
Graphene Oxide Synthesis, Properties and Characterization Techniques: A Comprehensive Review




Abstract
1. Introduction
1.1. Carbon and Allotropes
1.2. Fullerenes
1.3. Carbon Nanotubes (CNTs)
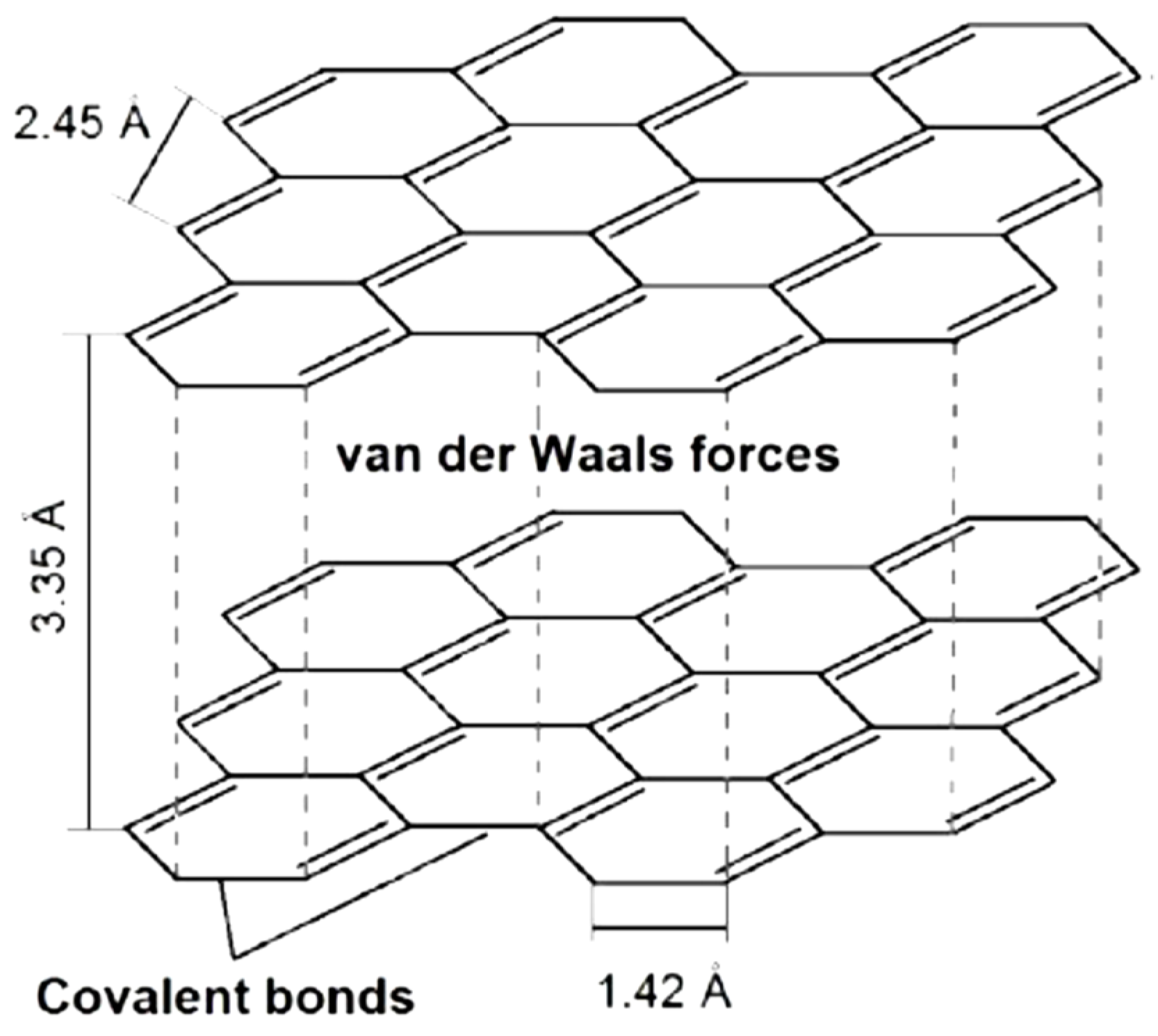
1.4. Graphites
1.5. Graphenes
1.5.1. Graphene Properties
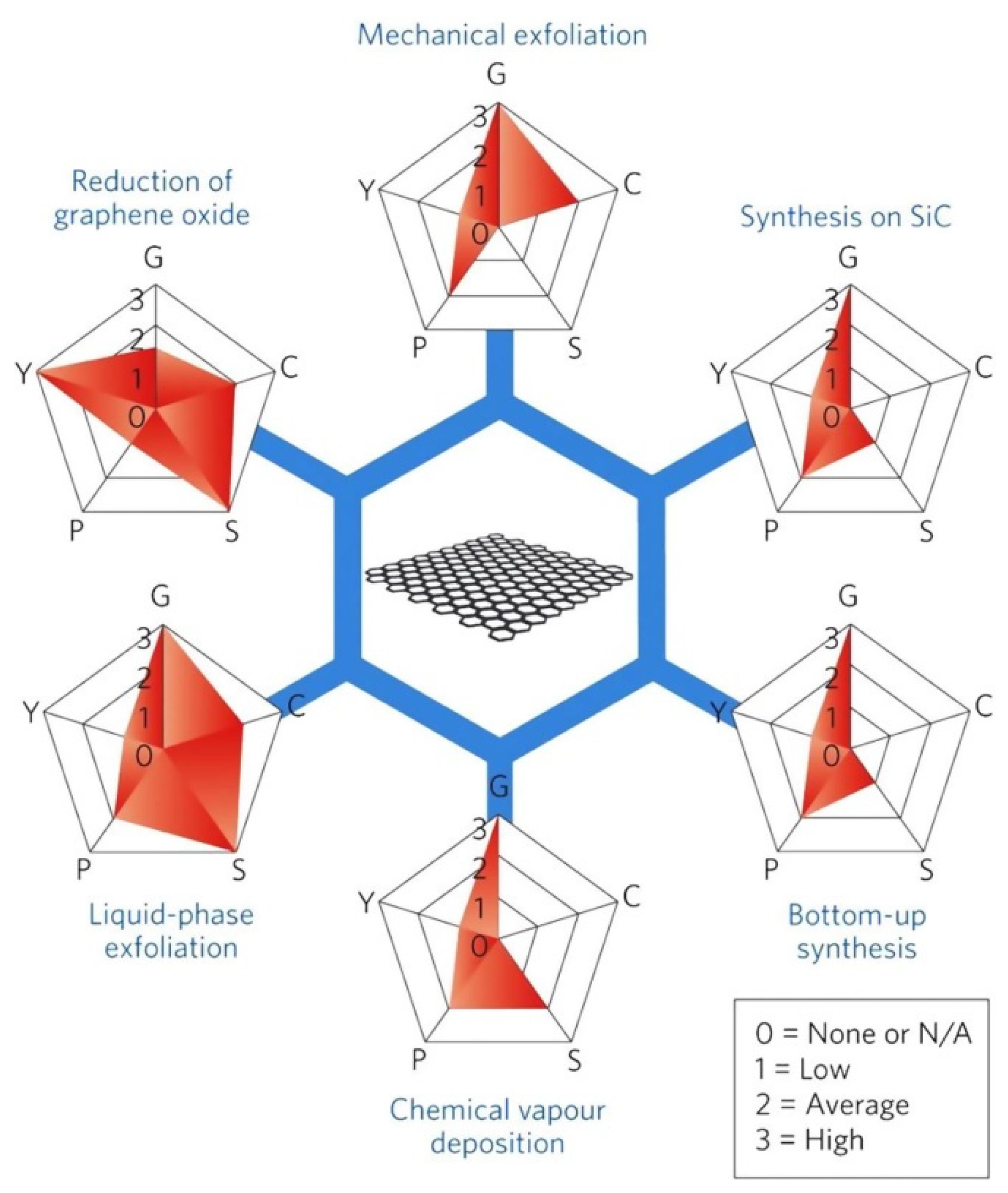
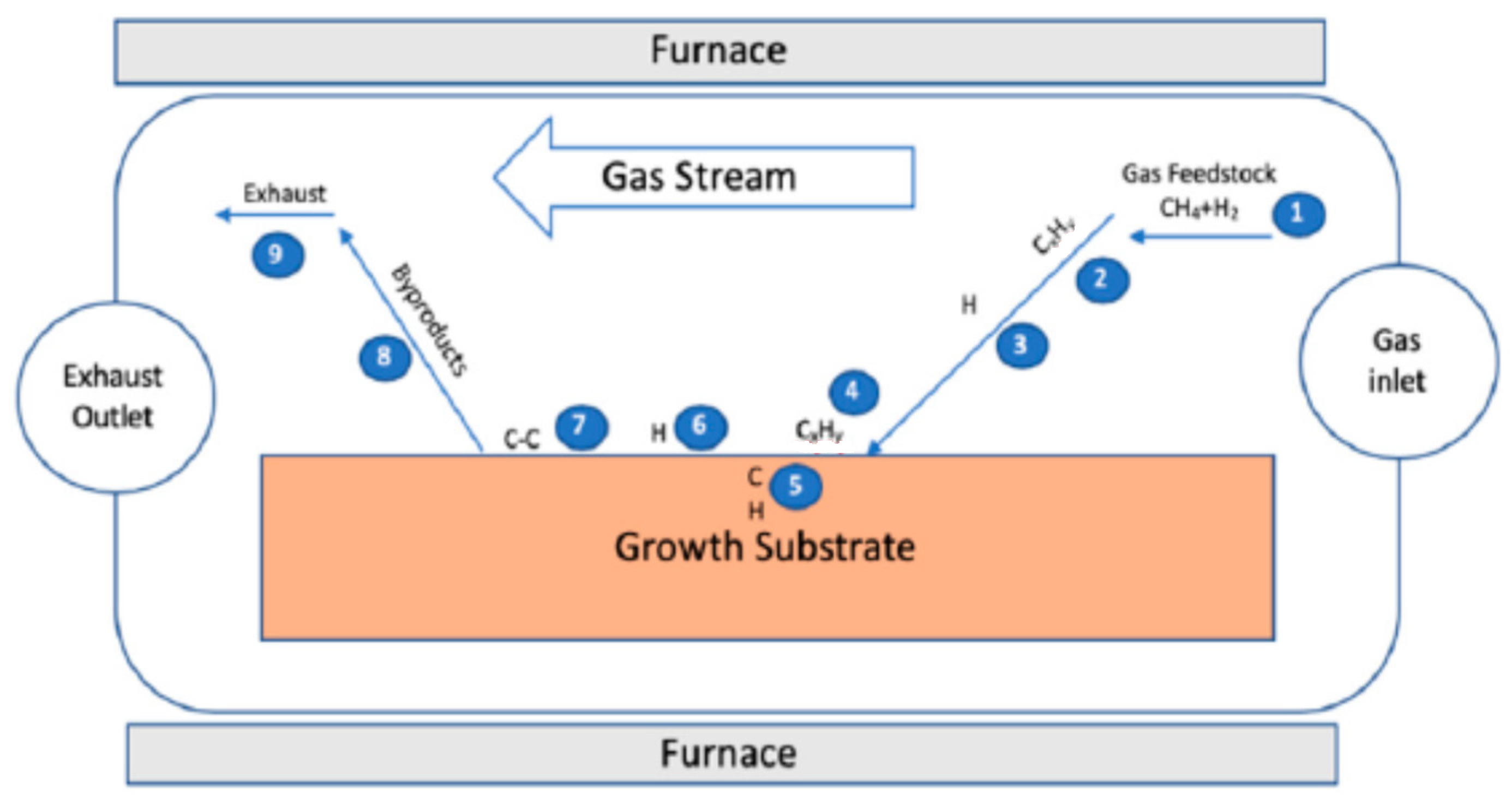
1.5.2. Graphene Synthesis
2. Graphene Oxide (GO)
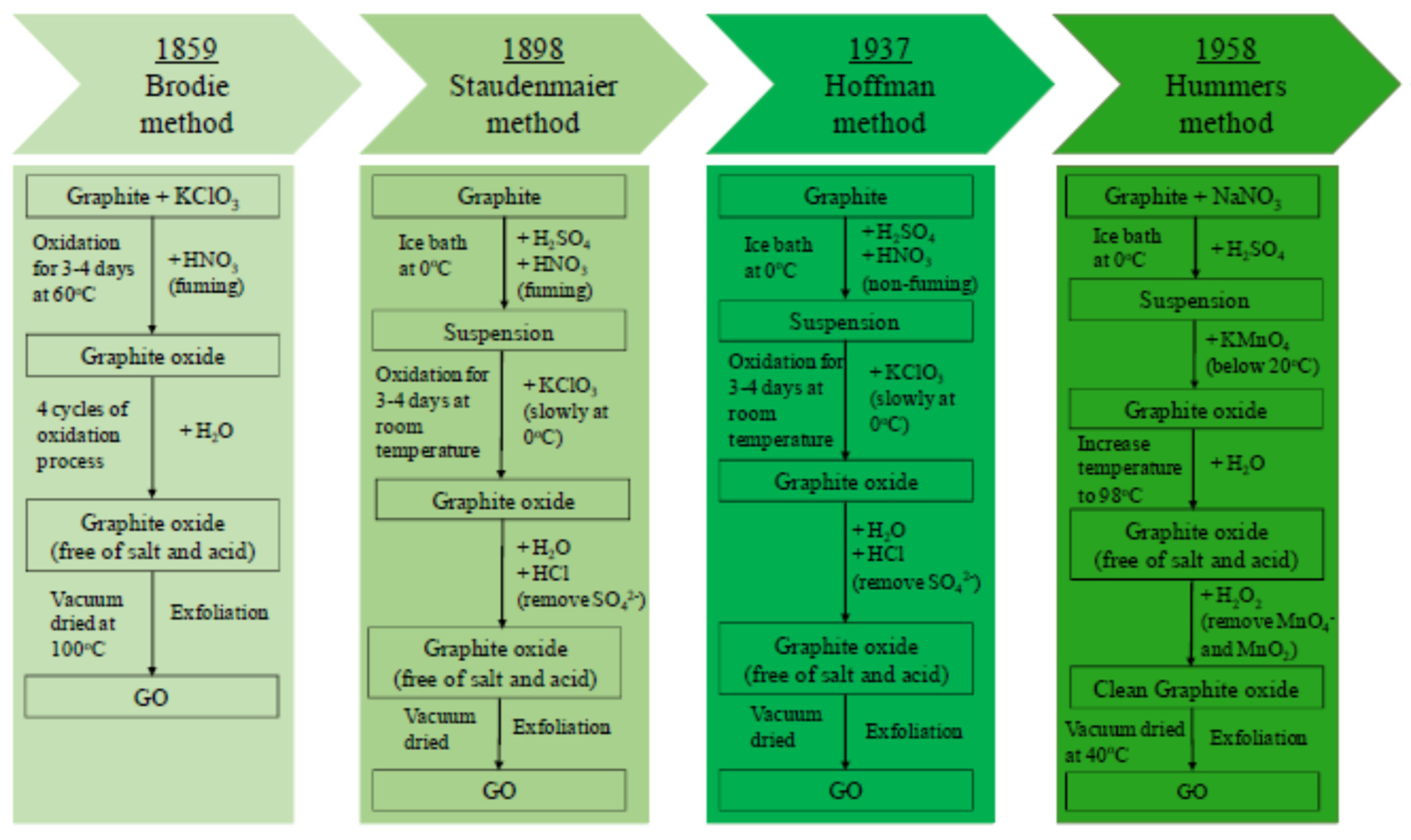
2.1. GO Synthesis
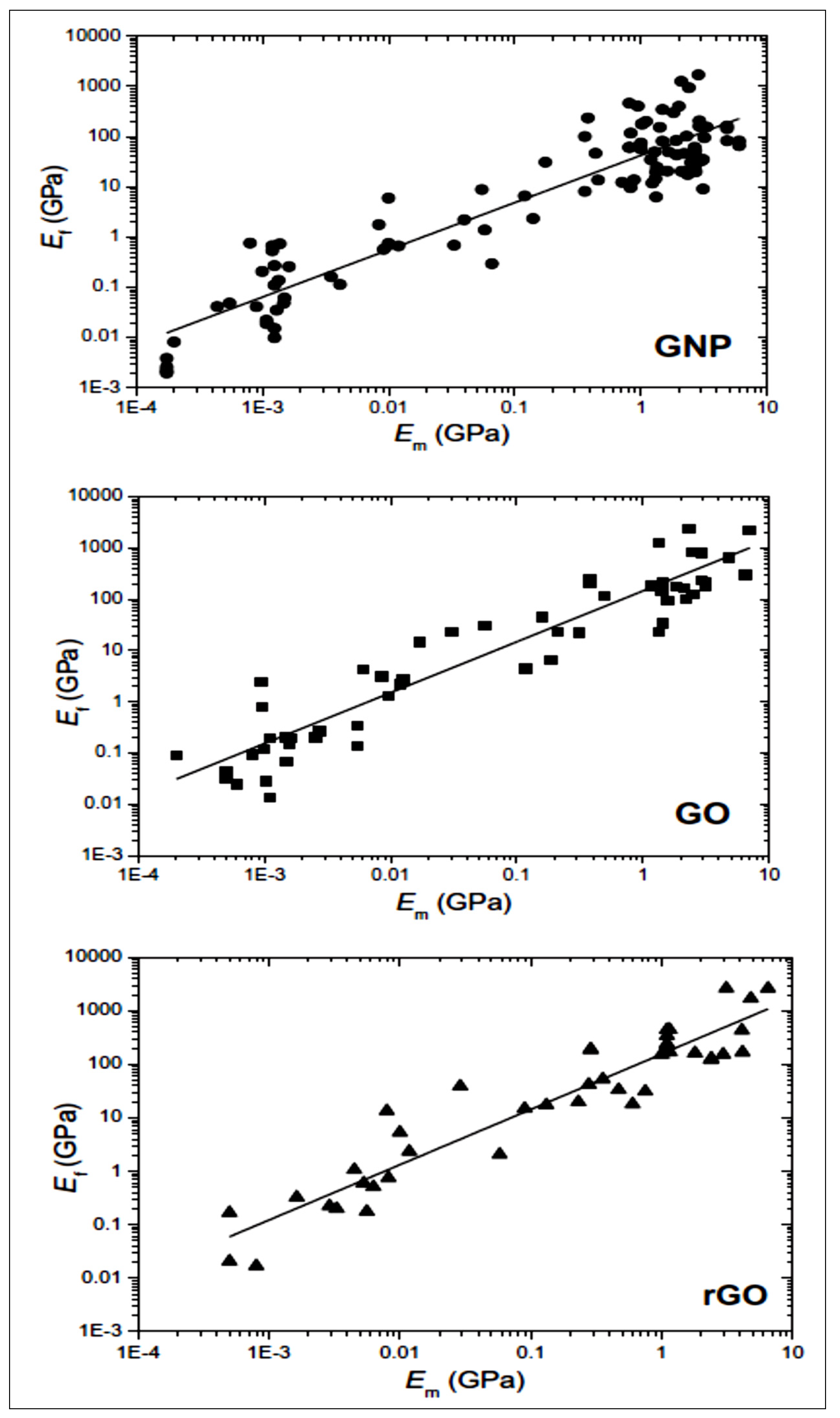
2.2. GO Properties
2.3. GO Composites
2.4. Reduced Graphene Oxide (rGO)
3. Characterization Techniques
3.1. X-ray Diffraction (XRD)
3.2. Scanning Electron Microscopy-Energy Dispersive X-ray Spectroscopy (SEM-EDS)
3.3. Fourier Transform Infrared Spectroscopy (FTIR)
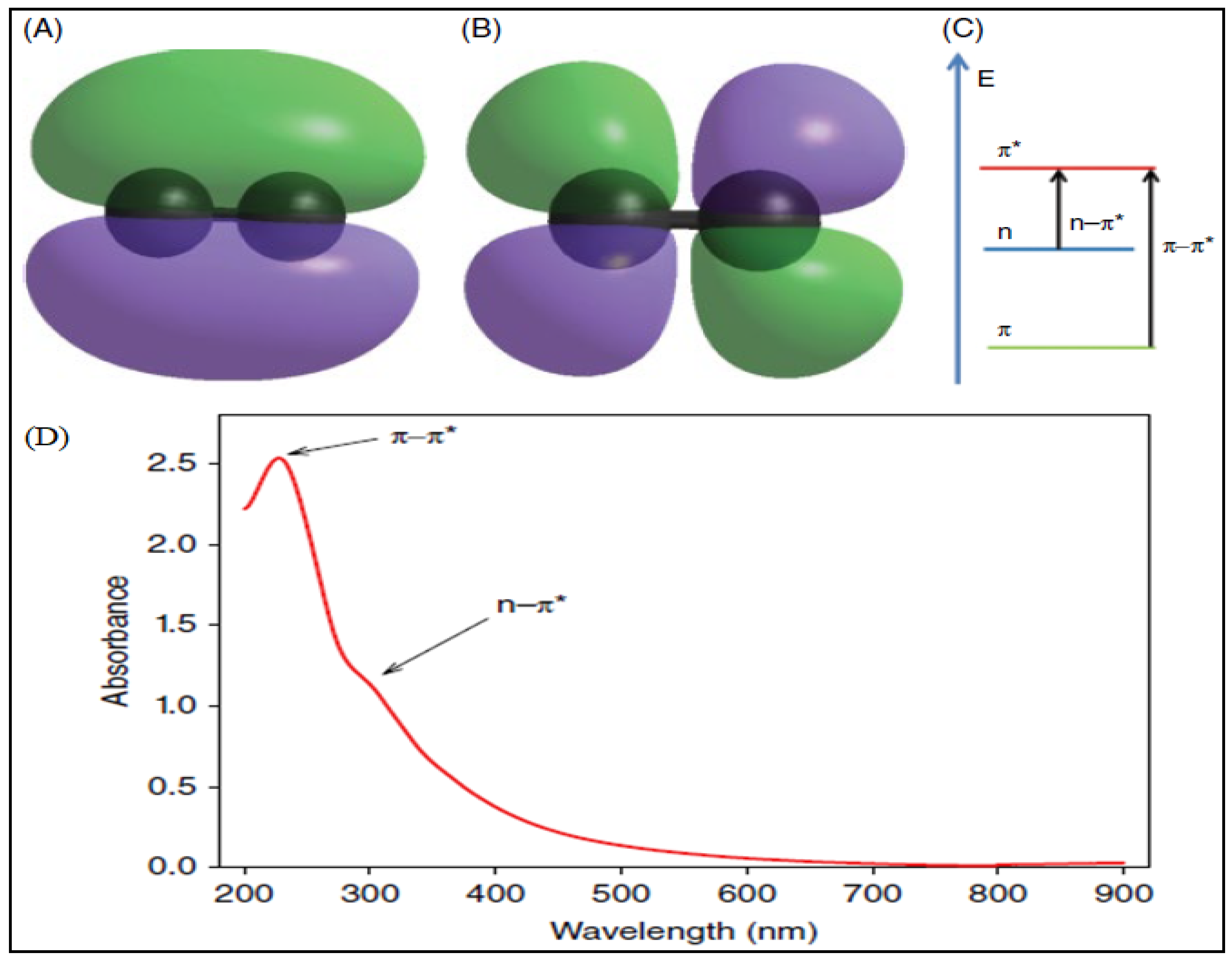
3.4. Ultraviolet-Visible Spectrophotometry (UV-Vis)
3.5. N2 Porosimetry
3.6. Atomic Force Microscopy (AFM)
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, Q.; Wu, Z.; Li, N.; Pu, Y.; Wang, B.; Zhang, T.; Tao, J. Advanced review of graphene-based nanomaterials in drug delivery systems: Synthesis, modification, toxicity and application. Mater. Sci. Eng. C 2017, 77, 1363–1375. [Google Scholar] [CrossRef]
- Kostarelos, K.; Novoselov, K. Graphene devices for life. Nat. Nanotechnol. 2014, 9, 744–745. [Google Scholar] [CrossRef] [PubMed]
- Schwarzschild, B. Nobel chemistry prize goes to Curl, Kroto and Smalley for discovering fullerenes. Phys. Today 1996, 49, 19. [Google Scholar] [CrossRef]
- Goodarzi, S.; Da Ros, T.; Conde, J.; Sefat, F.; Mozafari, M. Fullerene: Biomedical engineers get to revisit an old friend. Mater. Today 2017, 20, 460–480. [Google Scholar] [CrossRef]
- Bendicho, C.; Bendicho-Lavilla, C.; Lavilla, I. Nanoparticle-assisted chemical speciation of trace elements. TrAC Trends Anal. Chem. 2016, 77, 109–121. [Google Scholar] [CrossRef]
- Abella, L.; Wang, Y.; Rodríguez-Fortea, A.; Chen, N.; Poblet, J.M. Current status of oxide clusterfullerenes. Inorganica Chim. Acta 2017, 468, 91–104. [Google Scholar] [CrossRef]
- Balch, A.L.; Winkler, K. Two-component polymeric materials of fullerenes and the transition metal complexes: A bridge between metal–organic frameworks and conducting polymers. Chem. Rev. 2016, 116, 3812–3882. [Google Scholar] [CrossRef]
- Aderibigbe, B.; Aderibigbe, I.; Popoola, P. Design and biological evaluation of delivery systems containing bisphosphonates. Pharmaceutics 2016, 9, 2. [Google Scholar] [CrossRef]
- Pitorre, M.; Gonde, H.; Haury, C.; Messous, M.; Poilane, J.; Bousaud, D.; Kanber, E.; Ndombina, G.A.R.; Benoit, J.P.; Bastiat, G. Recent advances in nanocarrier-loaded gels: Which drug delivery technologies against which diseases? J. Control. Release 2017, 266, 140–155. [Google Scholar] [CrossRef]
- Gatti, T.; Menna, E.; Meneghetti, M.; Maggini, M.; Petrozza, A.; Lamberti, F. The Renaissance of fullerenes with perovskite solar cells. Nano Energy 2017, 41, 84–100. [Google Scholar] [CrossRef]
- Rice, A.M.; Dolgopolova, E.A.; Shustova, N.B. Fulleretic materials: Buckyball- and Buckybowl-based crystalline frameworks. Chem. Mater. 2017, 29, 7054–7061. [Google Scholar] [CrossRef]
- Mykhailiv, O.; Zubyk, H.; Plonska-Brzezinska, M. Carbon nano-onions: Unique carbon nanostructures with fascinating properties and their potential applications. Inorganica Chim. Acta 2017, 468, 49–66. [Google Scholar] [CrossRef]
- Abbas, I.A.; Al-Amer, A.M.; Laoui, T.; Al-Marri, M.J.; Nasser, M.S.; Khraisheh, M.; Atieh, M.A. Heavy metal removal from aqueous solution by advanced carbon nanotubes: Critical review of adsorption applications. Sep. Purif. Technol. 2016, 157, 141–161. [Google Scholar]
- Gupta, V.K.; Moradi, O.; Tyagi, I.; Agarwal, S.; Sadegh, H.; Shahryari-Ghoshekandi, R.; Makhlouf, A.S.H.; Goodarzi, M.; Garshasbi, A. Study on the removal of heavy metal ions from industry waste by carbon nanotubes: Effect of the surface modification: A review. Crit. Rev. Environ. Sci. Technol. 2015, 46, 93–118. [Google Scholar] [CrossRef]
- Hollanda, L.; Lobo, A.; Lancellotti, M.; Berni, E.; Corat, E.; Zanin, H. Graphene and carbon nanotube nanocomposite for gene transfection. Mater. Sci. Eng. C 2014, 39, 288–298. [Google Scholar] [CrossRef]
- Wang, H.; Kakade, B.A.; Tamaki, T.; Yamaguchi, T. Synthesis of 3D graphite oxide-exfoliated carbon nanotube carbon composite and its application as catalyst support for fuel cells. J. Power Sources 2014, 260, 338–348. [Google Scholar] [CrossRef]
- Kumar, R.; Chawla, J.; Kaur, I. Removal of cadmium ion from wastewater by carbon-based nanosorbents: A review. J. Water Health 2014, 13, 18–33. [Google Scholar] [CrossRef]
- Alsharef, J.; Taha, M.R.; Khan, T.A. Physical dispersion of nanocarbons in composites—A Review. J. Teknol. 2017, 79, 69–81. [Google Scholar] [CrossRef]
- Nupearachchi, C.; Mahatantila, K.; Vithanage, M. Application of graphene for decontamination of water; Implications for sorptive removal. Groundw. Sustain. Dev. 2017, 5, 206–215. [Google Scholar] [CrossRef]
- Zhang, H.; Chen, N.; Sun, C.; Luo, X. Investigations on physicochemical properties and electrochemical performance of graphite felt and carbon felt for iron-chromium redox flow battery. Int. J. Energy Res. 2020, 44, 3839–3853. [Google Scholar] [CrossRef]
- Zhang, H.; Tan, Y.; Luo, X.; Sun, C.; Chen, N. Polarization effects of a rayon and polyacrylonitrile based graphite felt for iron-chromium redox flow batteries. ChemElectroChem 2019, 6, 3175–3188. [Google Scholar] [CrossRef]
- Inagaki, M.; Kaburagi, Y.; Hishiyama, Y. Thermal management material: Graphite. Adv. Eng. Mater. 2014, 16, 494–506. [Google Scholar] [CrossRef]
- de Oliveira, E.H.C.; Fraga, D.M.D.S.M.; da Silva, M.P.; Fraga, T.J.M.; Carvalho, M.N.; Freire, E.M.P.D.L.; Ghislandi, M.G.; Sobrinho, M.A.D.M. Removal of toxic dyes from aqueous solution by adsorption onto highly recyclable xGnP® graphite nanoplatelets. J. Environ. Chem. Eng. 2019, 7, 103001. [Google Scholar] [CrossRef]
- Peierls, R. Quelques proprietes typiques des corps solides. Ann. L’institut Henri Poincare 1935, 5, 177–222. [Google Scholar]
- Landau, L.D. Zur theorie der phase numwandlungen II. Phys. Z. Sowjetunion 1937, 11, 26–35. [Google Scholar]
- Wallace, P.R. The band theory of graphite. Phys. Rev. 1947, 71, 622–634. [Google Scholar] [CrossRef]
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Zhang, Y.; Dubonos, S.V.; Grigorieva, I.V.; Firsov, A.A. Electric field effect in atomically thin carbon films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef] [PubMed]
- Molina, J.; Cases, F.; Moretto, L. Graphene-based materials for the electrochemical determination of hazardous ions. Anal. Chim. Acta 2016, 946, 9–39. [Google Scholar] [CrossRef]
- Randviir, E.P.; Brownson, D.; Banks, C.E. A decade of graphene research: Production, applications and outlook. Mater. Today 2014, 17, 426–432. [Google Scholar] [CrossRef]
- Compton, O.C.; Jain, B.; Dikin, D.A.; Abouimrane, A.; Amine, K.; Nguyen, S. Chemically active reduced graphene oxide with tunable C/O ratios. ACS Nano 2011, 5, 4380–4391. [Google Scholar] [CrossRef]
- Hussain, A.; Mehdi, S.M.; Abbas, N.; Hussain, M.; Naqvi, R.A. Synthesis of graphene from solid carbon sources: A focused review. Mater. Chem. Phys. 2020, 248, 122924. [Google Scholar] [CrossRef]
- Shen, Y.; Fang, Q.; Chen, B. Environmental applications of three-dimensional graphene-based macrostructures: Adsorption, transformation, and detection. Environ. Sci. Technol. 2014, 49, 67–84. [Google Scholar] [CrossRef]
- Del Castillo, E.; Cargnoni, F.; Soave, R.; Trioni, I.M. Spin-polarized charge transfer induced by transition metal adsorption on graphene. Phys. Scr. 2016, 91, 53007. [Google Scholar] [CrossRef]
- Lee, X.J.; Hiew, B.Y.Z.; Lai, K.C.; Lee, L.Y.; Gan, S.; Thangalazhy-Gopakumar, S.; Rigby, S. Review on graphene and its derivatives: Synthesis methods and potential industrial implementation. J. Taiwan Inst. Chem. Eng. 2018, 98, 163–180. [Google Scholar] [CrossRef]
- Papageorgiou, D.; Kinloch, I.A.; Young, R.J. Mechanical properties of graphene and graphene-based nanocomposites. Prog. Mater. Sci. 2017, 90, 75–127. [Google Scholar] [CrossRef]
- Singh, Z.S. Applications and toxicity of graphene family nanomaterials and their composites. Nanotechnol. Sci. Appl. 2016, 9, 15–28. [Google Scholar] [CrossRef]
- Wang, H.; Hu, Y.H. Effect of oxygen content on structures of graphite oxides. Ind. Eng. Chem. Res. 2011, 50, 6132–6137. [Google Scholar] [CrossRef]
- Karu, A.E.; Beer, M. Pyrolytic formation of highly crystalline graphite films. J. Appl. Phys. 1966, 37, 2179–2181. [Google Scholar] [CrossRef]
- Land, T.; Michely, T.; Behm, R.; Hemminger, J.; Comsa, G. STM investigation of single layer graphite structures produced on Pt(111) by hydrocarbon decomposition. Surf. Sci. 1992, 264, 261–270. [Google Scholar] [CrossRef]
- Somani, P.R.; Somani, S.P.; Umeno, M. Planer nano-graphenes from camphor by CVD. Chem. Phys. Lett. 2006, 430, 56–59. [Google Scholar] [CrossRef]
- Lim, J.Y.; Mubarak, N.; Abdullah, E.C.; Nizamuddin, S.; Khalid, M. Inamuddin Recent trends in the synthesis of graphene and graphene oxide based nanomaterials for removal of heavy metals—A review. J. Ind. Eng. Chem. 2018, 66, 29–44. [Google Scholar] [CrossRef]
- Nawz, T.; Safdar, A.; Hussain, M.; Lee, D.S.; Siyar, M. Graphene to advanced MoS2: A review of structure, synthesis, and optoelectronic device application. Crystals 2020, 10, 902. [Google Scholar] [CrossRef]
- Bae, S.; Kim, H.; Lee, Y.; Xu, X.; Park, J.-S.; Zheng, Y.; Balakrishnan, J.; Lei, T.; Kim, H.R.; Song, Y.I.; et al. Roll-to-roll production of 30-inch graphene films for transparent electrodes. Nat. Nanotechnol. 2010, 5, 574–578. [Google Scholar] [CrossRef]
- Saeed, M.; Alshammari, Y.; Majeed, S.A.; Al-Nasrallah, E. Chemical vapour deposition of graphene—Synthesis, characterisation, and applications: A review. Molecules 2020, 25, 3856. [Google Scholar] [CrossRef]
- Dimiev, A.; Kosynkin, D.V.; Alemany, L.B.; Chaguine, P.; Tour, J.M. Pristine graphite oxide. J. Am. Chem. Soc. 2012, 134, 2815–2822. [Google Scholar] [CrossRef]
- Brodie, B.C. On the atomic weight of graphite. Philos. Trans. 1859, 149, 249–259. [Google Scholar]
- Schafhaeutl, C. On the combination of carbon with silicon and iron, and other metals, forming the different species of cast iron, steel, and malleable iron. Philos. Mag. 1840, 16, 570–590. [Google Scholar]
- Staudenmaier, L. Verfahren zur darstellung der graphitsäure. Eur. J. Inorg. Chem. 1898, 31, 1481–1487. [Google Scholar] [CrossRef]
- Hummers, W.S.; Offeman, R.E. Preparation of graphitic oxide. J. Am. Chem. Soc. 1958, 80, 1339. [Google Scholar] [CrossRef]
- Charpy, G. Sur la formation de l’oxyde graphitique et la d’efnition du graphite. C. R. Hebd. Seances Acad. Sci. 1909, 148, 920–923. [Google Scholar]
- Scholz, W.; Boehm, H.P. Untersuchungen am graphitoxid. VI. Betrachtungen zur struktur des graphitoxids. J. Inorg. Gen. Chem. 1969, 369, 327–340. [Google Scholar] [CrossRef]
- Matuyama, E. Pyrolysis of graphitic acid. J. Phys. Chem. 1954, 58, 215–219. [Google Scholar] [CrossRef]
- Hiew, B.Y.Z.; Lee, L.Y.; Lee, X.J.; Gopakumar, S.T.; Gan, S.; Lim, S.S.; Pan, G.T.; Yang, T.C.K.; Chiu, W.S.; Khiew, P.S. Review on synthesis of 3D graphene-based configurations and theiradsorption performance for hazardous water pollutants. Process. Saf. Environ. Prot. 2018, 116, 262–286. [Google Scholar] [CrossRef]
- Marcano, D.C.; Kosynkin, D.V.; Berlin, J.M.; Sinitskii, A.; Sun, Z.; Slesarev, A.; Alemany, L.B.; Lu, W.; Tour, J.M. Improved synthesis of graphene oxide. ACS Nano 2010, 4, 4806–4814. [Google Scholar] [CrossRef]
- Dimiev, A.M.; Eigler, S. Graphene Oxide: Fundamentals and Applications, 1st ed.; John Wiley & Sons: Hoboken, NJ, USA, 2017; p. 454. [Google Scholar]
- Chen, J.; Yao, B.; Li, C.; Shi, G. An improved Hummers method for eco-friendly synthesis of graphene oxide. Carbon 2013, 64, 225–229. [Google Scholar] [CrossRef]
- Thiele, H. Graphit und graphitsäure. J. Inorg. Gen. Chem. 1930, 190, 145–160. [Google Scholar] [CrossRef]
- Lerf, A.; He, H.; Riedl, T.; Forster, M.; Klinowski, J. 13C and 1H MAS NMR studies of graphite oxide and its chemically modified derivatives. Solid State Ionics 1997, 101–103, 857–862. [Google Scholar] [CrossRef]
- Eigler, S.; Enzelberger-Heim, M.; Grimm, S.; Hofmann, P.; Kroener, W.; Geworski, A.; Dotzer, C.; Röckert, M.; Xiao, J.; Papp, C.; et al. Wet chemical synthesis of graphene. Adv. Mater. 2013, 25, 3583–3587. [Google Scholar] [CrossRef]
- Rouf, T.B.; Kokini, J.L. Biodegradable biopolymer–graphene nanocomposites. J. Mater. Sci. 2016, 51, 9915–9945. [Google Scholar] [CrossRef]
- Pei, S.; Cheng, H.M. The reduction of graphene oxide. Carbon 2012, 50, 3210–3228. [Google Scholar] [CrossRef]
- Zhao, B.; Liu, P.; Jiang, Y.; Pan, D.; Tao, H.; Song, J.; Fang, T.; Xu, W. Supercapacitor performances of thermally reduced graphene oxide. J. Power Sources 2012, 198, 423–427. [Google Scholar] [CrossRef]
- Chen, J.; Li, Y.; Huang, L.; Li, C.; Shi, G. High-yield preparation of graphene oxide from small graphite flakes via an improved Hummers method with a simple purification process. Carbon 2015, 81, 826–834. [Google Scholar] [CrossRef]
- Wang, Y.; Guo, L.; Qi, P.; Liu, X.; Wei, G. Synthesis of three-dimensional graphene-based hybrid materials for water purification: A review. Nanomaterials 2019, 9, 1123. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Chen, G.; Bao, J.; Xu, X. General preparation and shaping of multifunctional nanowire aerogels for pressure/gas/photo-sensing. Adv. Fiber Mater. 2021, in press. [Google Scholar] [CrossRef]
- Chang, D.; Liu, J.; Fang, B.; Xu, Z.; Li, Z.; Liu, Y.; Brassart, L.; Guo, F.; Gao, W.; Gao, C. Reversible fusion and fission of graphene oxide-based fibers. Science 2021, 372, 614–617. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Cheng, C.; Shen, K.; Zhang, T.; Wang, X.; Hsiao, B.S. Enhancing dehydration performance of isopropanol by introducing intermediate layer into sodium alginate nanofibrous composite pervaporation membrane. Adv. Fiber Mater. 2019, 1, 137–151. [Google Scholar] [CrossRef]
- Boehm, H.-P.; Clauss, A.; Hofmann, U. Graphite oxide and its membrane properties. J. Chim. Phys. 1961, 58, 141–147. [Google Scholar] [CrossRef]
- Nair, R.R.; Wu, H.A.; Jayaram, P.N.; Grigorieva, I.V.; Geim, A.K. Unimpeded permeation of water through helium-leak-tight graphene-based membranes. Science 2012, 335, 442–444. [Google Scholar] [CrossRef]
- Boehm, H.P.; Clauss, A.; Fischer, G.; Hofmann, U. Proceedings of the Fifth Conference on Carbon; Pergamon Press: Oxford, UK, 1962. [Google Scholar]
- Mohan, V.B.; Lau, K.-T.; Hui, D.; Bhattacharyya, D. Graphene-based materials and their composites: A review on production, applications and product limitations. Compos. Part B Eng. 2018, 142, 200–220. [Google Scholar] [CrossRef]
- Zhang, C.; Lv, W.; Xie, X.; Tang, D.; Liu, C.; Yang, Q.-H. Towards low temperature thermal exfoliation of graphite oxide for graphene production. Carbon 2013, 62, 11–24. [Google Scholar] [CrossRef]
- Gudarzi, M.M.; Moghadam, M.H.M.; Sharif, F. Spontaneous exfoliation of graphite oxide in polar aprotic solvents as the route to produce graphene oxide—organic solvents liquid crystals. Carbon 2013, 64, 403–415. [Google Scholar] [CrossRef]
- Botas, C.; Álvarez, P.; Blanco, C.; Santamaria, R.; Granda, M.; Gutiérrez, D.; Rodriguez-Reinoso, F.; Menéndez, R. Critical temperatures in the synthesis of graphene-like materials by thermal exfoliation–reduction of graphite oxide. Carbon 2012, 52, 476–485. [Google Scholar] [CrossRef]
- Kohlschütter, V.; Haenni, P. Zur kenntnis des graphitischen kohlenstoffs und der graphitsäure. J. Inorg. Gen. Chem. 1919, 105, 121–144. [Google Scholar] [CrossRef]
- Hofmann, U.; Frenzel, A. The reduction of graphite oxide with hydrogen sulphide. Kolloid-Zeitschrift 1934, 68, 149–151. [Google Scholar] [CrossRef]
- Vermisoglou, E.; Giannakopoulou, T.; Romanos, G.; Boukos, N.; Giannouri, M.; Lei, C.; Lekakou, C.; Trapalis, C. Non-activated high surface area expanded graphite oxide for supercapacitors. Appl. Surf. Sci. 2015, 358, 110–121. [Google Scholar] [CrossRef]
- Guan, F.; Han, Z.; Jin, M.; Wu, Z.; Chen, Y.; Chen, S.; Wang, H. Durable and flexible bio-assembled RGO-BC/BC bilayer electrodes for pressure sensing. Adv. Fiber Mater. 2021, 3, 128–137. [Google Scholar] [CrossRef]
- Zhang, Y.-L.; Guo, L.; Xia, H.; Chen, Q.-D.; Feng, J.; Sun, H.-B. Photoreduction of graphene oxides: Methods, properties, and applications. Adv. Opt. Mater. 2013, 2, 10–28. [Google Scholar] [CrossRef]
- Valipour, A.; Hamnabard, N.; Ahn, Y.-H. Performance evaluation of highly conductive graphene (RGOHI–AcOH) and graphene/metal nanoparticle composites (RGO/Ni) coated on carbon cloth for supercapacitor applications. RSC Adv. 2015, 5, 92970–92979. [Google Scholar] [CrossRef]
- Yang, H.; Cao, Y.; He, J.; Zhang, Y.; Jin, B.; Sun, J.-L.; Wang, Y.; Zhao, Z. Highly conductive free-standing reduced graphene oxide thin films for fast photoelectric devices. Carbon 2017, 115, 561–570. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, Y.; Lacey, S.D.; Xu, L.; Xie, H.; Li, T.; Danner, V.A.; Hu, L. Reduced graphene oxide film with record-high conductivity and mobility. Mater. Today 2017, 21, 186–192. [Google Scholar] [CrossRef]
- Yun, S.; Kang, S.-O.; Park, S.; Park, H.S. CO2-activated, hierarchical trimodal porous graphene frameworks for ultrahigh and ultrafast capacitive behavior. Nanoscale 2014, 6, 5296–5302. [Google Scholar] [CrossRef]
- Peng, W.; Li, H.; Liu, Y.; Song, S. A review on heavy metal ions adsorption from water by graphene oxide and its composites. J. Mol. Liq. 2017, 230, 496–504. [Google Scholar] [CrossRef]
- Harris, W.; White, N.G. X-ray diffraction techniques for soil mineral identification. In Methods of Soil Analysis Part 5—Mineralogical Methods, 5.5; Soil Science Society of America, Inc.: New York, NY, USA, 2008; pp. 81–115. [Google Scholar]
- Ioelovich, M. Crystallinity and hydrophility of chitin and chitosan. Res. Rev. J. Chem. 2014, 3, 7–14. [Google Scholar]
- Trikkaliotis, D.G.; Mitropoulos, A.C.; Kyzas, G.Z. Low-cost route for top-down synthesis of over- and low-oxidized graphene oxide. Colloids Surf. A Physicochem. Eng. Asp. 2020, 600, 124928. [Google Scholar] [CrossRef]
- Chen, H.; Du, W.; Liu, J.; Qu, L.; Li, C. Efficient room-temperature production of high-quality graphene by introducing removable oxygen functional groups to the precursor. Chem. Sci. 2018, 10, 1244–1253. [Google Scholar] [CrossRef] [PubMed]
- Johra, F.T.; Lee, J.-W.; Jung, W.-G. Facile and safe graphene preparation on solution based platform. J. Ind. Eng. Chem. 2014, 20, 2883–2887. [Google Scholar] [CrossRef]
- Gurzeda, B.; Buchwald, T.; Nocun, M.; Bakowicz, A.; Krawczyk, P. Graphene material preparation through thermal treatment of graphite oxide electrochemically synthesized in aqueous sulfuric acid. RSC Adv. 2017, 7, 19904–19911. [Google Scholar] [CrossRef]
- Kyzas, G.Z.; Bikiaris, D.N.; Deliyanni, E.A. Advanced low-swelling chitosan/graphite oxide-based biosorbents. Mater. Lett. 2014, 128, 46–49. [Google Scholar] [CrossRef]
- Ossonon, B.D.; Bélanger, D. Synthesis and characterization of sulfophenyl-functionalized reduced graphene oxide sheets. RSC Adv. 2017, 7, 27224–27234. [Google Scholar] [CrossRef]
- Dimiev, A.; Tour, J. Mechanism of graphene oxide formation. ACS Nano 2014, 8, 3060–3068. [Google Scholar] [CrossRef]
- Shen, B.; Lu, D.; Zhai, W.; Zheng, W. Synthesis of graphene by low-temperature exfoliation and reduction of graphite oxide under ambient atmosphere. J. Mater. Chem. C 2013, 1, 50–53. [Google Scholar] [CrossRef]
- Andersson, P.O.; Viberg, P.; Forsberg, P.; Nikolajeff, F.; Österlund, L.; Karlsson, M. Nanocrystalline diamond sensor targeted for selective CRP detection: An ATR-FTIR spectroscopy study. Anal. Bioanal. Chem. 2016, 408, 3675–3680. [Google Scholar] [CrossRef] [PubMed]
- Fale, P.L.V.; Chan, A.K.L. Preventing damage of germanium optical material in attenuated total reflection-Fourier transform infrared (ATR-FTIR) studies of living cells. Vib. Spectrosc. 2017, 91, 59–67. [Google Scholar] [CrossRef]
- Farinas, K.C.; Doh, L.; Venkatraman, S.; Potts, R.O. Characterization of solute diffusion in a polymer using ATR-FTIR spectroscopy and bulk transport techniques. Macromolecules 1994, 27, 5220–5222. [Google Scholar] [CrossRef]
- Ivanov, D.; Dubreuil, N.; Raussens, V.; Ruysschaert, J.M.; Goormaghtigh, E. Evaluation of the ordering of membranes in multilayer stacks built on an ATR-FTIR germanium crystal with atomic force microscopy: The case of the H+, K+-ATPase-containing gastric tubulovesicle membranes. Biophys. J. 2004, 87, 1307–1315. [Google Scholar] [CrossRef] [PubMed]
- Devouge, S.; Conti, J.; Goldsztein, A.; Gosselin, E.; Brans, A.; Voué, M.; De Coninck, J.; Homblé, F.; Goormaghtigh, E.; Marchand-Brynaert, J. Surface functionalization of germanium ATR devices for use in FTIR-biosensors. J. Colloid Interface Sci. 2009, 332, 408–415. [Google Scholar] [CrossRef]
- Guiliano, M.; Asia, L.; Onoratini, G.; Mille, G. Applications of diamond crystal ATR FTIR spectroscopy to the characterization of ambers. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2007, 67, 1407–1411. [Google Scholar] [CrossRef] [PubMed]
- Edelmann, A.; Diewok, J.; Baena, J.R.; Lendl, B. High-performance liquid chromatography with diamond ATR–FTIR detection for the determination of carbohydrates, alcohols and organic acids in red wine. Anal. Bioanal. Chem 2003, 376, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Sotomayor, F.J.; Cychosz, K.A.; Thommes, M. Characterization of micro/mesoporous materials by physisorption: Concepts and case studies. Acc. Mater. Surf. Res. 2018, 3, 34–50. [Google Scholar]
- Avila, A.; Bhushan, B. Electrical measurement techniques in atomic force microscopy. Crit. Rev. Solid State Mater. Sci. 2010, 35, 38–51. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Substrate | Condition | Size & Shape | Yield |
|---|---|---|---|---|
| Electron-beamirradiation | Amorphous carbon soot | - | Pristine dense-core spherical CNOs, ca. 6–50 nm | Low yield |
| Annealing | NDs | 800–2100 °C | Pristine dense-core spherical CNOs, 5–10 nm | High yield |
| Arc-discharge | Amorphous carbon soot Ni/Al catalyst | Arc discharge between 2 graphite electrodes, water 5000–6000 K under water | High-quality spherical hollowcore CNOs, 15–25 nm Pristine hollow-core spherical CNOs 5–50 nm | Largequantities Low yield |
| Chemical vapour deposition | CH4, N2 CH4 (60 mL min−1) N2 (540 mL min−1) | Ni/Al catalyst, Ni content >60 wt% | Pristine hollow-core spherical CNOs, 5–50 nm | Largequantities |
| Ion implantation | carbon into Cu, Ni substrate | 600–1000 °C | Pristine dense-core spherical CNOs, 15–200 nm | Low yield |
| Ag substrate | >600 °C | Pristine dense-core spherical CNOs, 5–100 nm | Low yield | |
| Pyrolysis | Phenol-formaldehyde | Ferric nitrate catalyst, 1000 °C | Pristine dense-core CNOs, ca. 40 nm | Largequantities |
| Propane | Propane/Oxygen flame (1.8 stoichiometric coefficient), Al | CNOs with many by-product, 10–25 nm | Low yield | |
| Plastic wastes (polyethylene, styrene, ethylene terephthalate) | - | CNOs with many by-product, 50–70 nm | Low yield |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trikkaliotis, D.G.; Christoforidis, A.K.; Mitropoulos, A.C.; Kyzas, G.Z. Graphene Oxide Synthesis, Properties and Characterization Techniques: A Comprehensive Review. ChemEngineering 2021, 5, 64. https://doi.org/10.3390/chemengineering5030064
Trikkaliotis DG, Christoforidis AK, Mitropoulos AC, Kyzas GZ. Graphene Oxide Synthesis, Properties and Characterization Techniques: A Comprehensive Review. ChemEngineering. 2021; 5(3):64. https://doi.org/10.3390/chemengineering5030064
Chicago/Turabian StyleTrikkaliotis, Dimitrios G., Achilleas K. Christoforidis, Athanasios C. Mitropoulos, and George Z. Kyzas. 2021. "Graphene Oxide Synthesis, Properties and Characterization Techniques: A Comprehensive Review" ChemEngineering 5, no. 3: 64. https://doi.org/10.3390/chemengineering5030064
APA StyleTrikkaliotis, D. G., Christoforidis, A. K., Mitropoulos, A. C., & Kyzas, G. Z. (2021). Graphene Oxide Synthesis, Properties and Characterization Techniques: A Comprehensive Review. ChemEngineering, 5(3), 64. https://doi.org/10.3390/chemengineering5030064