Abstract
Mutual solubilities of water with n-alkanes, cycloalkanes, iso-alkanes (branched alkanes), alkenes, alkynes, alkadienes, and alkylbenzenes were calculated at 298 K for 153 systems not yet measured. Recommended data for 64 systems reported in the literature were compared with the predicted values. The solubility of the hydrocarbons in water was calculated with a thermodynamically based equation, which depends on specific properties of the hydrocarbon. The concentration in the second coexisting liquid phase (water in hydrocarbon) was calculated using liquid-liquid equilibrium with an equation of state, which takes into account the self-association of water and co-association of water with π-bonds of the hydrocarbons.
1. Introduction
An example of liquid-liquid equilibrium, LLE, in a hydrocarbon + water system is shown in Figure 1.
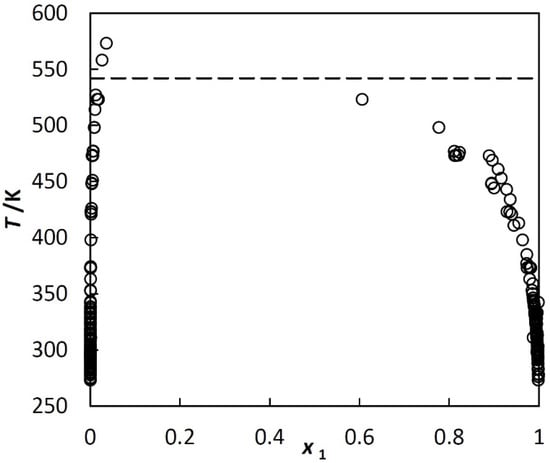
Figure 1.
Liquid-liquid equilibrium in benzene (1) + water (2) system under saturated vapor pressure; experimental mole fractions of benzene at various temperatures taken from different literature sources. Dashed line corresponds to the three-phase critical temperature.
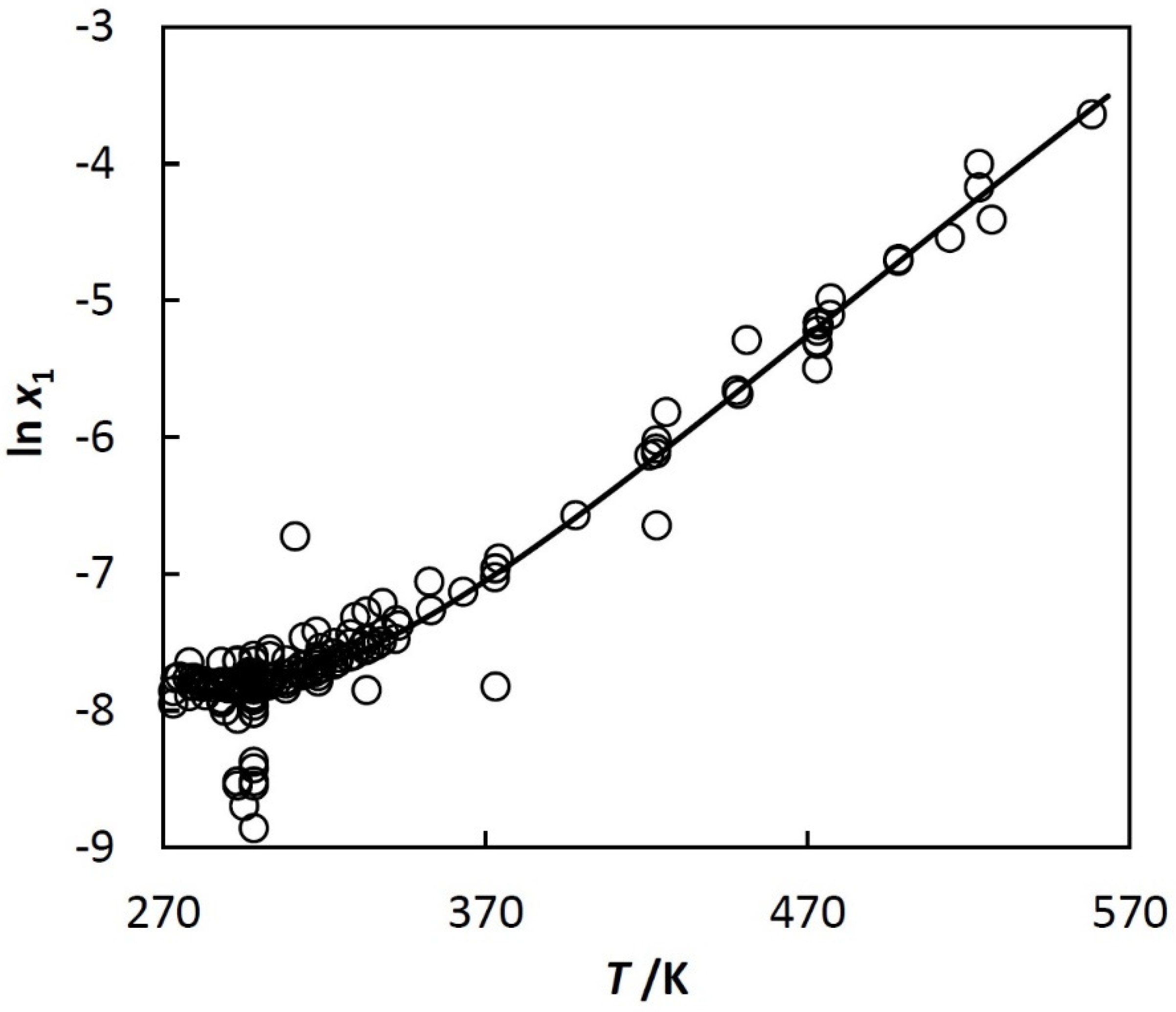
Figure 1 is typical for systems investigated in this paper. The two liquids and vapor exist up to the three-phase critical temperature, T3c. At T3c, hydrocarbon-rich phase disappears, but water saturated with the hydrocarbon and the vapor still exists. The solubilities in the coexisting phases are strongly asymmetric. The solubility of the hydrocarbon in the water-rich phase is very low. It exhibits a minimum at room temperature, which is shown in Figure 2. The water in hydrocarbon solubility is rapidly increasing, as a monotonic function of temperature. Unlike the benzene + water system, shown in Figure 1, many other systems are represented only by a few and often scattered experimental points.
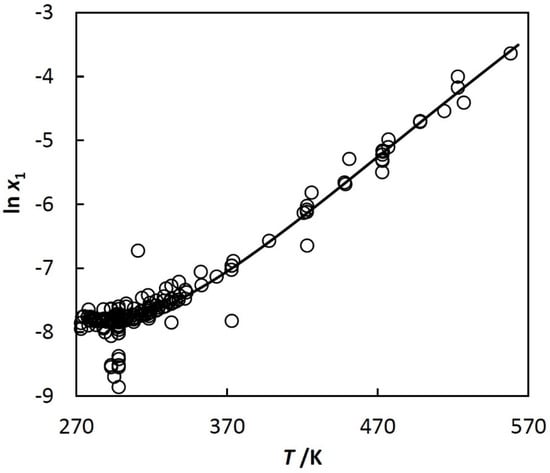
Figure 2.
Solubility of benzene (1) in water (2), natural logarithm of mole fraction of benzene (1) in water (2), ln x1, vs. temperature, T.
Since these systems are of significant importance for chemical and related industry and environment protection, much effort has been made to develop suitable correlation and prediction methods. Among the correlation methods, the work of Tsonopoulos [1,2] results in suitable correlation equations. However, this excellent method has two disadvantages. First, both branches are correlated independently by different equations that are not thermodynamically interrelated. The second disadvantage is that these equations cannot be extended for systems for which no experimental data exist. Various attempts have been made to develop predictive methods. Among others, group contribution methods and equations of state incorporated with an association term were tested. The UNIFAC and the ASOG methods are the best-known group-contribution methods. We have calculated LLE in the representative group of selected water + hydrocarbon systems with below mentioned versions of the UNIFAC method and the ASOG method [3]. The original UNIFAC method and its modifications (Dortmund and Lyngby) are mostly intended to represent primarily VLE data and are not suitable for the quantitative prediction of LLE in hydrocarbon + water systems. Magnussen et al. [4] developed the UNIFAC-LLE method. This version was examined by Gupte and Danner [5] but reported results for hydrocarbon + water systems are rather ambiguous (the number of systems examined is small, and the precision of the reported deviations is insufficient for interpretation). According to our calculations, this method is also not suitable for the quantitative prediction of LLE in hydrocarbon + water systems. Hooper et al. [6] developed the modified UNIFAC method aimed at the prediction of liquid-liquid equilibria for water-organic liquid systems over a wide temperature range. This modified UNIFAC successfully represents the hydrocarbon-rich phase but cannot correctly predict the minimum solubility of hydrocarbons in water. Voutsas and Tassios [7] examined and modified this version of the UNIFAC method. They introduced the Flory–Huggins combinatorial contribution with adjusted r and q structural parameters for water and developed new interaction parameters for the H2O-CH2 and H2O-ACH groups. This is probably the best attempt. However, while this is the first time this method predicted the minima fairly well, it still does not reproduce all the features of aqueous solutions of hydrocarbons. The main disadvantage is the inability to model alkene, alkyne, and alkylbenzene groups. The method was originally tested only for five systems with relatively low hydrocarbons, from n-pentane to n-octane, cyclohexane, and benzene. When we performed calculations for higher hydrocarbons, increased positive deviations in both solubilities were observed. We proposed [3] new and improved interaction parameters for water with the main groups CH2 and ACH.
In 2015, Kang et al. [8,9] published a comprehensive, newly modified UNIFAC parameter matrix, based on the same formulations as those used for the modified UNIFAC (Dortmund). In most cases, the authors used the same main- and subgroups and van der Waals properties as in modified UNIFAC (Dortmund). Parameters were fitted using critically evaluated phase equilibrium data stored in the NIST databank (including selected data for LLE). Critical evaluation of selected data was performed with the use of algorithms developed at NIST. Unfortunately, for the interaction parameters of the H2O group with the main-groups CH2, ACH, and ACCH2, the values are the same as previously in the modified UNIFAC (Dortmund) [10]. New interaction parameters have been proposed only for the interaction between the main-groups C=C and H2O.
Among the equations of state, the CPA [11,12,13], SAFT [12,14,15,16] APACT [14], PHCT [14], and Peng–Robinson [14,17] equations were applied to these systems. According to authors of these papers, these highly complex equations are not able to satisfactorily represent the experimental data and are not superior to the simple cubic Peng–Robinson equation with the simple van der Waals-type mixing rules. Since in all above equations of state some adjustable parameters must be fitted to experimental data, none of these equations can be considered as purely predictive as UNIFAC. Results of application are far from a satisfactory representation of data, particularly concerning the solubility of hydrocarbons in water. Vega et al. [15] obtained the solubility minima of n-alkanes in water using Soft-SAFT. However, these predicted minima did not quantitatively agree with recommended values, the representation of the other data points was not satisfactory, and these predictions cannot be regarded as qualitative. An exception is the successful application of the PC-SAFT to binary mixtures of n-alkanes and water as reported by Haarmann et al. [18].
A brief overview of the different methods (e.g., different versions of the SAFT equation) can be found in the works of Gupte and Danner [5], Vega et al. [15], Oracz and Góral [3], Safamirzaei and Modarress [19], Oliveira et al. [13], or Haarmann et al. [18] cited above.
Other noteworthy methods are: the COSMO-RS method used by Klamt [20], the use of artificial neural networks by Safamirzaei and Modarress [19] (as an example of using ANNs for these problems), or the use of molecular dynamics methods by Morgado et al. [21].
The conclusion is that the reported predictive methods are of very limited value. Some of them fairly well predict the solubility of water in hydrocarbons, but they are usually unsatisfactory for predicting the solubility of hydrocarbons in water.
In our work, we analyzed all available binary solubility data for systems of water with n-alkanes, cycloalkanes, iso-alkanes (branched alkanes), alkenes, alkynes, alkadienes and alkylbenzenes. The resulting method of the LLE prediction was described in papers [22,23,24,25]. In this method, the calculation of such solubilities as those shown in Figure 1 consists of two steps:
- Solubilities of hydrocarbons in water are approximated with a thermodynamically based equation described in the next section. The two coefficients of this equation are linearly dependent on the excluded volume of the hydrocarbon molecule, which enables prediction.
- Solubilities of water in hydrocarbons are calculated using liquid–liquid equilibrium calculations. The input data for these calculations are concentrations in the second liquid phase predicted by the previously mentioned smoothing equation.
In these two steps, an extensive body of experimental data is described with a few adjustable parameters, providing a framework for the comparison of experimental data and supporting the recognition of systematic error. During the adjustment of the parameters to a large number of experimental points, the experimental errors partially compensate each other, which makes the calculated solubilities more reliable than individual experimental points. Error analysis is given in papers [22,23,24,25]. The method approximates numerical experimental values with analytical equations, allowing for temperature interpolation and extrapolation, which is important for engineering practice. It is also possible to make extrapolations to other systems not investigated experimentally.
The calculated solubilities were used as the reference values for the evaluation of experimental data in volume 81 of the IUPAC Solubility Data Series [26,27,28,29,30,31,32,33,34,35,36,37]. Based on all available literature data, the experimental recommended data for the mutual solubility of water with hydrocarbons for 64 systems were proposed. In this paper, this set of recommended data is appended with mutual solubilities at 298.15 K for 153 systems, which were not investigated experimentally. The prediction can also be applied at other temperatures and for other hydrocarbons using programs and databases described in this paper. We believe that our work reduces the necessity for further measurements in this field. (However, some independent measurements would be useful for testing our predictions).
2. Solubility of Hydrocarbons in Water
To show more detail, the left part of Figure 1 is plotted in Figure 2 using other coordinates. The shape of the curve shown in Figure 2 is typical for n-alkanes, cycloalkanes, iso-alkanes (branched alkanes), alkenes, alkynes, alkadienes, and alkylbenzenes, but their solubilities are quite different. The reference data in [25] and [23] show that, for example, the mole fraction of benzene in water at 298.15 K is 4·10−4, whereas for decane it decreases to 3·10−9.
The mole fraction of the hydrocarbon in water, xh, at temperature T along the three-phase equilibrium line is described by a thermodynamic equation:
where Δslnhh is the partial molar enthalpy of solution of the hydrocarbon. The calorimetric measurements suggest that Δslnhh is a linearly increasing function of temperature going through zero at a temperature Tmin. In this case, Δslnhh can be approximated with the equation:
Integration of Equation (1) with Δslnhh as expressed with Equation (2) yields Equation (3)
where xh,min is the minimum value of xh at T = Tmin. The function f(T/Tmin) results from the integration of Equation (2):
For alkylbenzenes, instead of Equation (2), a logarithmic function was assumed, which leads to Equation (5):
Equation (3) was used for approximation of experimental solubility data of the hydrocarbons. An example is shown in Figure 2, where the approximating curve is calculated with Equations (3) and (5). At the beginning of the investigation, ln xh,min, Tmin, and Ch in Equation (3) were treated as adjustable parameters. After the analysis of experimental data, it was found (see papers [22,23,24,25]) that Tmin is constant for a given class of hydrocarbons, whereas ln xh,min and Ch depend linearly on the excluded volume, bh, of the hydrocarbon. The excluded volume is used in equations of state of van der Waals type. Here, the Redlich–Kwong Equation of state (RK EoS) is used, where b is calculated with:
In the above equation Tc is the critical temperature and Pc is the critical pressure of the corresponding substance.
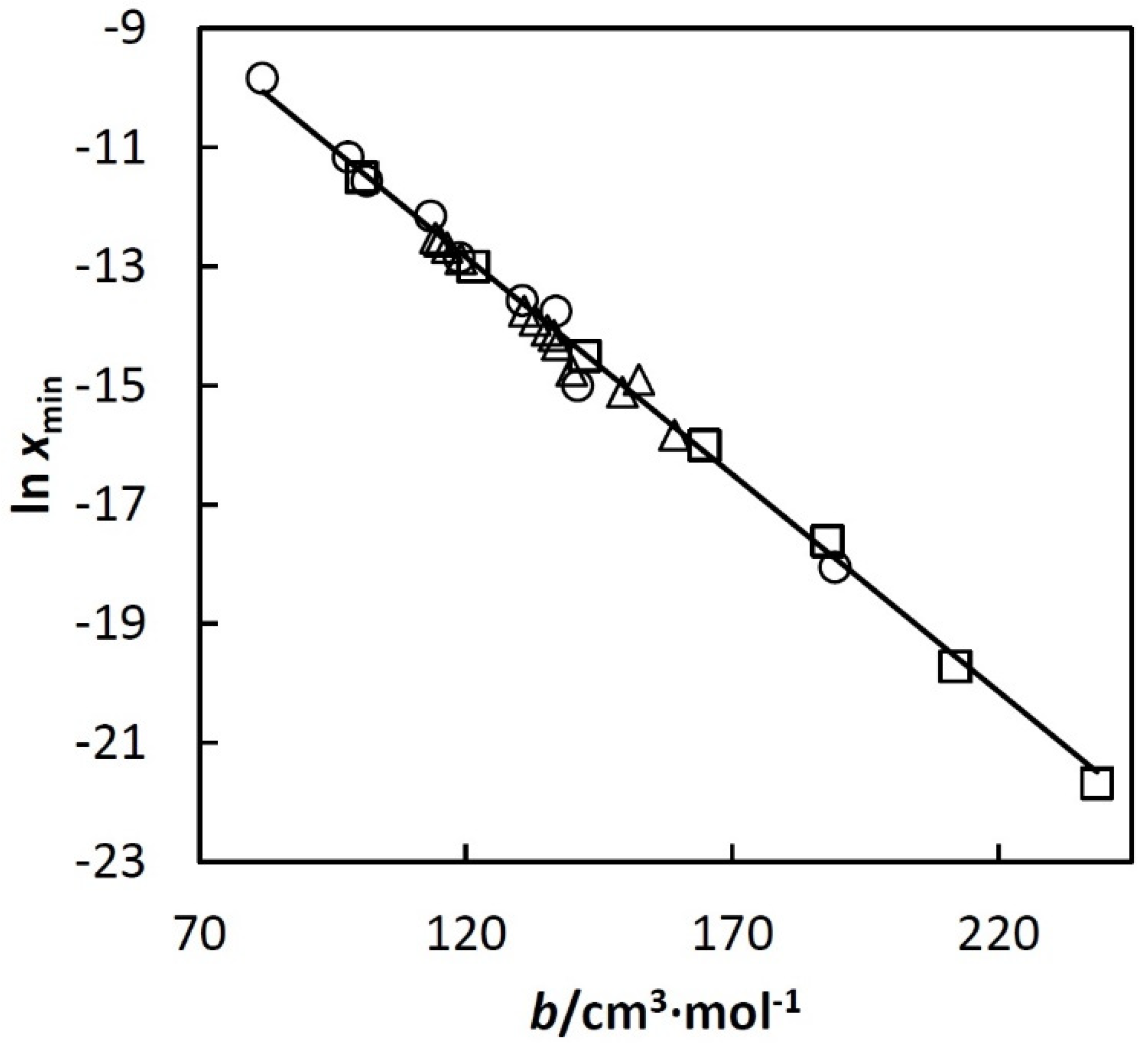
An example of the mentioned linear dependence is shown in Figure 3 (Others are given in papers [22,23,24,25].)
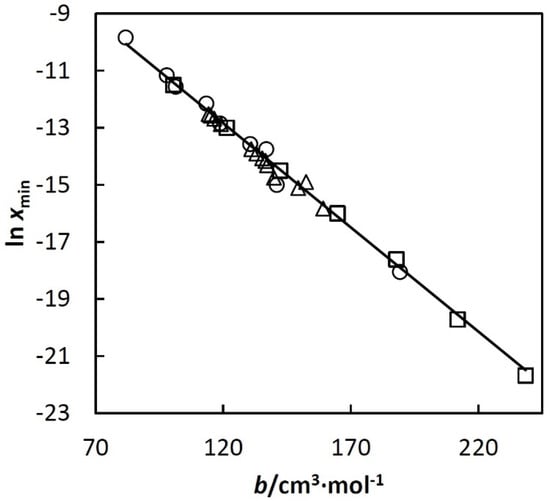
Figure 3.
ln xmin used in Equation (3) vs. excluded volume of the corresponding alkane, b (bh). Where symbols represent: □, n-alkanes; ○, cycloalkanes; ∆, isoalkanes. The alkanes are listed below in increasing order of b within each group: cyclopentane, cyclohexane, methylcyclopentane, cycloheptane, methylcyclohexane, cyclooctane, ethylcyclohexane, propylcyclopentane, pentylcyclopentane, pentane, hexane, heptane, octane, nonane, decane, undecane, 2,2- and 2,3-dimethylbutane, 3-methylpentane, 2-methylpentane, 3,3-, 2,3-, 2,2- and 2,4-dimethylpentane, 3-methylhexane, 2-methylhexane, 2,3,4-trimethylpentane, 2,2,4-trimethylpentane and 3-methylheptane.
The linear dependences introduced into Equation (3) give a general formula for n–alkanes, branched alkanes, and cycloalkanes as well as unsaturated hydrocarbons:
where Lh is the number of π–bonds in the hydrocarbon, e.g., Lh = 1 for alkenes, Lh = 2 for alkadienes and alkynes, and Lh = 4 for alkadiynes. If Lh = 0, then Equation (7) describes the solubility of n–alkanes, iso-alkanes, and cycloalkanes.
The analogous linear relations for ln xh,min and Ch, which hold for alkylbenzenes, introduced into Equation (3) give:
The coefficients cπ and c1–c7 in Equations (7) and (8) as well as the corresponding values of Tmin were obtained from simultaneous regression of all experimental solubility data reported in literature for given class of hydrocarbons. Prior to regression, the plots of the solubility data were inspected in order to remove evidently outlying experimental points. After the regression, the most outlying point was removed and the remaining points were regressed once more, yielding new a estimation of the standard deviation. This procedure was repeated until the deviation of the most outlying point in the remaining data set did not exceed three times the estimated standard deviation.
The following values of the parameters in Equation (7) were obtained: c1 = −4.08, c2/mol·cm−3 = −0.073, c3/mol·cm−3 = 0.376, cπ = 1.10, or cπ = 0.79 for conjugated π-bonds; Tmin/K = 298 (cycloalkanes); Tmin/K = 306 (other hydrocarbons).
The values of the coefficients in Equation (8) are as follows c4 = −2.605; c5/mol·cm−3 = −0.063; c6 = 7.98; c7/mol·cm−3 = 0.267; Tmin/K = 290.
Equations (7) and (8) with the obtained coefficients c1–c7 are valid at least up to T3c. Error analysis is given in papers [22,23,24,25]. It shows that standard deviation of the calculated xh depends on T and bh but in all cases it is below 10% of the calculated xh. Equations (7) and (8) approximate together almost 1000 experimental points for 64 systems using a relatively small number of adjustable parameters. The analysis of the deviations shows that they are randomly distributed and can be ascribed mainly to error of the experimental data. No trends were observed. In such a situation, the experimental random errors compensate each other during the regression, which makes the resulting Equations (7) and (8) more accurate than individual experimental points. Linear dependences such as that shown in Figure 3 were very useful for disclosing systematic errors of hydrocarbon solubility data.
There were no cases when measurements from different laboratories agreed with each other but not did not agree with the calculated values. As a rule, discrepancies occurred in systems represented by a few experimental points measured by one laboratory. Based on tests described in papers [22,23,24,25], we conclude that the observed discrepancies between experimental and calculated values result mainly from errors in the experimental data.
Additionally, the accuracy of the prediction also depends on the accuracy of the excluded volume, b, used as the input data in the Equations (7) or (8). The influence of b can be easily estimated from these equations. To estimate this, we calculated values of b from experimental Tc and Pc for n-alkanes from pentane to hexadecane. These values of b were plotted vs. the number of carbon atoms. From this plot, we estimated the standard deviation of b for this series is below 1 cm3·mol−1. This corresponds to a standard deviation of xh at room temperature equal to about 7% whereas the estimated standard deviation of the experimental mole fractions is about 30%. However, for some hydrocarbons, error in b can be much greater, so values of b calculated from experimental or predicted critical parameters must be selected with care before using them in the Equations (7) or (8).
The above parameters, and estimates of accuracy, are for mixtures with of n-alkanes, cycloalkanes, iso-alkanes, alkenes, alkadienes, alkynes, and alkylbenzenes having the number of carbon atoms between five and eleven. This limit is particularly important for alkanes. For heavier alkanes, the observed linearity shown in Figure 3 changes rapidly.
3. Solubility of Water in Hydrocarbons
Component concentrations in two coexisting liquid phases are related to each other by thermodynamic constraints of phase equilibrium. Hence, the solubility in one phase can be calculated using the concentration of the second liquid phase as the input data. To make such calculations, Economou and Tsonopoulos [14] applied equations of state SAFT and APACT for water + alkane systems. They started from the experimental solubility of water in hydrocarbons and tried to calculate the solubility curve of the alkanes in analogy to that shown in Figure 2. In conclusion, the authors wrote: “We have tested several models of hydrogen bonding for water as well as different mixing rules and found that none of these theories provide a quantitative estimate of the n-alkane solubility in water nor do they predict the n-alkane solubility minimum”.
In this work, the solubility of water in hydrocarbons was calculated by a method developed by Góral [38]. The method of correlation is called EoSC (Equation of State + Chemical term). In this method, a pure liquid is described by the cubic equation of state (EoS) with properly adjusted “effective” parameters. This approach is widely used in methods, which extends cubic EoS to associating mixtures using complicated mixing rules for the parameters in EoS. In the EoSC, the classical mixing rules are not modified. Instead, the chemical potential of the component derived from the cubic EoS is appended with a chemical term, which accounts for hydrogen bonding. It is defined as an excess function resulting from a change of the association of the pure substance when it is transferred to a mixture. Application of the EoSC for water systems is described in papers [22,23,24,25]. The input information for the LLE correlation is the solubility of a hydrocarbon in water, xh, calculated with Equation (7) or Equation (8). The output is the solubility of water, xw, in the hydrocarbon as a function of temperature. It is calculated from the constraints of chemical potentials at equilibrium:
where , , and are the chemical potentials of hydrocarbon and water in the first and the second phase, respectively. Equations (9a) and(9b) contain one adjustable binary parameter, Θ, in the physical part of the EoSC. Otherwise, the chemical potentials are based on a model of association. Once the parameters of the model are fixed, Equations (9a) and (9b) at given temperature and under the saturated vapor pressure contain three variables, xh, xw, and, where the hydrocarbon solubility, xh, is known from the smoothing equations (Equations (7) or (8)) described in the previous section. Thus, two unknown quantities, Θ and xw-mole fractions of water in the hydrocarbon, can be found by solving Equations (9a) and (9b). Such treatment ensures internal thermodynamic consistency between both branches of the solubility curve. Additional comments are presented in Appendix A.
The standard deviation of the calculated mole fraction of water in hydrocarbon does not exceed 5% at room temperatures and increases to about 10% at elevated temperatures, say sixty degrees below T3c.
4. Calculated Solubility Values
The calculated values for 217 systems at 298 K are listed below in Table 1. In the same table, the experimental values are given for 64 systems. This set of recommended data was selected from all available data reported in the literature. For 153 other systems, only the predicted solubilities given in Table 1 are available. In this paper, prediction is limited to these hydrocarbons, for which critical parameters have been measured. The prediction of the mutual solubility in other hydrocarbon + water systems is also possible, but one has to estimate values of the critical constants using one of the methods published in literature.

Table 1.
Calculated mutual solubilities in hydrocarbon + water systems at 298.00 K, xh–mole fraction of hydrocarbon in water, xw–mole fraction of water in the hydrocarbon. Whenever possible the recommended experimental data taken from [23,24,25] and/or [26,27,28,29,30,31,32,33,34,35,36,37] are reported in the second row. Aqueous solubilities for six hydrocarbons at 298.15 K measured by Dohányosová et al. [39] are included for comparison. For five of them, there was no prior experimental data. It should be noted that these data were not critically evaluated.
The estimated standard deviation of xh or xw resulting from accuracy of the method is estimated to within 5% of the calculated mole fraction.
Author Contributions
Conceptualization, M.G. and P.O.; methodology, M.G. and P.O.; software, M.G. and P.O.; writing—original draft preparation, M.G. and P.O.; writing—review and editing, P.O.; visualization, M.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Conflicts of Interest
The authors declare no conflict of interest.
Appendix A
The model of association takes into account auto-association of water and weak co–association between water and π-bonds in the unsaturated and aromatic hydrocarbons. The model of the association assumes that each active site of the donor type can interact with any active site of the acceptor type and that the equilibrium constant depends only on the type of the interacting sites. This assumption leads to a mixture of various pure and mixed associates. The kind and concentration of the hydrogen-bonded clusters depends on the chemical equilibrium in the mixture. Activities of the reacting species, in equations of the chemical equilibrium, were approximated with an expression resulting from the equation of state, which replaced molar concentration widely used in literature. The chemical equilibrium in water + alkane systems is described by equilibrium constant of auto-association of water (K22). The temperature dependency of K22 is described by three parameters: the equilibrium constant at the reference temperature, the enthalpy of hydrogen bond formation, and the corresponding heat capacity. Additionally, the excluded volume of water used in the chemical part is shifted by −6.5 cm3·mol−1 with respect to value calculated with Equation (6) and used in the physical part. Altogether, this model of auto-association of water contains four parameters, which were kept constant for all the investigated systems. With these four parameters, LLE calculations were performed for water + hydrocarbon systems, including n–alkanes, iso-alkanes (branched alkanes), as well as cycloalkanes, in the temperature range from 273.15 K up to tabout 60 K below T3c. The calculated solubility of water as a function of temperature was compared with experimental points for 21 systems reported in literature.
Water is more soluble in unsaturated hydrocarbons than in alkanes. This shift of water solubility depends on the number of π–bonds in the hydrocarbon molecule. To account for this phenomenon, it was assumed that each π–bond can co–associate with free hydrogen atom of water. Each hydrogen bond of this kind was described with the same equilibrium constant. Altogether, the model of the co-association uses two additional parameters in addition to the previously used parameters of water, which remained unchanged. These parameters were used for the calculation of solubility curves of water in unsaturated hydrocarbons. The calculated values were compared with experimental data for all 24 systems available in the reported in literature.
For alkylbenzene, it was assumed that each aromatic ring can co-associate with the free hydrogen atom of water. The hydrogen bond of this kind was described by a temperature dependent equilibrium constant of the co-association, which was described with three parameters. Solubility of water in the alkylbenzenes was used only at the beginning of this investigation to fix these three parameters. They were kept constant for all investigated mixtures. The parameters of the auto-association of water were unchanged. The solubility of water in alkylbenzenes was reported in literature for 15 systems consisting of 405 experimental points. The most outlying 36 points were rejected from further investigations. The remaining 369 points were compared with the calculated values.
Altogether, the model of association for all types of systems investigated here uses nine physically meaningful constants. With these constants, the solubility of water in 60 hydrocarbons in large temperature intervals was calculated and compared with experimental data. These comparisons and additional tests show good agreement between experimental and predicted data. They are described in papers [22,23,24,25]. Within the same class of mixtures, both positive and negative deviations are observed. Calculations of the water solubility were performed with physically meaningful values of the equilibrium constants of association common to the given class of systems, which reveals systems with deviation. We conclude that these deviations result mainly from errors in the experimental data. For more details, refer to [22,23,24,25].
References
- Tsonopoulos, C. Thermodynamic analysis of the mutual solubilities of normal alkanes and water. Fluid Phase Equilibria 1999, 156, 21–33. [Google Scholar] [CrossRef]
- Tsonopoulos, C. Thermodynamic analysis of the mutual solubilities of normal alkanes and water. Fluid Phase Equilibria 2001, 186, 185–206. [Google Scholar] [CrossRef]
- Oracz, P.; Góral, M. Application of the Unified Functional Activity Coefficient (UNIFAC) and Analytical Solution of Groups (ASOG) for the Calculation of Mutual Solubilities in Water Systems of Alkanes, Arenes, and Alkanols. J. Chem. Eng. Data 2011, 56, 4853–4861. [Google Scholar] [CrossRef]
- Magnussen, T.; Rasmussen, P.; Fredenslund, A. UNIFAC parameter table for prediction of liquid-liquid equilibria. Ind. Eng. Chem. Process Des. Dev. 1981, 20, 331–339. [Google Scholar] [CrossRef]
- Gupte, P.A.; Danner, R.P. Prediction of Liquid-Liquid Equilibria with UNIFAC: A Critical Evaluation. Ind. Eng. Chem. Res. 1987, 26, 2036–2042. [Google Scholar] [CrossRef]
- Hooper, H.; Michel, S.; Prausnitz, J.M. Correlation of liquid-liquid equilibria for some water-organic liquid systems in the region 20–250 °C. Ind. Eng. Chem. Res. 1988, 27, 2182–2187. [Google Scholar] [CrossRef]
- Voutsas, E.C.; Tassios, D.P. An Analysis of the UNIFAC-Type Group-Contribution Models at the Highly Dilute Region. 2. Empirical Improvements with Application to Water/Hydrocarbon Mixtures. Ind. Eng. Chem. Res. 1997, 36, 4973–4976. [Google Scholar] [CrossRef]
- Kang, J.W.; Diky, V.; Frenkel, M. New modified UNIFAC parameters using critically evaluated phase equilibrium data. Fluid Phase Equilibria 2015, 388, 128–141. [Google Scholar] [CrossRef]
- Kang, J.W.; Diky, V.; Frenkel, M. Corrigendum to “New modified UNIFAC parameters using critically evaluated phase equilibrium data” [Fluid Phase Equilib. 388 (2015) 128-141]. Fluid Phase Equilibria 2017, 440, 122–123. [Google Scholar] [CrossRef]
- Gmehling, J.; Li, J.; Schiller, M. A Modified UNIFAC Model. 2. Present Parameter Matrix and Results. Ind. Eng. Chem. Res. 1993, 32, 178–193. [Google Scholar] [CrossRef]
- Yakoumis, I.V.; Kontogeorgis, G.M.; Voutsas, E.C.; Hendriks, E.M.; Tassios, D.P. Prediction of Phase Equilibria in Binary Aqueous Systems Containing Alkanes, Cycloalkanes, and Alkenes with the Cubic-plus-Association Equation of State. Ind. Eng. Chem. Res. 1998, 37, 4175–4182. [Google Scholar] [CrossRef]
- Voutsas, E.C.; Boulougouris, G.C.; Economou, I.G.; Tassios, D.P. Water/Hydrocarbon Phase Equilibria Using the Thermodynamic Perturbation Theory. Ind. Eng. Chem. Res. 2000, 39, 797–804. [Google Scholar] [CrossRef]
- Oliveira, M.B.; Coutinho, J.A.P.; Queimad, A.J. Mutual solubilities of hydrocarbons and water with the CPA EoS. Fluid Phase Equilibria 2007, 258, 58–66. [Google Scholar] [CrossRef] [Green Version]
- Economou, I.G.; Tsonopoulos, C. Association models and mixing rules in equations of state for water/hydrocarbon mixtures. Chem. Eng. Sci. 1997, 52, 511–525. [Google Scholar] [CrossRef]
- Vega, L.F.; Llovell, F.; Blas, F.J. Capturing the Solubility Minima of n-Alkanes in Water by Soft-SAFT. J. Phys. Chem. B 2009, 113, 7621–7630. [Google Scholar] [CrossRef]
- Ahmed, S.; Ferrando, N.; de Hemptinne, J.-C.; Simonin, J.-P.; Bernard, O.; Baudouin, O. A New PC-SAFT Model for Pure Water, Water–Hydrocarbons, and Water–Oxygenates Systems and Subsequent Modeling of VLE, VLLE, and LLE. J. Chem. Eng. Data 2016, 61, 4178–4190. [Google Scholar] [CrossRef]
- Landra, C.; Satyro, M.A. Mutual Solubility of Water and Hydrocarbons. J. Chem. Eng. Data 2016, 61, 525–534. [Google Scholar] [CrossRef]
- Haarmann, N.; Enders, S.; Sadowski, G. Modeling binary mixtures of n-alkanes and water using PC-SAFT. Fluid Phase Equilibria 2018, 470, 203–211. [Google Scholar] [CrossRef]
- Safamirzaei, M.; Modarress, H. Modeling and predicting solubility of n-alkanes in water. Fluid Phase Equilibria Equilibria 2011, 309, 53–61. [Google Scholar] [CrossRef]
- Klamt, A. Prediction of the mutual solubilities of hydrocarbons and water with COSMO-RS. Fluid Phase Equilibria 2003, 206, 223–235. [Google Scholar] [CrossRef] [Green Version]
- Morgado, P.; Barras, J.; Duarte, P.; Filipe, E.J.M. Solubility of water in n-alkanes: New experimental measurements and molecular dynamics simulations. Fluid Phase Equilibria 2020, 503, 112322. [Google Scholar] [CrossRef]
- Mączyński, A.; Góral, M.; Wiśniewska–Gocłowska, B.; Skrzecz, A.; Shaw, D. Mutual Solubilities of Water and Alkanes. Mon. Chem. 2003, 134, 633–653. [Google Scholar] [CrossRef]
- Maczynski, A.; Wisniewska–Goclowska, B.; Goral, M. Recommended Liquid-Liquid Equilibrium Data, Part 1: Binary C5–C11 Alkane—Water Systems. J. Phys. Chem. Ref. Data 2003, 33, 549–577. [Google Scholar] [CrossRef]
- Goral, M.; Wisniewska–Goclowska, B.; Maczynski, A. Recommended Liquid-Liquid Equilibrium Data, Part 2: Binary Unsaturated hydrocarbon—Water Systems. J. Phys. Chem. Ref. Data 2004, 33, 579–591. [Google Scholar] [CrossRef] [Green Version]
- Goral, M.; Wisniewska–Goclowska, B.; Maczynski, A. Recommended Liquid-Liquid Equilibrium Data, Part 3: Binary Alkylbenzene—Water Systems. J. Phys. Chem. Ref. Data 2004, 33, 1159–1190. [Google Scholar] [CrossRef]
- Maczynski, A.; Shaw, D.; Goral, M.; Wisniewska–Goclowska, B.; Skrzecz, A.; Owczarek, I.; Blazej, K.; Haulait–Pirson, M.-C.; Kapuku, F.; Hefter, G.T.; et al. IUPAC-NIST Solubility Data Series, Hydrocarbons with Water and Seawater–Revised and Updated, Part 1. C5 Hydrocarbons with Water. J. Phys. Chem. Ref. Data 2005, 34, 441–476. [Google Scholar] [CrossRef] [Green Version]
- Maczynski, A.; Shaw, D.; Goral, M.; Wisniewska–Goclowska, B.; Skrzecz, A.; Owczarek, I.; Blazej, K.; Haulait–Pirson, M.-C.; Hefter, G.T.; Szafranski, A.; et al. IUPAC-NIST Solubility Data Series, Hydrocarbons with Water and Seawater–Revised and Updated, Part 2. Benzene with Water and Heavy Water. J. Phys. Chem. Ref. Data 2005, 34, 477–552. [Google Scholar] [CrossRef] [Green Version]
- Maczynski, A.; Shaw, D.; Goral, M.; Wisniewska–Goclowska, B.; Skrzecz, A.; Owczarek, I.; Blazej, K.; Haulait–Pirson, M.-C.; Hefter, G.T.; Maczynska, Z.; et al. IUPAC-NIST Solubility Data Series, Hydrocarbons with Water and Seawater–Revised and Updated, Part 3. C6H8–C6H12 Hydrocarbons with Water and Heavy Water. J. Phys. Chem. Ref. Data 2005, 34, 657–708. [Google Scholar] [CrossRef] [Green Version]
- Maczynski, A.; Shaw, D.; Goral, M.; Wisniewska–Goclowska, B.; Skrzecz, A.; Owczarek, I.; Blazej, K.; Haulait–Pirson, M.-C.; Hefter, G.T.; Kapuku, F.; et al. IUPAC-NIST Solubility Data Series, Hydrocarbons with Water and Seawater–Revised and Updated, Part 4. C6H14 Hydrocarbons with Water. J. Phys. Chem. Ref. Data 2005, 34, 709–753. [Google Scholar] [CrossRef] [Green Version]
- Maczynski, A.; Shaw, D.; Goral, M.; Wisniewska–Goclowska, B.; Skrzecz, A.; Owczarek, I.; Blazej, K.; Haulait–Pirson, M.-C.; Hefter, G.T.; Kapuku, F.; et al. IUPAC-NIST Solubility Data Series, Hydrocarbons with Water and Seawater–Revised and Updated, Part 5. C7 Hydrocarbons with Water and Heavy Water. J. Phys. Chem. Ref. Data 2005, 34, 1399–1488. [Google Scholar] [CrossRef] [Green Version]
- Shaw, D.; Maczynski, A.; Goral, M.; Wisniewska–Goclowska, B.; Skrzecz, A.; Owczarek, I.; Blazej, K.; Haulait–Pirson, M.-C.; Hefter, G.T.; Maczynska, Z.; et al. IUPAC-NIST Solubility Data Series, Hydrocarbons with Water and Seawater–Revised and Updated, Part 6. C8H8–C8H10 Hydrocarbons with Water. J. Phys. Chem. Ref. Data 2005, 34, 1489–1553. [Google Scholar] [CrossRef] [Green Version]
- Shaw, D.; Maczynski, A.; Goral, M.; Wisniewska–Goclowska, B.; Skrzecz, A.; Owczarek, I.; Blazej, K.; Haulait–Pirson, M.-C.; Hefter, G.T.; Kapuku, F.; et al. IUPAC-NIST Solubility Data Series, Hydrocarbons with Water and Seawater–Revised and Updated, Part 7. C8H12–C8H18 Hydrocarbons with Water. J. Phys. Chem. Ref. Data 2005, 34, 2261–2298. [Google Scholar] [CrossRef] [Green Version]
- Shaw, D.; Maczynski, A. Erratum: IUPAC-NIST Solubility Data Series. 81. Hydrocarbons with Water and Seawater Revised and Updated. Part 7. C8H12–C8H18 Hydrocarbons with Water. J. Phys. Chem. Ref. Data 2005, 34, 2261. [Google Scholar] [CrossRef]
- Shaw, D.; Maczynski, A.; Goral, M.; Wisniewska–Goclowska, B.; Skrzecz, A.; Owczarek, I.; Blazej, K.; Haulait–Pirson, M.-C.; Hefter, G.T.; Kapuku, F.; et al. IUPAC-NIST Solubility Data Series, Hydrocarbons with Water and Seawater–Revised and Updated, Part 8. C9 Hydrocarbons with Water. J. Phys. Chem. Ref. Data 2005, 34, 2299–2345. [Google Scholar] [CrossRef] [Green Version]
- Shaw, D.; Maczynski, A.; Goral, M.; Wisniewska–Goclowska, B.; Skrzecz, A.; Owczarek, I.; Blazej, K.; Haulait–Pirson, M.-C.; Hefter, G.T.; Kapuku, F.; et al. IUPAC-NIST Solubility Data Series, Hydrocarbons with Water and Seawater–Revised and Updated, Part 9. C10 Hydrocarbons with Water. J. Phys. Chem. Ref. Data 2006, 35, 93–151. [Google Scholar] [CrossRef] [Green Version]
- Shaw, D.; Maczynski, A.; Goral, M.; Wisniewska–Goclowska, B.; Skrzecz, A.; Owczarek, I.; Blazej, K.; Haulait–Pirson, M.-C.; Hefter, G.T.; Kapuku, F.; et al. IUPAC-NIST Solubility Data Series, Hydrocarbons with Water and Seawater–Revised and Updated, Part 10. C11 and C12 Hydrocarbons with Water and Heavy Water. J. Phys. Chem. Ref. Data 2006, 35, 153–203. [Google Scholar] [CrossRef] [Green Version]
- Shaw, D.; Maczynski, A.; Goral, M.; Wisniewska–Goclowska, B.; Skrzecz, A.; Owczarek, I.; Blazej, K.; Haulait–Pirson, M.-C.; Hefter, G.T.; Kapuku, F.; et al. IUPAC-NIST Solubility Data Series, Hydrocarbons with Water and Seawater–Revised and Updated, Part 11. C13–C36 Hydrocarbons with Water. J. Phys. Chem. Ref. Data 2006, 35, 687–784. [Google Scholar] [CrossRef] [Green Version]
- Góral, M. Cubic equation of state for calculation of phase equilibria in association systems. Fluid Phase Equilibria 1996, 118, 27–59. [Google Scholar] [CrossRef]
- Dohányosová, P.; Sarraute, S.; Dohnal, V.; Majer, V.; Gomes, M.C. Aqueous Solubility and Related Thermodynamic Functions of Nonaromatic Hydrocarbons as a Function of Molecular Structure. Ind. Eng. Chem. Res. 2004, 43, 2805–2815. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).