Solid Polymer Electrolytes Derived from Crosslinked Polystyrene Nanoparticles Covalently Functionalized with a Low Lattice Energy Lithium Salt Moiety

 ,
, 

Abstract
1. Introduction
2. Experimental
2.1. Materials and Characterization
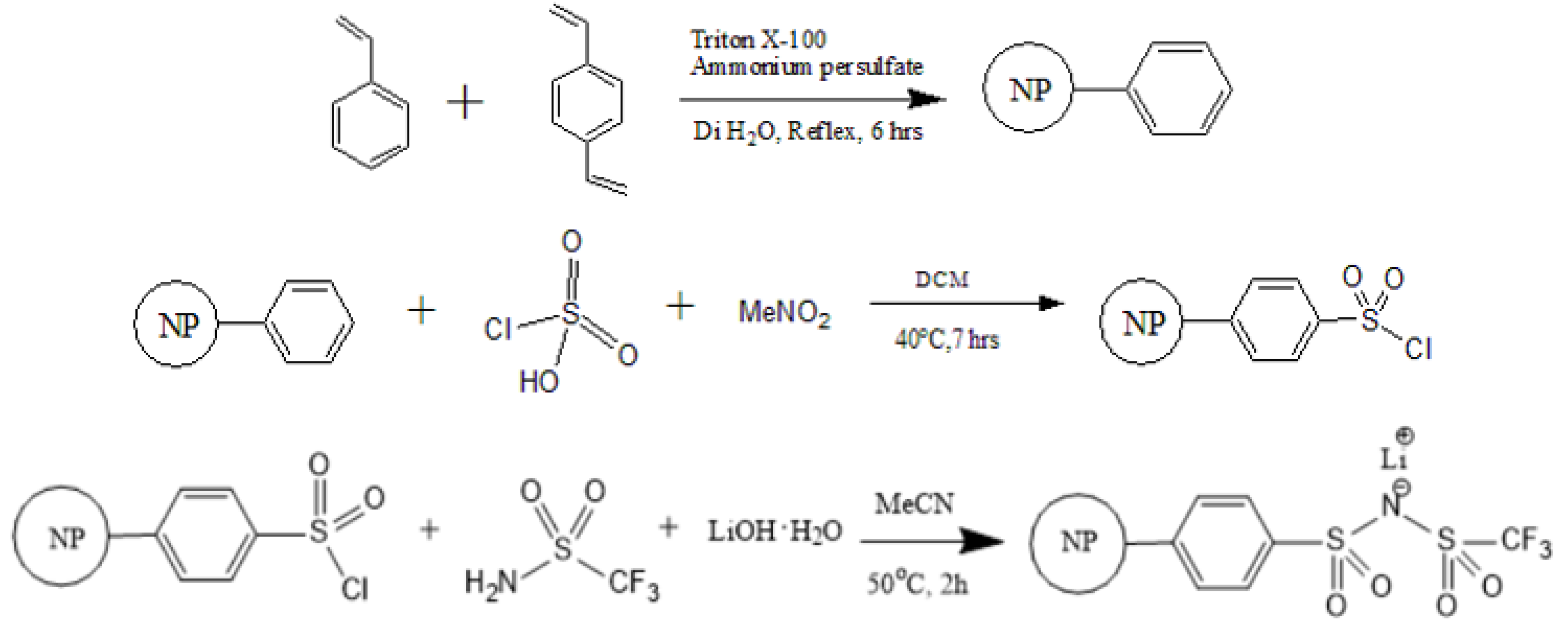
2.2. Synthesis of PS–SO2Cl
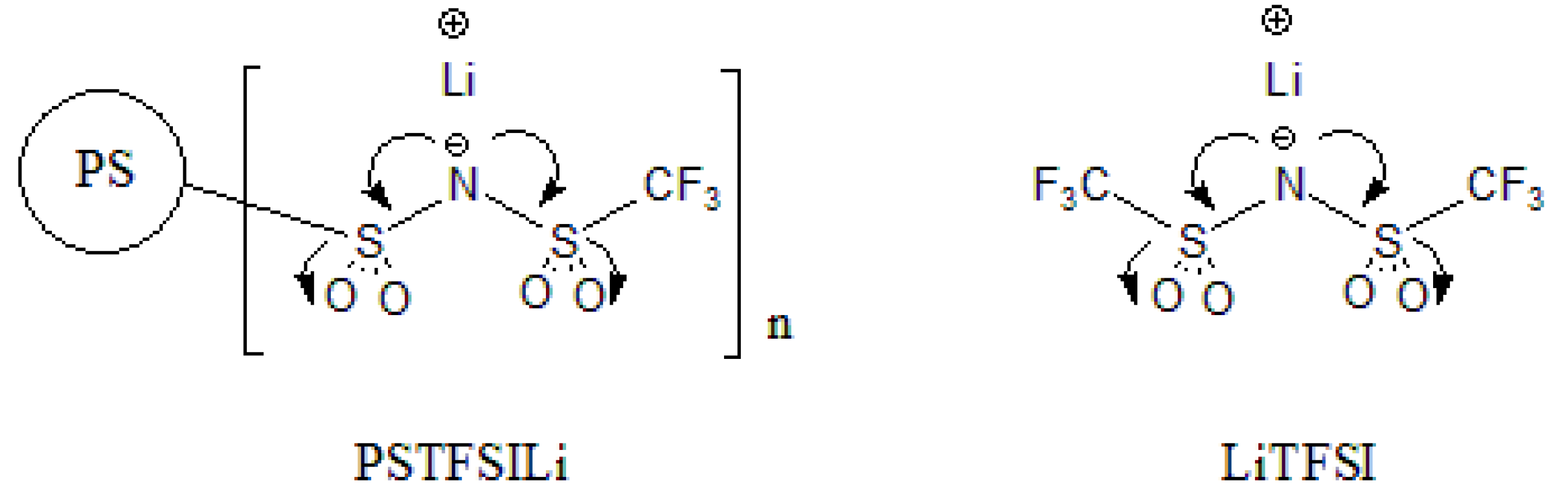
2.3. Synthesis of PSTFSILi
2.4. Synthesis of PSPhSILi
2.5. Synthesis of PSDTTOLi
2.6. Thin Film Processing and Cell Fabrication
3. Results and Discussion
3.1. Synthesis of PS-Bound Lithium Salts
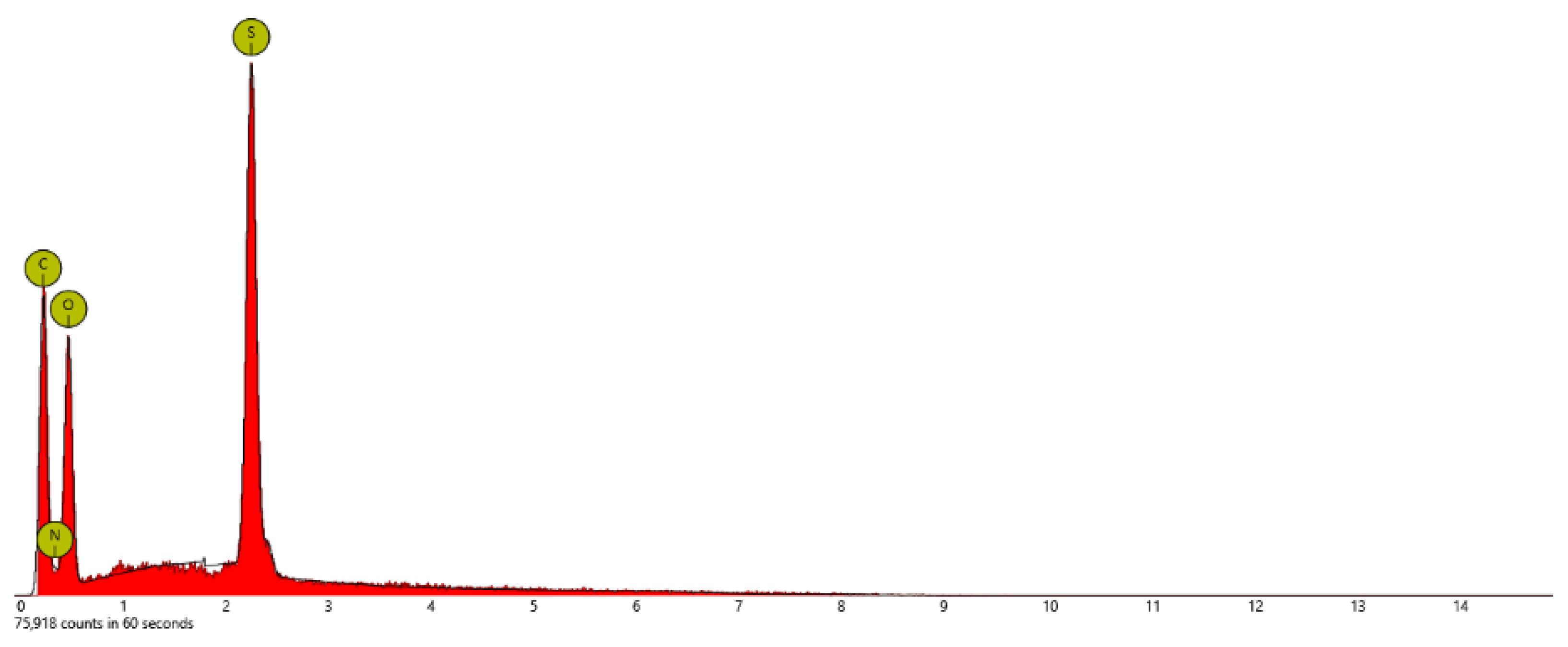
3.2. Structural Characterization
3.3. SPE Formulations
3.4. Thermogravimetric Analysis (TGA)
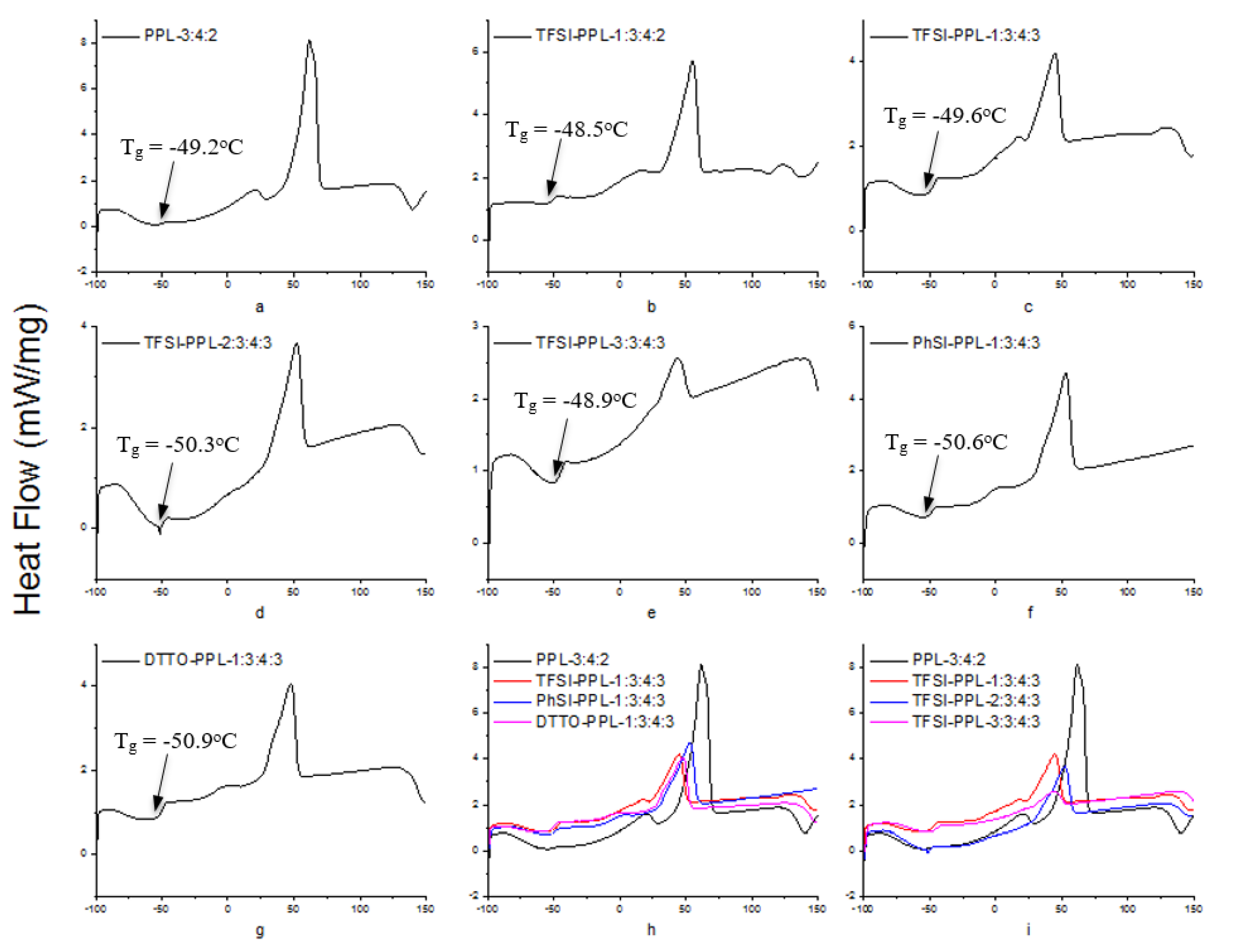
3.5. Differential Scanning Calorimetry
3.6. Ionic Conductivity
3.7. Cyclic Voltammetry
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Polu, A.R.; Rhee, H.-W. Nanocomposite solid polymer electrolytes based on poly(ethylene oxide)/POSS-PEG (n = 13.3) hybrid nanoparticles for lithium ion batteries. J. Ind. Eng. Chem. 2015, 31, 323–329. [Google Scholar] [CrossRef]
- Wang, Z.; Fu, Y.; Zhang, Z.; Yuan, S.; Amine, K.; Battaglia, V.; Liu, G. Application of Stabilized Lithium Metal Powder (SLMP®) in graphite anode—A high efficient prelithiation method for lithium-ion batteries. J. Power Sources 2014, 260, 57–61. [Google Scholar] [CrossRef]
- Zhou, J. Ionic conductivity of composite electrolytes based on oligo(ethylene oxide) and fumed oxides. Solid State Ion. 2004, 166, 275–293. [Google Scholar] [CrossRef]
- Cheng, Q.; Cui, Z.; Li, J.; Qin, S.; Yan, F.; Li, J. Preparation and performance of polymer electrolyte based on poly(vinylidene fluoride)/polysulfone blend membrane via thermally induced phase separation process for lithium ion battery. J. Power Sources 2014, 266, 401–413. [Google Scholar] [CrossRef]
- Wright, P.V. Electrical conductivity in ionic complexes of poly(ethylene oxide). Br. Polym. J. 1975, 7, 319–327. [Google Scholar] [CrossRef]
- Fenton, D.E.; Parker, J.M.; Wright, P.V. Complexes of alkali metal ions with poly(ethylene oxide). Polymer 1973, 14, 589. [Google Scholar] [CrossRef]
- Wang, X.; Gong, C.; He, D.; Xue, Z.; Chen, C.; Liao, Y.; Xie, X. Gelled microporous polymer electrolyte with low liquid leakage for lithium-ion batteries. J. Membr. Sci. 2014, 454, 298–304. [Google Scholar] [CrossRef]
- Marzantowicz, M.; Dygas, J.R.; Krok, F.; Łasińska, A.; Florjańczyk, Z.; Zygadło-Monikowska, E.; Affek, A. Crystallization and melting of PEO:LiTFSI polymer electrolytes investigated simultaneously by impedance spectroscopy and polarizing microscopy. Electrochim. Acta 2005, 50, 3969–3977. [Google Scholar] [CrossRef]
- Marzantowicz, M.; Dygas, J.R.; Krok, F.; Tomaszewska, A.; Florjańczyk, Z.; Zygadło-Monikowska, E.; Lapienis, G. Star-branched poly(ethylene oxide) LiN(CF3SO2)2: A promising polymer electrolyte. J. Power Sources 2009, 194, 51–57. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, C.; Zheng, L.; Xu, F.; Feng, W.; Li, H.; Huang, X.; Armand, M.; Nie, J.; Zhou, Z. Lithium bis(fluorosulfonyl)imide/poly(ethylene oxide) polymer electrolyte. Electrochim. Acta 2014, 133, 529–538. [Google Scholar] [CrossRef]
- Aricò, A.S.; Bruce, P.; Scrosati, B.; Tarascon, J.-M.; van Schalkwijk, W. Nanostructured materials for advanced energy conversion and storage devices. Nat. Mater. 2005, 4, 366–377. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Fan, H.; Fan, L.-Z.; Shi, Q. Preparation and performance of a non-ionic plastic crystal electrolyte with the addition of polymer for lithium ion batteries. Electrochim. Acta 2013, 114, 720–725. [Google Scholar] [CrossRef]
- Tillmann, S.D.; Isken, P.; Lex-Balducci, A. Gel polymer electrolyte for lithium-ion batteries comprising cyclic carbonate moieties. J. Power Sources 2014, 271, 239–244. [Google Scholar] [CrossRef]
- Xing, Y.; Wu, Y.; Wang, H.; Yang, G.; Li, W.; Xu, L.; Jiang, X. Preparation of hybrid polymer based on polyurethane lithium salt and polyvinylidene fluoride as electrolyte for lithium-ion batteries. Electrochim. Acta 2014, 136, 513–520. [Google Scholar] [CrossRef]
- MacFarlane, D.R.; Tachikawa, N.; Forsyth, M.; Pringle, J.M.; Howlett, P.C.; Elliott, G.D.; Davis, J.H.; Watanabe, M.; Simon, P.; Angell, C.A. Energy applications of ionic liquids. Energy Environ. Sci. 2014, 7, 232–250. [Google Scholar] [CrossRef]
- Lu, Y.; Moganty, S.S.; Schaefer, J.L.; Archer, L.A. Ionic liquid-nanoparticle hybrid electrolytes. J. Mater. Chem. 2012, 22, 4066. [Google Scholar] [CrossRef]
- Caimi, S.; Klaue, A.; Wu, H.; Morbidelli, M. Effect of SiO2 Nanoparticles on the Performance of PVdF-HFP/Ionic Liquid Separator for Lithium-Ion Batteries. Nanomaterials 2018, 8, 926. [Google Scholar] [CrossRef]
- Wang, W.; Alexandridis, P. Composite polymer electrolytes: Nanoparticles affect structure and properties. Polymers (Basel) 2016, 8, 387. [Google Scholar] [CrossRef]
- Liu, W.; Liu, N.; Sun, J.; Hsu, P.-C.; Li, Y.; Lee, H.-W.; Cui, Y. Ionic conductivity enhancement of polymer electrolytes with ceramic nanowire fillers. Nano Lett. 2015, 15, 2740–2745. [Google Scholar] [CrossRef]
- Mandal, B.K.; Sooksimuang, T.; Griffin, B.; Padhi, A.; Filler, R. New lithium salts for rechargeable battery electrolytes. Solid State Ion. 2004, 175, 267–272. [Google Scholar] [CrossRef]
- Kim, M.S.; Kim, S.K.; Lee, J.Y.; Cho, S.H.; Lee, K.-H.; Kim, J.; Lee, S.-S. Synthesis of polystyrene nanoparticles with monodisperse size distribution and positive surface charge using metal stearates. Macromol. Res. 2008, 16, 178–181. [Google Scholar] [CrossRef]
- Brijmohan, S.B.; Swier, S.; Weiss, R.A.; Shaw, M.T. Synthesis and characterization of cross-linked sulfonated polystyrene nanoparticles. Ind. Eng. Chem. Res. 2005, 44, 8039–8045. [Google Scholar] [CrossRef]
- Feng, S.; Shi, D.; Liu, F.; Zheng, L.; Nie, J.; Feng, W.; Huang, X.; Armand, M.; Zhou, Z. Single lithium-ion conducting polymer electrolytes based on poly[(4-styrenesulfonyl)(trifluoromethanesulfonyl)imide] anions. Electrochim. Acta 2013, 93, 254–263. [Google Scholar] [CrossRef]
- Ulaganathan, M.; Nithya, R.; Rajendr, S. Surface analysis studies on polymer electrolyte membranes using scanning electron microscope and atomic force microscope. In Scanning Electron Microscopy; Kazmiruk, V., Ed.; InTech: London, UK, 2012; ISBN 978-953-51-0092-8. [Google Scholar]
- Paillard, E.; Iojoiu, C.; Alloin, F.; Guindet, J.; Sanchez, J.-Y. Poly(oxyethylene) electrolytes based on lithium pentafluorobenzene sulfonate. Electrochim. Acta 2007, 52, 3758–3765. [Google Scholar] [CrossRef]
- Joge, P.; Kanchan, D.K.; Sharma, P.; Jayswal, M.; Avasthi, D.K. Effect of swift heavy O7+ ion radiations on conductivity of lithium based polymer blend electrolyte. Radiat. Phys. Chem. 2014, 100, 74–79. [Google Scholar] [CrossRef]
- Costa, C.M.; Gomez Ribelles, J.L.; Lanceros-Méndez, S.; Appetecchi, G.B.; Scrosati, B. Poly(vinylidene fluoride)-based, co-polymer separator electrolyte membranes for lithium-ion battery systems. J. Power Sources 2014, 245, 779–786. [Google Scholar] [CrossRef]
- Chen, R.; Zhu, L.; Wu, F.; Li, L.; Zhang, R.; Chen, S. Investigation of a novel ternary electrolyte based on dimethyl sulfite and lithium difluoromono(oxalato)borate for lithium ion batteries. J. Power Sources 2014, 245, 730–738. [Google Scholar] [CrossRef]
- Sun, X.-G.; Reeder, C.L.; Kerr, J.B. Synthesis and characterization of network type single ion conductors. Macromolecules 2004, 37, 2219–2227. [Google Scholar] [CrossRef]
- Xiao, S.Y.; Yang, Y.Q.; Li, M.X.; Wang, F.X.; Chang, Z.; Wu, Y.P.; Liu, X. A composite membrane based on a biocompatible cellulose as a host of gel polymer electrolyte for lithium ion batteries. J. Power Sources 2014, 270, 53–58. [Google Scholar] [CrossRef]
- Yoshizawa, M.; Ohno, H. Synthesis of molten salt-type polymer brush and effect of brush structure on the ionic conductivity. Electrochim. Acta 2001, 46, 1723–1728. [Google Scholar] [CrossRef]
- Bouchet, R.; Maria, S.; Meziane, R.; Aboulaich, A.; Lienafa, L.; Bonnet, J.-P.; Phan, T.N.T.; Bertin, D.; Gigmes, D.; Devaux, D.; et al. Single-ion BAB triblock copolymers as highly efficient electrolytes for lithium-metal batteries. Nat. Mater. 2013, 12, 452–457. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Casselman, M.D.; Li, Y.; Wei, A.; Abraham, D.P. Perfluoroalkyl-substituted ethylene carbonates: Novel electrolyte additives for high-voltage lithium-ion batteries. J. Power Sources 2014, 246, 184–191. [Google Scholar] [CrossRef]
- Chakrabarti, A.; Filler, R.; Mandal, B.K. Synthesis and properties of a new class of fluorine-containing dilithium salts for lithium-ion batteries. Solid State Ion. 2010, 180, 1640–1645. [Google Scholar] [CrossRef]
- Olgun, U.; Gülfen, M. Synthesis of fluorescence poly(phenylenethiazolo[5,4-d]thiazole) copolymer dye: Spectroscopy, cyclic voltammetry and thermal analysis. Dye. Pigment. 2014, 102, 189–195. [Google Scholar] [CrossRef]
- Sharma, P.K.; Gupta, G.; Singh, V.V.; Tripathi, B.K.; Pandey, P.; Boopathi, M.; Singh, B.; Vijayaraghavan, R. Synthesis and characterization of polypyrrole by cyclic voltammetry at different scan rate and its use in electrochemical reduction of the simulant of nerve agents. Synth. Met. 2010, 160, 2631–2637. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | At. % C | At % N | At. % O | At. % S | At. % F |
|---|---|---|---|---|---|
| PSTFSILi | 48.9 | 13.5 | 26.4 | 3.1 | 8.2 |
| PSPhSILi | 58.4 | 5.7 | 26.2 | 9.7 | - |
| PSDTTOLi | 40.4 | - | 26.9 | 26.9 | - |
| Components Weight [mg] | Abbreviation of the Formulation | |||
|---|---|---|---|---|
| PSTFSILi | PEO | PEGDM | LiTFSI | |
| 0 | 75 | 100 | 50 | PPL = 3:4:2 |
| 25 | 75 | 100 | 25 | TFSI–PPL = 1:3:4:1 |
| 25 | 75 | 100 | 50 | TFSI–PPL = 1:3:4:2 |
| 25 | 75 | 100 | 75 | TFSI–PPL = 1:3:4:3 |
| 25 | 37.5 | 50 | 12.5 | TFSI–PPL = 2:3:4:1 |
| 25 | 37.5 | 50 | 25 | TFSI–PPL = 2:3:4:2 |
| 25 | 37.5 | 50 | 37.5 | TFSI–PPL = 2:3:4:3 |
| 25 | 25 | 37.5 | 8.3 | TFSI–PPL = 3:3:4:1 |
| 25 | 25 | 37.5 | 16.7 | TFSI–PPL = 3:3:4:2 |
| 25 | 25 | 37.5 | 25 | TFSI–PPL = 3:3:4:3 |
| Components Weight [mg] | Abbreviation of the Formulation | |||
|---|---|---|---|---|
| PSTFSILi | PEO | PEGDM | LiTFSI | |
| 25 | 75 | 100 | 25 | PhSI–PPL = 1:3:4:1 |
| 25 | 75 | 100 | 50 | PhSI–PPL = 1:3:4:2 |
| 25 | 75 | 100 | 75 | PhSI–PPL = 1:3:4:3 |
| 25 | 37.5 | 50 | 12.5 | PhSI–PPL = 2:3:4:1 |
| 25 | 37.5 | 50 | 25 | PhSI–PPL = 2:3:4:2 |
| 25 | 37.5 | 50 | 37.5 | PhSI–PPL = 2:3:4:3 |
| 25 | 25 | 37.5 | 8.3 | PhSI–PPL = 3:3:4:1 |
| 25 | 25 | 37.5 | 16.7 | PhSI–PPL = 3:3:4:2 |
| 25 | 25 | 37.5 | 25 | PhSI–PPL = 3:3:4:3 |
| Components Weight [mg] | Abbreviation of the Formulation | |||
|---|---|---|---|---|
| PSTFSILi | PEO | PEGDM | LiTFSI | |
| 25 | 75 | 100 | 25 | DTTO–PPL = 1:3:4:1 |
| 25 | 75 | 100 | 50 | DTTO–PPL = 1:3:4:2 |
| 25 | 75 | 100 | 75 | DTTO–PPL = 1:3:4:3 |
| 25 | 37.5 | 50 | 12.5 | DTTO–PPL = 2:3:4:1 |
| 25 | 37.5 | 50 | 25 | DTTO–PPL = 2:3:4:2 |
| 25 | 37.5 | 50 | 37.5 | DTTO–PPL = 2:3:4:3 |
| 25 | 25 | 37.5 | 8.3 | DTTO–PPL = 3:3:4:1 |
| 25 | 25 | 37.5 | 16.7 | DTTO–PPL = 3:3:4:2 |
| 25 | 25 | 37.5 | 25 | DTTO–PPL = 3:3:4:3 |
| Sample | Decomposition Temperature, Td [°C] | Percentage Weight Loss [%] at Various Temperature | |||
|---|---|---|---|---|---|
| 200 °C | 300 °C | 400 °C | 500 °C | ||
| PSTFSILi | 376 | 4.5 | 6.6 | 11.1 | 29.9 |
| PSPhSILi | 450 | 0.7 | 2.1 | 2.7 | 22.5 |
| PSDTTOLi | 449 | 0.5 | 1.5 | 4.8 | 23.6 |
| Sample | a | b | c | d | e | f | g |
|---|---|---|---|---|---|---|---|
| Tg [°C] | −49.2 | −48.5 | −49.6 | −50.3 | −48.9 | −50.6 | −50.9 |
| Tm [°C] | 61.7 | 55.2 | 44.8 | 52.2 | 43.7 | 52.8 | 47.7 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mei, X.; Zhao, W.; Ma, Q.; Yue, Z.; Dunya, H.; He, Q.; Chakrabarti, A.; McGarry, C.; Mandal, B.K. Solid Polymer Electrolytes Derived from Crosslinked Polystyrene Nanoparticles Covalently Functionalized with a Low Lattice Energy Lithium Salt Moiety. ChemEngineering 2020, 4, 44. https://doi.org/10.3390/chemengineering4030044
Mei X, Zhao W, Ma Q, Yue Z, Dunya H, He Q, Chakrabarti A, McGarry C, Mandal BK. Solid Polymer Electrolytes Derived from Crosslinked Polystyrene Nanoparticles Covalently Functionalized with a Low Lattice Energy Lithium Salt Moiety. ChemEngineering. 2020; 4(3):44. https://doi.org/10.3390/chemengineering4030044
Chicago/Turabian StyleMei, Xinyi, Wendy Zhao, Qiang Ma, Zheng Yue, Hamza Dunya, Qianran He, Amartya Chakrabarti, Christopher McGarry, and Braja K. Mandal. 2020. "Solid Polymer Electrolytes Derived from Crosslinked Polystyrene Nanoparticles Covalently Functionalized with a Low Lattice Energy Lithium Salt Moiety" ChemEngineering 4, no. 3: 44. https://doi.org/10.3390/chemengineering4030044
APA StyleMei, X., Zhao, W., Ma, Q., Yue, Z., Dunya, H., He, Q., Chakrabarti, A., McGarry, C., & Mandal, B. K. (2020). Solid Polymer Electrolytes Derived from Crosslinked Polystyrene Nanoparticles Covalently Functionalized with a Low Lattice Energy Lithium Salt Moiety. ChemEngineering, 4(3), 44. https://doi.org/10.3390/chemengineering4030044