D.; Schmidt, J. Lignin and Lignans: Advances in Chemistry; Taylor & Francis: Oxfordshire, UK, 2010. [Google Scholar]
- Sethupathy, S.; Murillo Morales, G.; Gao, L.; Wang, H.; Yang, B.; Jiang, J.; Sun, J.; Zhu, D. Lignin valorization: Status, challenges and opportunities. Bioresour. Technol. 2022, 347, 126696. [Google Scholar] [CrossRef]
- Pinto, P.C.R.; Borges da Silva, E.A.; Rodrigues, A.E. Lignin as source of fine chemicals: Vanillin and syringaldehyde. In Biomass Conversion; Baskar, C., Baskar, S., Dhillon, R.S., Eds.; Springer: Berlin/Heidelberg, Germany, 2012; pp. 381–420. [Google Scholar]
- Berlin, A.; Balakshin, M. Chapter 18—Industrial lignins: Analysis, properties, and applications. In Bioenergy Research: Advances and Applications; Gupta, V.K., Tuohy, M.G., Kubicek, C.P., Saddler, J., Xu, F., Eds.; Elsevier: Amsterdam, The Netherlands, 2014; pp. 315–336. [Google Scholar] [CrossRef]
- Costa, C.A.E.; Pinto, P.C.R.; Rodrigues, A.E. Evaluation of chemical processing impact on E. globulus wood lignin and comparison with bark lignin. Ind. Crops Prod. 2014, 61, 479–491. [Google Scholar] [CrossRef]
- Rodrigues, J.S.; Lima, V.; Araújo, L.C.P.; Botaro, V.R. Lignin fractionation methods: Can lignin fractions be separated in a true industrial process? Ind. Eng. Chem. Res. 2021, 60, 10863–10881. [Google Scholar] [CrossRef]
- Costa, C.A.E.; Pinto, P.C.R.; Rodrigues, A.E. Lignin fractionation from E. globulus kraft liquor by ultrafiltration in a three stage membrane sequence. Sep. Purif. Technol. 2018, 192, 140–151. [Google Scholar] [CrossRef]
- Lin, S.Y.; Dence, C.W. Methods in Lignin Chemistry; Springer: Berlin/Heidelberg, Germany, 1992. [Google Scholar]
- Toledano, A.; Serrano, L.; Garcia, A.; Mondragon, I.; Labidi, J. Comparative study of lignin fractionation by ultrafiltration and selective precipitation. Chem. Eng. J. 2010, 157, 93–99. [Google Scholar] [CrossRef]
- Jiang, X.; Savithri, D.; Du, X.; Pawar, S.; Jameel, H.; Chang, H.-m.; Zhou, X. Fractionation and characterization of kraft lignin by sequential precipitation with various organic solvents. ACS Sustain. Chem. Eng. 2017, 5, 835–842. [Google Scholar] [CrossRef]
- Domínguez-Robles, J.; Tamminen, T.; Liitiä, T.; Peresin, M.S.; Rodríguez, A.; Jääskeläinen, A.-S. Aqueous acetone fractionation of kraft, organosolv and soda lignins. Int. J. Biol. Macromol. 2018, 106, 979–987. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Kim, J.-Y.; Youn, H.J.; Choi, J.W. Fractionation of lignin macromolecules by sequential organic solvents systems and their characterization for further valuable applications. Int. J. Biol. Macromol. 2018, 106, 793–802. [Google Scholar] [CrossRef]
- Jääskeläinen, A.S.; Liitiä, T.; Mikkelson, A.; Tamminen, T. Aqueous organic solvent fractionation as means to improve lignin homogeneity and purity. Ind. Crops Prod. 2017, 103, 51–58. [Google Scholar] [CrossRef]
- Boeriu, C.G.; Fiţigău, F.I.; Gosselink, R.J.A.; Frissen, A.E.; Stoutjesdijk, J.; Peter, F. Fractionation of five technical lignins by selective extraction in green solvents and characterisation of isolated fractions. Ind. Crops Prod. 2014, 62, 481–490. [Google Scholar] [CrossRef]
- Sadeghifar, H.; Ragauskas, A. Perspective on technical lignin fractionation. ACS Sustain. Chem. Eng. 2020, 8, 8086–8101. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Li, M.; Wyman, C.E.; Cai, C.M.; Ragauskas, A.J. Fast fractionation of technical lignins by organic cosolvents. ACS Sustain. Chem. Eng. 2018, 6, 6064–6072. [Google Scholar] [CrossRef]
- Jin, H.; Shi, H.; Jia, W.; Sun, Y.; Sheng, X.; Guo, Y.; Li, H.; Sun, H. Green solvents-based molecular weight controllable fractionation process for industrial alkali lignin at room temperature. Int. J. Biol. Macromol. 2022, 207, 531–540. [Google Scholar] [CrossRef] [PubMed]
- Ponnuchamy, V.; Gordobil, O.; Diaz, R.H.; Sandak, A.; Sandak, J. Fractionation of lignin using organic solvents: A combined experimental and theoretical study. Int. J. Biol. Macromol. 2021, 168, 792–805. [Google Scholar] [CrossRef] [PubMed]
- Sivagurunathan, P.; Raj, T.; Mohanta, C.S.; Semwal, S.; Satlewal, A.; Gupta, R.P.; Puri, S.K.; Ramakumar, S.S.V.; Kumar, R. 2G waste lignin to fuel and high value-added chemicals: Approaches, challenges and future outlook for sustainable development. Chemosphere 2021, 268, 129326. [Google Scholar] [CrossRef] [PubMed]
- Pinto, P.C.R.; Costa, C.E.; Rodrigues, A.E. Oxidation of lignin from Eucalyptus globulus pulping liquors to produce syringaldehyde and vanillin. Ind. Eng. Chem. Res. 2013, 52, 4421–4428. [Google Scholar] [CrossRef]
- Casimiro, F.M.; Costa, C.A.E.; Vega-Aguilar, C.; Rodrigues, A.E. Hardwood and softwood lignins from sulfite liquors: Structural characterization and valorization through depolymerization. Int. J. Biol. Macromol. 2022, 215, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Costa, C.A.E.; Pinto, P.C.R.; Rodrigues, A.E. Radar tool for lignin classification on the perspective of its valorization. Ind. Eng. Chem. Res. 2015, 54, 7580–7590. [Google Scholar] [CrossRef]
- Pinto, P.C.; Borges da Silva, E.A.; Rodrigues, A.E. Comparative study of SPE and liquid-liquid extraction of lignin oxidation products for HPLC-UV quantification. Ind. Eng. Chem. Res. 2010, 49, 12311–12318. [Google Scholar] [CrossRef]
- Casimiro, F.M.; Costa, C.A.E.; Botelho, C.M.; Barreiro, M.F.; Rodrigues, A.E. Kinetics of oxidative degradation of lignin-based phenolic compounds in batch reactor. Ind. Eng. Chem. Res. 2019, 58, 16442–16449. [Google Scholar] [CrossRef]
- Pinto, P.C.; Borges da Silva, E.A.; Rodrigues, A.E. Insights into oxidative conversion of lignin to high-added-value phenolic aldehydes. Ind. Eng. Chem. Res. 2011, 50, 741–748. [Google Scholar] [CrossRef]
- Sadeghifar, H.; Wells, T.; Le, R.K.; Sadeghifar, F.; Yuan, J.S.; Jonas Ragauskas, A. Fractionation of organosolv lignin using acetone:water and properties of the obtained fractions. ACS Sustain. Chem. Eng. 2017, 5, 580–587. [Google Scholar] [CrossRef]
- Ringena, O.; Lebioda, S.; Lehnen, R.; Saake, B. Size-exclusion chromatography of technical lignins in dimethyl sulfoxide/water and dimethylacetamide. J. Chromatogr. A 2006, 1102, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Constant, S.; Wienk, H.L.J.; Frissen, A.E.; Peinder, P.d.; Boelens, R.; van Es, D.S.; Grisel, R.J.H.; Weckhuysen, B.M.; Huijgen, W.J.J.; Gosselink, R.J.A.; et al. New insights into the structure and composition of technical lignins: A comparative characterisation study. Green Chem. 2016, 18, 2651–2665. [Google Scholar] [CrossRef]
- Balakshin, M.Y.; Capanema, E.A.; Santos, R.B.; Chang, H.; Jameel, H. Structural analysis of hardwood native lignins by quantitative 13C NMR spectroscopy. Holzforschung 2015, 70, 95–108. [Google Scholar] [CrossRef]
- Capanema, E.A.; Balakshin, M.Y.; Kadla, J.F. A comprehensive approach for quantitative lignin characterization by NMR spectroscopy. J. Agric. Food Chem. 2004, 52, 1850–1860. [Google Scholar] [CrossRef]
- Argyropoulos, D.S.; Abacherli, A.; Rincón, A.G.; Arx, U.V. Quantitative 31P nuclear magnetic resonance (NMR) spectra of lignin. In Analytical Methods for Lignin Characterisation; International Lignin Institute: Lausanne, Switzerland, 2009. [Google Scholar]
- Araújo, L.C.P.; Yamaji, F.M.; Lima, V.H.; Botaro, V.R. Kraft lignin fractionation by organic solvents: Correlation between molar mass and higher heating value. Bioresour. Technol. 2020, 314, 123757. [Google Scholar] [CrossRef]
- Tarabanko, V.E.; Tarabanko, N. Catalytic oxidation of lignins into the aromatic aldehydes: General process trends and development prospects. Int. J. Mol. Sci. 2017, 18, 2421. [Google Scholar] [CrossRef]
- Tarabanko, V.E.; Petukhov, D.V.; Selyutin, G.E. New mechanism for the catalytic oxidation of lignin to vanillin. Kinet. Catal. 2004, 45, 569–577. [Google Scholar] [CrossRef]
- Tsutsumi, Y.; Kondo, R.; Sakai, K.; Imamura, H. The difference of reactivity between syringyl lignin and guaiacyl lignin in alkaline systems. Holzforschung 1995, 49, 423–428. [Google Scholar] [CrossRef]
- Sultanov, V.S.; Wallis, A.F.A. Reactivities of guaiacyl and syringyl lignin model phenols towards oxidation with oxygen-alkali. J. Wood Chem. Technol. 1991, 11, 291–305. [Google Scholar] [CrossRef]
- Gierer, J.; Imsgard, F.; Norén, I. Studies on the degradation of phenolic lignin units of the b aryl ether type with oxygen in alkaline media. Acta Chem. Scand. B 1977, 31, 561–572. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lignins, % w/w lignin (Dry Weight) | |||||
|---|---|---|---|---|---|
| LKL | IF 60:40 | IF 30:70 | IF 15:85 | SF 15:85 | |
| Inorganics content | 17.5 | 74.1 | 0.94 | 0.37 | 1.40 |
| Lignin | Mw (g·mol−1) | Mw/Mn |
|---|---|---|
| LKL | 69,233 | 1.17 |
| IF 60:40 | 48,966 | 1.23 |
| IF 30:70 | 121,989 | 1.27 |
| IF 15:85 | 78,538 | 1.21 |
| SF 15:85 | 54,476 | 1.15 |
| Lignin | Products, % w/wlignin 1 | |||||
|---|---|---|---|---|---|---|
| Hy | VA | SA | V | Sy | η NOtotal | |
| LKL | 0.13 | 0.34 | 2.79 | 2.58 | 10.5 | 16.3 |
| IF 60:40 | 0.00 | 0.07 | 1.41 | 1.02 | 3.66 | 6.15 |
| IF 30:70 | 0.03 | 0.44 | 1.30 | 3.34 | 8.82 | 13.9 |
| IF 15:85 | 0.11 | 0.39 | 2.42 | 2.85 | 8.46 | 14.2 |
| SF 15:85 | 0.08 | 0.32 | 1.24 | 2.32 | 8.16 | 12.1 |
| Assignments (Spectroscopic Range) | Amount (Number per Aromatic Ring) | |||
|---|---|---|---|---|
| LKL | IF 30:70 | IF 15:85 | SF 15:85 | |
| Cβ in β-5 and β-β structures (δ 51.0–53.8 ppm) | 0.12 | 0.12 | 0.13 | 0.11 |
| Aromatic OCH3 (δ 54.3–57.3 ppm) | 1.29 | 1.40 | 1.44 | 1.43 |
| Cγ in β-O-4 structures without Cα=O (δ 59.3–60.8 ppm) | 0.14 | 0.19 | 0.16 | 0.13 |
| Cγ in β-5 and β-O-4 structures with Cα=O; Cγ in β-1 (δ 62.5–63.8 ppm) | 0.12 | 0.07 | 0.07 | 0.05 |
| Cα in β-O-4 structures; Cγ in pinoresinol/syringaresinol and β-β structures (δ 70.0–76.0 ppm) | 0.65 | 0.40 | 0.35 | 0.30 |
| Cβ in β-O-4 structures; Cα in β-5 and β-β structures (δ 80.0–90.0 ppm) | 0.38 | 0.46 | 0.40 | 0.32 |
| Aromatic CAr-H (δ 103.0–123.0 ppm) | 1.93 | 1.94 | 1.87 | 1.88 |
| Aromatic CAr-C (δ 123.0–137.0 ppm) | 1.64 | 1.76 | 1.70 | 1.76 |
| Aromatic CAr-O (δ 137.0–156.0 ppm) | 2.34 | 2.40 | 2.36 | 2.32 |
| Lignins | β-O-4 Structures | DC (%) | S:G:H | S/G |
|---|---|---|---|---|
| LKL | 26 | 34 | 62:26:12 | 2.35 |
| IF 30:70 | 33 | 36 | 63:30:07 | 2.10 |
| IF 15:85 | 27 | 39 | 66:27:07 | 2.46 |
| SF 15:85 | 20 | 40 | 68:27:05 | 2.49 |
| Assignments (Spectroscopic Range) | Amount (mmol/g lignin) | |||
|---|---|---|---|---|
| LKL | IF 30:70 | IF 15:85 | SF 15:85 | |
| Aliphatic OH (δ 150.2–145.8 ppm) | 3.59 | 1.84 | 1.38 | 1.03 |
| Carboxylic acids (δ 135.0–133.0 ppm) | 2.34 | 0.18 | 0.20 | 0.14 |
| Total phenolic units | 7.72 | 3.98 | 4.11 | 4.33 |
| Condensed phenolic units (δ 144.4–143.2 ppm; δ 141.6–139.6 ppm) | 2.04 | 0.79 | 0.83 | 0.81 |
| Non-condensed phenolic units | ||||
| S phenolic units (δ 143.2–141.6 ppm) | 4.59 | 2.31 | 2.43 | 2.64 |
| G phenolic units (δ 139.6–136.9 ppm) | 1.04 | 0.86 | 0.84 | 0.87 |
| H phenolic units (δ 136.8–136.3 ppm) | 0.04 | 0.02 | 0.01 | 0.01 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Costa, C.A.E.; Casimiro, F.M.; Vega-Aguilar, C.; Rodrigues, A.E. Lignin Valorization for Added-Value Chemicals: Kraft Lignin versus Lignin Fractions. ChemEngineering 2023, 7, 42. https://doi.org/10.3390/chemengineering7030042
Costa CAE, Casimiro FM, Vega-Aguilar C, Rodrigues AE. Lignin Valorization for Added-Value Chemicals: Kraft Lignin versus Lignin Fractions. ChemEngineering. 2023; 7(3):42. https://doi.org/10.3390/chemengineering7030042
Chicago/Turabian StyleCosta, Carina A. E., Filipa M. Casimiro, Carlos Vega-Aguilar, and Alírio E. Rodrigues. 2023. "Lignin Valorization for Added-Value Chemicals: Kraft Lignin versus Lignin Fractions" ChemEngineering 7, no. 3: 42. https://doi.org/10.3390/chemengineering7030042
APA StyleCosta, C. A. E., Casimiro, F. M., Vega-Aguilar, C., & Rodrigues, A. E. (2023). Lignin Valorization for Added-Value Chemicals: Kraft Lignin versus Lignin Fractions. ChemEngineering, 7(3), 42. https://doi.org/10.3390/chemengineering7030042