
Improved Catalytic Activity of the High-Temperature Water Gas Shift Reaction on Metal-Exsolved La0.9Ni0.05Fe0.95O3 by Controlling Reduction Time


Abstract
1. Introduction
2. Materials and Methods
2.1. Catalyst Preparation
2.2. Catalytic Activity Test
2.3. Catalyst Characterizations
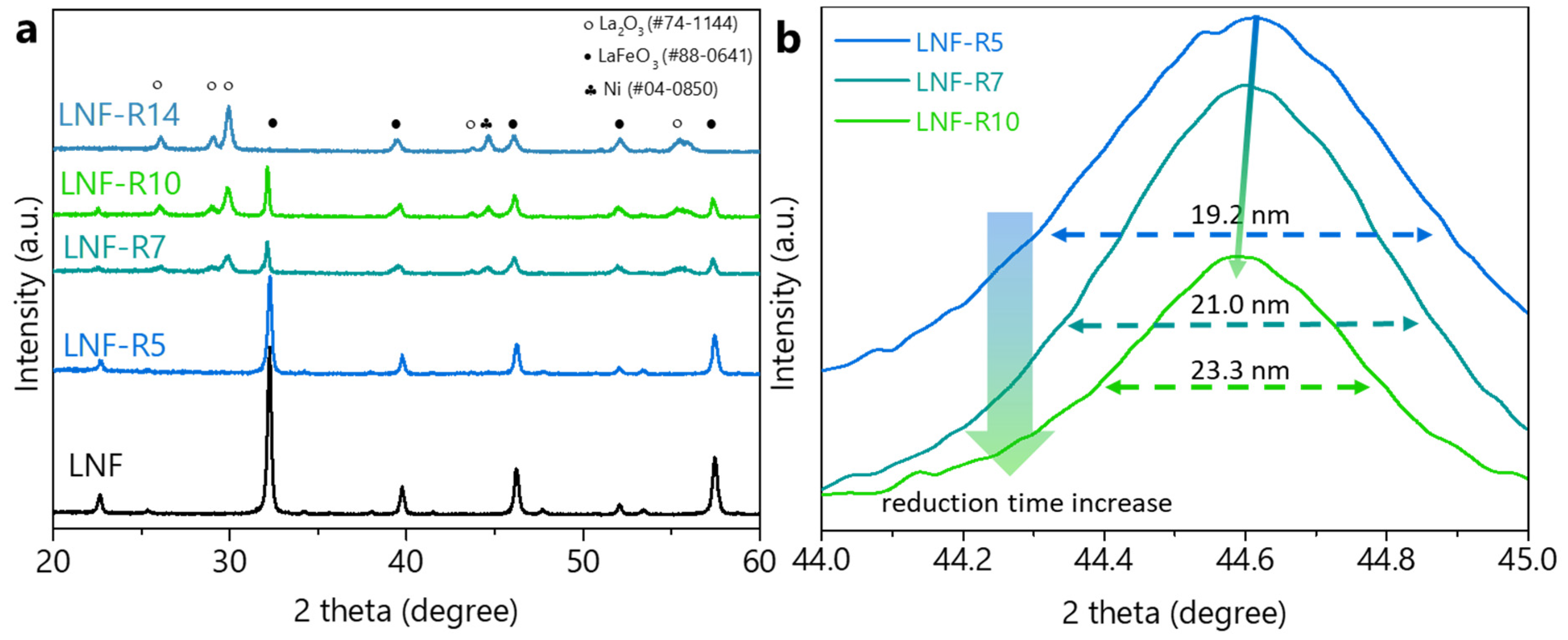
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chiriac, R.; Racovitza, A.; Podevin, P.; Descombes, G. On the possibility to reduce CO2 emissions of heat engines fuelled partially with hydrogen produced by waste heat recovery. Int. J. Hydrog. Energy 2015, 40, 15856–15863. [Google Scholar] [CrossRef]
- Ebrahimi, P.; Kumar, A.; Khraisheh, M. A review of recent advances in water-gas shift catalysis for hydrogen production. Emergent Mater. 2020, 3, 881–917. [Google Scholar] [CrossRef]
- Pal, D.B.; Chand, R.; Upadhyay, S.N.; Mishra, P.K. Performance of water gas shift reaction catalysts: A review. Renew. Sustain. Energy Rev. 2018, 93, 549–565. [Google Scholar] [CrossRef]
- Han, S. Biohydrogen production by anaerobic fermentation of food waste. Int. J. Hydrog. Energy 2004, 29, 569–577. [Google Scholar] [CrossRef]
- Lay, J.-J.; Lee, Y.-J.; Noike, T. Feasibility of biological hydrogen production from organic fraction of municipal solid waste. Water Res. 1999, 33, 2579–2586. [Google Scholar] [CrossRef]
- Mizuno, O.; Dinsdale, R.; Hawkes, F.R.; Hawkes, D.L.; Noike, T. Enhancement of hydrogen production from glucose by nitrogen gas sparging. Bioresour. Technol. 2000, 73, 59–65. [Google Scholar] [CrossRef]
- Chen, L.; Qi, Z.; Zhang, S.; Su, J.; Somorjai, G.A. Catalytic Hydrogen Production from Methane: A Review on Recent Progress and Prospect. Catalysts 2020, 10, 858. [Google Scholar] [CrossRef]
- Hairuddin, A.A.; Yusaf, T.; Wandel, A.P. A review of hydrogen and natural gas addition in diesel HCCI engines. Renew. Sustain. Energy Rev. 2014, 32, 739–761. [Google Scholar] [CrossRef]
- Maneerung, T.; Hidajat, K.; Kawi, S. K-doped LaNiO3 perovskite for high-temperature water-gas shift of reformate gas: Role of potassium on suppressing methanation. Int. J. Hydrog. Energy 2017, 42, 9840–9857. [Google Scholar] [CrossRef]
- Jha, A.; Jeong, D.-W.; Lee, Y.-L.; Jang, W.-J.; Shim, J.-O.; Jeon, K.-W.; Rode, C.V.; Roh, H.-S. Chromium free high temperature water–gas shift catalyst for the production of hydrogen from waste derived synthesis gas. Appl. Catal. A Gen. 2016, 522, 21–31. [Google Scholar] [CrossRef]
- Meshkani, F.; Rezaei, M. Hydrogen production by high temperature water gas shift reaction over highly active and stable chromium free Fe-Al-Ni catalysts. Int. J. Hydrog. Energy 2015, 40, 10867–10875. [Google Scholar] [CrossRef]
- Ramírez Reina, T.; González Castaño, M.; Palma, S.; Ivanova, S.; Odriozola, J.A. Heterogeneous Gold Catalysts and Catalysis; The Royal Society of Chemistry: London, UK, 2014; pp. 111–139. [Google Scholar]
- Ashok, J.; Wai, M.H.; Kawi, S. Nickel-based Catalysts for High-temperature Water Gas Shift Reaction-Methane Suppression. ChemCatChem 2018, 10, 3927–3942. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, J.K.; Liu, J.; Curcio, A.; Jang, J.-S.; Kim, I.-D.; Ciucci, F.; Jung, W. Nanoparticle Ex-solution for Supported Catalysts: Materials Design, Mechanism and Future Perspectives. ACS Nano 2021, 15, 81–110. [Google Scholar] [CrossRef] [PubMed]
- Neagu, D.; Oh, T.-S.; Miller, D.N.; Ménard, H.; Bukhari, S.M.; Gamble, S.R.; Gorte, R.J.; Vohs, J.M.; Irvine, J.T.S. Nano-socketed nickel particles with enhanced coking resistance grown in situ by redox exsolution. Nat. Commun. 2015, 6, 8120. [Google Scholar] [CrossRef] [PubMed]
- Yentekakis, I.V.; Goula, G.; Hatzisymeon, M.; Betsi-Argyropoulou, I.; Botzolaki, G.; Kousi, K.; Kondarides, D.I.; Taylor, M.J.; Parlett, C.M.A.; Osatiashtiani, A.; et al. Effect of support oxygen storage capacity on the catalytic performance of Rh nanoparticles for CO2 reforming of methane. Appl. Catal. B Environ. 2019, 243, 490–501. [Google Scholar] [CrossRef]
- Papargyriou, D.; Miller, D.N.; Sirr Irvine, J.T. Exsolution of Fe-Ni alloy nanoparticles from (La,Sr)(Cr,Fe,Ni)O3 perovskites as potential oxygen transport membrane catalysts for methane reforming. J. Mater. Chem. A 2019, 7, 15812–15822. [Google Scholar] [CrossRef]
- Jang, W.-J.; Shim, J.-O.; Jeon, K.-W.; Na, H.-S.; Kim, H.-M.; Lee, Y.-L.; Roh, H.-S.; Jeong, D.-W. Design and scale-up of a Cr-free Fe-Al-Cu catalyst for hydrogen production from waste-derived synthesis gas. Appl. Catal. B Environ. 2019, 249, 72–81. [Google Scholar] [CrossRef]
- Nishihata, Y.; Mizuki, J.; Akao, T.; Tanaka, H.; Uenishi, M.; Kimura, M.; Okamoto, T.; Hamada, N. Self-regeneration of a Pd-perovskite catalyst for automotive emissions control. Nature 2002, 418, 164–167. [Google Scholar] [CrossRef] [PubMed]
- Neagu, D.; Tsekouras, G.; Miller, D.N.; Ménard, H.; Irvine, J.T.S. In situ growth of nanoparticles through control of non-stoichiometry. Nat. Chem. 2013, 5, 916–923. [Google Scholar] [CrossRef]
- Sun, Y.-F.; Li, J.-H.; Cui, L.; Hua, B.; Cui, S.-H.; Li, J.; Luo, J.-L. A-site-deficiency facilitated in situ growth of bimetallic Ni–Fe nano-alloys: A novel coking-tolerant fuel cell anode catalyst. Nanoscale 2015, 7, 11173–11181. [Google Scholar] [CrossRef]
- Kwon, O.; Sengodan, S.; Kim, K.; Kim, G.; Jeong, H.Y.; Shin, J.; Ju, Y.W.; Han, J.W.; Kim, G. Exsolution trends and co-segregation aspects of self-grown catalyst nanoparticles in perovskites. Nat. Commun. 2018, 8, 15967. [Google Scholar] [CrossRef]
- Zhang, J.; Gao, M.-R.; Luo, J.-L. In Situ Exsolved Metal Nanoparticles: A Smart Approach for Optimization of Catalysts. Chem. Mater. 2020, 32, 5424–5441. [Google Scholar] [CrossRef]
- Gao, Y.; Chen, D.; Saccoccio, M.; Lu, Z.; Ciucci, F. From material design to mechanism study: Nanoscale Ni exsolution on a highly active A-site deficient anode material for solid oxide fuel cells. Nano Energy 2016, 27, 499–508. [Google Scholar] [CrossRef]
- Tang, C.; Kousi, K.; Neagu, D.; Portolés, J.; Papaioannou, E.I.; Metcalfe, I.S. Towards efficient use of noble metals via exsolution exemplified for CO oxidation. Nanoscale 2019, 11, 16935–16944. [Google Scholar] [CrossRef] [PubMed]
- Opitz, A.K.; Nenning, A.; Rameshan, C.; Kubicek, M.; Götsch, T.; Blume, R.; Hävecker, M.; Knop-Gericke, A.; Rupprechter, G.; Klötzer, B.; et al. Surface Chemistry of Perovskite-Type Electrodes during High Temperature CO2 Electrolysis Investigated by Operando Photoelectron Spectroscopy. ACS Appl. Mater. Interfaces 2017, 9, 35847–35860. [Google Scholar] [CrossRef] [PubMed]
- Celorrio, V.; Dann, E.; Calvillo, L.; Morgan, D.J.; Hall, S.R.; Fermin, D.J. Oxygen Reduction at Carbon-Supported Lanthanides: The Role of the B-Site. ChemElectroChem 2016, 3, 283–291. [Google Scholar] [CrossRef]
- Yang, J.; Hu, S.; Fang, Y.; Hoang, S.; Li, L.; Yang, W.; Liang, Z.; Wu, J.; Hu, J.; Xiao, W.; et al. Oxygen Vacancy Promoted O2 Activation over Perovskite Oxide for Low-Temperature CO Oxidation. ACS Catal. 2019, 9, 9751–9763. [Google Scholar] [CrossRef]
- Moulder, J.F.; Stickle, W.F.; Sobol, P.E.; Bomben, K.D. Handbook of X-ray Photoelectron Spectroscopy; Physical Electronics, Inc.: Eden Praire, MN, USA, 1995. [Google Scholar]
- Ha, M.N.; Lu, G.; Liu, Z.; Wang, L.; Zhao, Z. 3DOM-LaSrCoFeO6–δ as a highly active catalyst for the thermal and photothermal reduction of CO2 with H2O to CH4. J. Mater. Chem. A 2016, 4, 13155–13165. [Google Scholar] [CrossRef]
- Deka, D.J.; Kim, J.; Gunduz, S.; Aouine, M.; Millet, J.-M.M.; Co, A.C.; Ozkan, U.S. Investigation of Hetero-phases Grown via In-situ Exsolution on a Ni-doped (La,Sr)FeO3 Cathode and the Resultant Activity Enhancement in CO2 Reduction. Appl. Catal. B Environ. 2021, 285, 119917. [Google Scholar] [CrossRef]
- Steiger, P.; Delmelle, R.; Foppiano, D.; Holzer, L.; Heel, A.; Nachtegaal, M.; Kröcher, O.; Ferri, D. Structural Reversibility and Nickel Particle stability in Lanthanum Iron Nickel Perovskite-Type Catalysts. ChemSusChem 2017, 10, 2505–2517. [Google Scholar] [CrossRef]
- Nakamura, T.; Petzow, G.; Gauckler, L.J. Stability of the perovskite phase LaBO3 (B = V, Cr, Mn, Fe, Co, Ni) in reducing atmosphere. Mater. Res. Bull. 1979, 14, 649–659. [Google Scholar] [CrossRef]
- Crespin, M.; Levitz, P.; Gatineau, L. Reduced forms of LaNiO3 perovskite. Part 1—Evidence for new phases: La2Ni2O5 and LaNiO2. J. Chem. Soc. Faraday Trans. 2 1983, 79, 1181–1194. [Google Scholar] [CrossRef]
- Koponen, M.J.; Suvanto, M.; Kallinen, K.; Kinnunen, T.-J.J.; Härkönen, M.; Pakkanen, T.A. Structural transformations in cubic structure of Mn/Co perovskites in reducing and oxidizing atmospheres. Solid State Sci. 2006, 8, 450–456. [Google Scholar] [CrossRef]
- Zheng, Y.; Wang, W.; Jiang, D.; Zhang, L.; Li, X.; Wang, Z. Ultrathin mesoporous Co3O4 nanosheets with excellent photo-/thermo-catalytic activity. J. Mater. Chem. A 2016, 4, 105–112. [Google Scholar] [CrossRef]
- Wu, S.-L.; Kuo, J.-H.; Wey, M.-Y. Design of catalysts comprising a nickel core and ceria shell for hydrogen production from plastic waste gasification: An integrated test for anti-coking and catalytic performance. Catal. Sci. Technol. 2020, 10, 3975–3984. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, Y.; Li, Q.; Wang, Z.; Zhang, Z. Identification of active oxygen species for soot combustion on LaMnO3 perovskite. Catal. Sci. Technol. 2012, 2, 1822–1824. [Google Scholar] [CrossRef]
- Iwamoto, M.; Yoda, Y.; Yamazoe, N.; Seiyama, T. Study of metal oxide catalysts by temperature programmed desorption. 4. Oxygen adsorption on various metal oxides. J. Phys. Chem. 1978, 82, 2564–2570. [Google Scholar] [CrossRef]
- Papaioannou, E.I.; Neagu, D.; Ramli, W.K.W.; Irvine, J.T.S.; Metcalfe, I.S. Sulfur-Tolerant, Exsolved Fe–Ni Alloy Nanoparticles for CO Oxidation. Top. Catal. 2018, 62, 1149–1156. [Google Scholar] [CrossRef]
- Huang, R.; Kim, K.; Kim, H.J.; Jang, M.G.; Han, J.W. Size-Controlled Pd Nanoparticles Loaded on Co3O4 Nanoparticles by Calcination for Enhanced CO Oxidation. ACS Appl. Nano Mater. 2019, 3, 486–495. [Google Scholar] [CrossRef]
- Tian, Z.; Wang, C.; Yue, J.; Zhang, X.; Ma, L. Effect of a potassium promoter on the Fischer-Tropsch synthesis of light olefins over iron carbide catalysts encapsulated in graphene-like carbon. Catal. Sci. Technol. 2019, 9, 2728–2741. [Google Scholar] [CrossRef]
- Pendyala, V.R.R.; Graham, U.M.; Jacobs, G.; Hamdeh, H.H.; Davis, B.H. Fischer-Tropsch Synthesis: Deactivation as a Function of Potassium Promoter Loading for Precipitated Iron Catalyst. Catal. Lett. 2014, 144, 1704–1716. [Google Scholar] [CrossRef]
- Pandey, A.; Jain, G.; Vyas, D.; Irusta, S.; Sharma, S. Nonreducible, Basic La2O3 to Reducible, Acidic La2–xSbxO3 with Significant Oxygen Storage Capacity, Lower Band Gap, and Effect on the Catalytic Activity. J. Phys. Chem. C 2017, 121, 481–489. [Google Scholar] [CrossRef]
- Thalinger, R.; Opitz, A.K.; Kogler, S.; Heggen, M.; Stroppa, D.; Schmidmair, D.; Tappert, R.; Fleig, J.; Klötzer, B.; Penner, S. Water-Gas Shift and Methane Reactivity on Reducible Perovskite-Type Oxides. J. Phys. Chem. C 2015, 119, 11739–11753. [Google Scholar] [CrossRef]
- Sehested, J.; Dahl, S.; Jacobsen, J.; Rostrup-Nielsen, J.R. Methanation of CO over Nickel: Mechanism and Kinetics at High H2/CO Ratios. J. Phys. Chem. B 2005, 109, 2432–2438. [Google Scholar] [CrossRef]
- Senanayake, S.D.; Evans, J.; Agnoli, S.; Barrio, L.; Chen, T.-L.; Hrbek, J.; Rodriguez, J.A. Water-Gas Shift and CO Methanation Reactions over Ni–CeO2(111) Catalysts. Top. Catal. 2011, 54, 34–41. [Google Scholar] [CrossRef]
- Roiaz, M.; Monachino, E.; Dri, C.; Greiner, M.; Knop-Gericke, A.; Schlögl, R.; Comelli, G.; Vesselli, E. Reverse Water-Gas Shift or Sabatier Methanation on Ni(110)? Stable Surface Species at Near-Ambient Pressure. J. Am. Chem. Soc. 2016, 138, 4146–4154. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reduction Time | Surface Area | NP Size 1 | NP Size 2 | NP Population 3 |
|---|---|---|---|---|
| (h) | (10–1 cm2/g) | (nm) | (nm) | (μm–2) |
| 5 | 3.3 | 19.7 | 19.2 | 32.6 |
| 7 | 4.9 | 20.5 | 21.0 | 44.3 |
| 10 | 8.3 | 23.6 | 23.3 | 51.6 |
| Sample | Ni0/(Ni0 + Ni2+) | T50 1 | Xco Decreased 2 |
|---|---|---|---|
| (%) | (°C) | (%) | |
| LNF–R5 | 36.2 | 445 | 23.0 |
| LNF–R7 | 38.2 | 430 | 26.3 |
| LNF–R10 | 43.5 | 403 | 24.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, R.; Han, J.W. Improved Catalytic Activity of the High-Temperature Water Gas Shift Reaction on Metal-Exsolved La0.9Ni0.05Fe0.95O3 by Controlling Reduction Time. ChemEngineering 2021, 5, 28. https://doi.org/10.3390/chemengineering5020028
Huang R, Han JW. Improved Catalytic Activity of the High-Temperature Water Gas Shift Reaction on Metal-Exsolved La0.9Ni0.05Fe0.95O3 by Controlling Reduction Time. ChemEngineering. 2021; 5(2):28. https://doi.org/10.3390/chemengineering5020028
Chicago/Turabian StyleHuang, Rui, and Jeong Woo Han. 2021. "Improved Catalytic Activity of the High-Temperature Water Gas Shift Reaction on Metal-Exsolved La0.9Ni0.05Fe0.95O3 by Controlling Reduction Time" ChemEngineering 5, no. 2: 28. https://doi.org/10.3390/chemengineering5020028
APA StyleHuang, R., & Han, J. W. (2021). Improved Catalytic Activity of the High-Temperature Water Gas Shift Reaction on Metal-Exsolved La0.9Ni0.05Fe0.95O3 by Controlling Reduction Time. ChemEngineering, 5(2), 28. https://doi.org/10.3390/chemengineering5020028