1. Introduction
Despite decades of study, the best possible choices for uranium-based used nuclear fuel (UNF) processing and storage are unclear, and the strategy for handling it still remains an open question in many countries. While international research effort moves towards a closed fuel cycle with advanced UNF reprocessing and new reactor designs, some countries developed, and are now starting to implement, final disposal facilities for once-through fuel. Uranium and plutonium recovery from UNF by the hydrometallurgical PUREX-process is the only time-proven technology used in industrial scales and in commercial use [
1]. Many novel separation technologies, both hydro- and pyrometallurgical, are maturing in laboratory use and are meant for not only the recovery of uranium and plutonium, but that of the other fissile actinides as well. In the international partitioning and transmutation (P&T) research effort, it is recognized that in addition to the recovery of uranium and plutonium, the recovery and transmutation of minor actinides americium, curium and neptunium decreases radiotoxicity and heat generation of the UNF, thus lowering the requirements for disposal of the remainder. In P&T, these minor actinides are to be transmuted into short-lived nuclides in future reactors. One critical problem is that of the partitioning of trivalent minor actinides from the fission products. The UNF inventories have trivalent lanthanides that are chemically very similar to the trivalent actinides and move with them through any conventional separation schemes. Highly selective technologies are crucial for this separation, as these lanthanides with their large neutron cross-sections would act as efficient neutron poisoners if still present in the fabricated minor actinide containing fuels or transmutation targets.
The majority of current hydrometallurgical partitioning processes are based on solvent extraction. Such processes use organic solvents or extraction reagents, and result in the generation of large volumes of liquid radioactive secondary waste. The use of organic solvents and materials is limited by their inherent radiolytic instabilities and toxicity. The ideal alternative would be a technology that relies only on inorganic materials and aqueous streams. Such is the motivation behind the work presented here. We study inorganic ion exchangers for the actinide-lanthanide separations in mineral acids. The applications would be in advanced UNF separation schemes, e.g., as an integral part of a novel process, or as a supportive process to handle the secondary waste of the PUREX process or its derivatives. Inorganic materials can withstand great doses of radiation and high temperatures, and can be chemically stable in strong acids. Ion exchangers can offer efficient uptake with large capacity selectively for specific elements or groups of elements. One drawback for ion exchange is the pH dependency: waste stream pH has to be maintained in an optimal working range.
Zirconium(IV) phosphates, among other metal(IV) phosphates, have been widely studied since the 1960s [
2,
3] for multiple purposes such as ion exchange [
4], catalysis [
5,
6], drug delivery systems [
7] and surface-modified multifunctional materials [
8]. In past decades, there has been an increasing amount of zirconium phosphonate research as well, as zirconium phosphate offers a highly versatile precursor for functionalization and hybridization into inorganic-organic frameworks, thus increasing the possibilities even further. Ion exchange studies for zirconium phosphate include light elements as well as heavy elements: from the first alkali metals all the way to actinides [
9,
10,
11,
12,
13,
14,
15]. α-Zirconium phosphate [
16] is known to have great selectivity towards trivalent cations. Amongst the physicochemically similar trivalent metals europium(III) and americium(III), the selectivity of α-zirconium phosphate has been reported higher for europium [
14,
15]. We have further investigated this, and have provided promising evidence of semi-crystalline α-zirconium phosphate as a selective cation exchanger for americium-europium separation [
17,
18]. We have also demonstrated its flexibility in the low-pH range through some delicate synthesis modifications: its acidity can be engineered which affects the optimal pH range for the separation [
18]. Past studies are focused on the material characteristics and development, and uptake or separation capabilities of several zirconium phosphate materials in simple media. Here, we further the research by studying some remaining critical questions: how the salinity affects the uptake or separation of americium and europium. Molar quantities of mono- and divalent cations are added to the mixture along with nanomolar quantities of Am and Eu. Further, the Am uptake is studied by relatively extreme levels of Eu presence (Eu:Am from 1:39 to 10,000:1) to simulate the presence of higher quantities of lanthanides in the separation application. Finally, dynamic column separation studies are reported for the first time for these analytes, in two modes: a load-elution experiment and a constant equimolar feed experiment. From these experiments, we gain insight into the capacity and separation capabilities in dynamic conditions.
2. Material and Methods
Synthesis and comprehensive material characterization for the semi-crystalline α-zirconium phosphate with crystallites in nanoscale is described in our earlier publication [
18] with the product designation ZrP25. The product will be called ZrP in this text for brevity. In summary, the ZrP was prepared with a hydrogen fluoride free precipitation reaction followed by reflux for 25 h under normal pressure and 80 ± 1 °C temperature. The characterization includes elemental analysis, solid-state
31P magic angle spinning nuclear magnetic resonance, Fourier transform infrared, thermogravimetric analysis, powder X-ray diffraction, scanning electron microscopy for imaging, and the determination of acid dissociation constants by base titration. Here, we have used the term semi-crystalline, although many of the characterization techniques reveal a rather crystalline structure, e.g., from the NMR we conclude the presence of mainly one type of phosphate group, unlike the less-crystalline products that were studied. Still, some degree of variation is reported, and the product could be also described as nanocrystalline.
Distribution coefficient (Kd) determinations: Samples of 20 mg of ZrP were placed in plastic 20 mL scintillation tubes and equilibrated with 10 mL volume of a given solution for 3 days in a slow rotary mixer with 10 rotations per minute. The solutions always contained 30 Bq∙mL−1 carrier-free Eu-152 and/or 30 Bq∙mL−1 carrier-free Am-241 tracer, in nitric acid with and without varying amounts of NaNO3, NaCl, or SrCl2. After equilibration, supernatant solution was separated from the slurry with 0.2 µm membrane filter for measurement. The Kd determinations were based on quantitative radiometric detection and comparison of equilibrium activity concentration in the supernatant with that of the equal method blank. For all distribution coefficient determinations, three full procedure repetitions were done for both the samples and the method blanks, unless otherwise stated. In the context of this text a method blank, or just blank, does not mean a sample without analytes, rather it means a sample solution with the analytes but without any ion exchanger. Blanks were treated comprehensively in equal ways to actual samples, including rotation, filtering, etc. The activity taken up by the exchanger in a given sample solution was determined as the equilibrium activity concentration of a blank solution minus the same of the sample supernant. The Kd was calculated as the ratio of equilibrium activity concentration (Bq∙kg−1) in the exchanger to the one in the solution (Bq∙L−1), having then the unit L∙kg−1.
Effect of salt concentration on Eu(III) distribution: Three salts of sodium and strontium were selected to see the possible effect of the concentration or the cation/anion. The salts solutions originated from anhydrous or hexahydrate (for strontium chloride) powders. Concentrations of the salts NaCl, SrCl2 and NaNO3 were varied. For the first study, 0.1 mol∙L−1 and 0.01 mol∙L−1 solutions were prepared for each of the three salts. For the second study for NaNO3 only, nine solutions from 0.001 mol∙L−1 to 4 mol∙L−1 were prepared. Strong nitrate concentrations are of interest since commonly the actinide-lanthanide separations are done in strong nitric acids. An equivalent amount of Eu(III)-152 tracer was then added to all samples, and the Kd(Eu) values for ZrP were determined as explained above.
Effect of Eu(III) concentration on Am(III) distribution: The Kd values were determined as explained above. A set amount of Am(III) was placed together with varying Eu(III) concentrations in nitric acid with 0.1 mol∙L−1 NaNO3 in pH 1.5, to create a series of samples with molar ratio Eu:Am from 10,000:1 to 1:40. For Eu:Am molar ratios 1:1 to 1:40, Eu was added as a carrier-free Eu(III)-152 tracer, and for molar ratios 10:1 to 10,000:1, Eu was added as inactive Eu(III) nitrate.
Kinetic experiments: This experiment was equal to the Kd determinations explained above, with the exception of the variable duration for the equilibration. The experiment was done with a binary solution containing Eu and Am tracers, and with separate solutions of each on their own. For selected samples, from the 0.2 µm-filtered solutions, aliquots were taken and further filtered with Amicon Ultra-4 Ultracel 3K filtering devices (Merck KGaA, Darmstadt, Germany), in a centrifuge at 3000 × g r.c.f. The equilibration time in the binary experiment was from 30 s to 24 h (seven different durations), while in the experiment with separate solutions it was from 30 s to seven days (13 different durations).
Breakthrough experiment: A constant feed of inactive Eu(III) was pumped through a fixed bed column containing 472 mg of ZrP. The bed volume was 1.1 mL and the feed rate was 2 bed volumes per hour. The feed consisted of 0.001 mol∙L−1 Eu(III) in 0.002 mol∙L−1 HNO3 with 0.1 mol∙L−1 NaNO3. The eluate was collected in fractions from which Eu concentrations were quantitatively determined by microwave plasma atomic emission spectrometer (Agilent 4200 MP-AES) at 381.967 nm with La at 394.910 nm as the internal standard.
Load-elution Eu-Am column separation experiment: Low-pressure Bio-Rad Econo plastic columns were used along with Tygon plastic tubing. First, the column was packed with 681 mg ZrP in a slurry containing 0.1 mol∙L−1 nitric acid and 0.1 mol∙L−1 NaNO3. The column material was then conditioned first with 20 mL of 0.1 mol∙L−1 nitric acid plus 0.1 mol∙L−1 NaNO3 and then continuously with 0.002 mol∙L−1 nitric acid plus 0.1 mol∙L−1 NaNO3 until eluate pH reached 2.5. Loading: the column was loaded with a solution that contained 2000 Bq carrier-free Eu-152 and 2000 Bq carrier-free Am-241 in 5 mL 0.002 mol∙L−1 nitric acid plus 0.1 mol∙L−1 NaNO3. Finally, the column was rinsed with the same volume of equal but non-traced solution. Elution: separation was started with 0.18 mol∙L−1 nitric acid plus 0.1 mol∙L−1 NaNO3 with the rate of 8 bed volumes per hour. The eluate was collected in 50 min fractions. The radioactivity in the fractions was quantitatively determined with the use of germanium gamma detector. During the loading and rinsing steps, no detectable activities were eluted. Later in the experiment, elution was expedited by changing the nitric acid strength to 1.0 mol∙L−1 along with doubling the speed to 16 bed volumes per hour, while halving the fraction collection time to 25 min.
Constant feed equimolar Eu-Am column separation experiment: The feed solution contained 0.1 mol∙L−1 NaNO3 in 0.1 mol∙L−1 nitric acid, along with 30 Bq∙mL−1 carrier-free Eu-152 and 30 Bq∙mL−1 carrier-free Am-241. A trace amount of Eu(III) nitrate was added to the feed to make it equimolar for the analytes: c(Eu total) = c(Am-241) = 9.8 × 10−10 mol∙L−1. A low-pressure Bio-Rad Econo column was packed with 443 mg of ZrP. The bed volume was 1.0 mL. The above solution was fed into the column at constant rate of 5.0 bed volumes per hour, and the eluate was collected and measured in 1-h fractions. The activity concentrations were compared with those of the original feed solution and the experiment was continued until the concentrations reached the feed levels.
3. Results and Discussion
3.1. Binary Batch Uptake Experiments
The effect of pH on Eu(III) and Am(III) trace ion exchange by zirconium phosphate was studied with and without the background electrolyte, 0.1 mol∙L
−1 NaNO
3. As reported in our earlier studies [
17,
18], zirconium phosphates have a preference for Eu(III) over Am(III). Distribution of analyte species between solution and exchanger is described with the distribution coefficient (
Kd), which can be determined utilizing radiometric measurements as follows.
where
A0 is the equilibrium radioactivity concentration in method blank without the ion exchanger, and
A is that of a sample with the ion exchanger. The use of method blanks is explained in the experimental section. Both with and without NaNO
3, the separation factor is highest at pH 1 (
Table 1), where Am uptake is negligible while Eu uptake is 85% with NaNO
3 and 79% without NaNO
3. The highest separation factor reported here (400 in pH 1) is the highest we have recorded so far for any of our α- or γ-zirconium phosphates, or titanium phosphates synthesized in the past. The separation factors are positively and surprisingly high considering the physicochemical similarity of Eu(III) and Am(III). In nitric acid in our experimental conditions, they are both present as free ions, M
3+, and as nitrates, M(NO
3)
2+ [
17]. Electron configurations for the ions are [Xe]4f
6 for Eu
3+ and [Rn]5f
6 for Am
3+, and the Shannon ionic radii are 94.7 pm (coordination number = 6) and 106.6 pm (8) for Eu
3+, and 97.5 pm (6) and 109 pm (8) for Am
3+ [
19]. We suggest that both the order of preference of Eu over Am and the high separation factors result from the combination of the following three. First, the Pearson’s hard-soft-acid-base (HSAB) principle: a hard base like the phosphate binds more strongly into a harder acid, Eu. Second, in our conditions, only the species M
3+ and M(NO
3)
2+ are present for M = Eu, Am, and a considerably larger fraction of Am than Eu is present as the larger divalent ion M(NO
3)
2+, which is exchanged less into the solid phase, if at all [
17]. This study was done in the past, and includes species such as nitrates with one or more complexing nitrates, and carbonates. Third, the narrow structure may act as an ion sieve thus preferring the smaller Eu with the higher charge density.
In some data points with extremely high uptake, there is no Eu left in the solution to be quantitatively measured within a reasonable detection time, leaving both the
Kd(Eu) and the separation factor undetermined, though high. The
Kd values were found to be generally higher in solutions with NaNO
3, as were the separation factors. This phenomenon was then further studied with varying salt concentrations. The relative uncertainties for
Kd values are sometimes high due to the way
Kd is determined: if the values are either very high or very low, the measurement is close to the background, or to the method blank. The uncertainties for the values presented in
Table 1 are between 6% to 9% for Eu, and between 5% to 9%, for Am, apart from the two extreme cases for Am in pH 0.5: 60% to 70%.
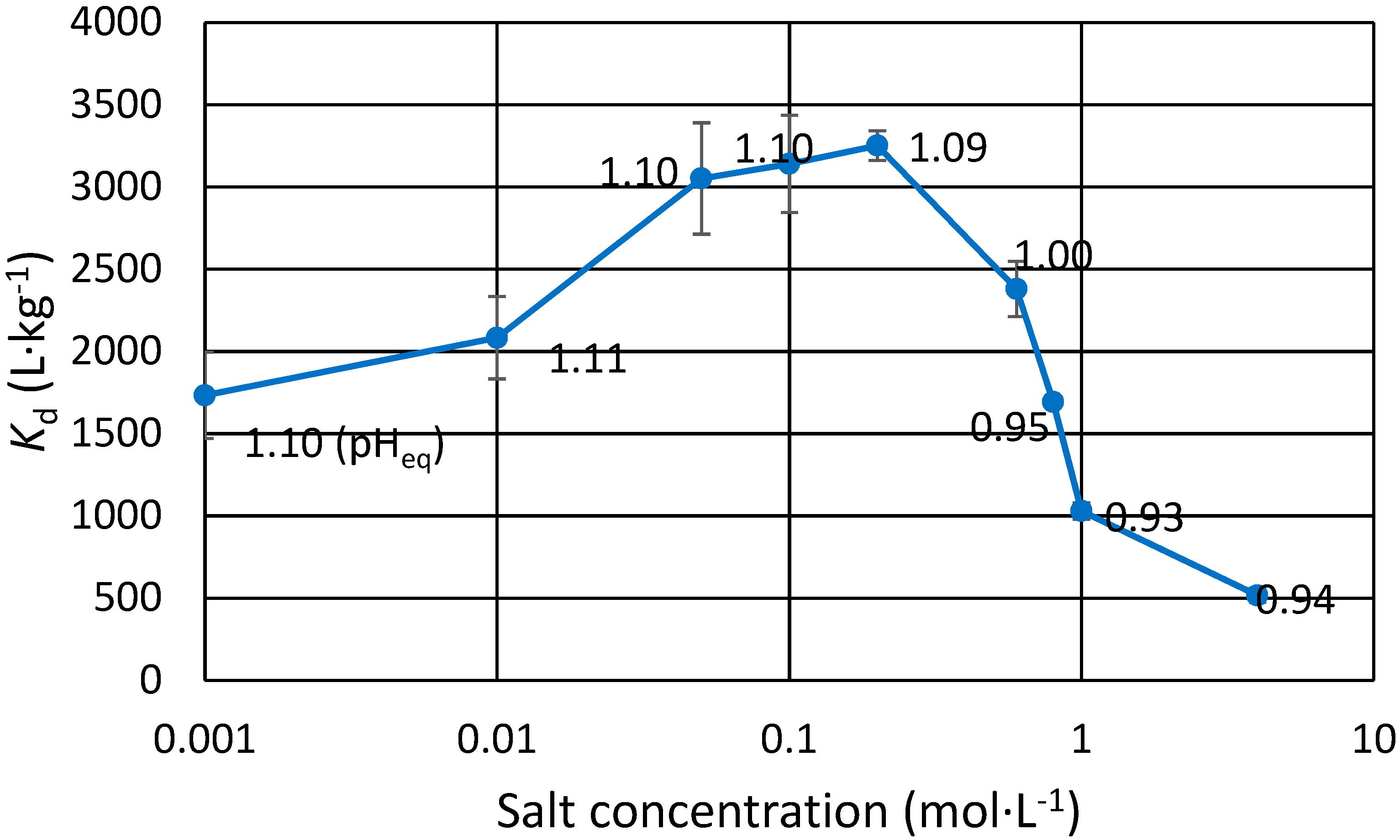
3.2. Effect of Na and Sr Salts on Eu(III) Uptake
The effect of mono and divalent cation concentration on trace Eu(III) exchange was studied using NaCl, SrCl
2 and NaNO
3 (
Table 2 and
Figure 1). Eu uptake increases as the Na concentration increases in dilute salt concentrations for both the chloride and the nitrate. This is not seen for Sr, however the
Kd is still 25% higher in Sr containing solutions than without any salt (1900 L∙kg
−1, pH
eq 1.01). If salt concentration is too high, i.e., >1 mol∙L
−1, less Eu(III) is taken up by the exchanger. In the highly saline samples, pH dropped drastically as H
+ was released through exchange for the cation. In these samples, small amount of dilute NaOH was added to balance their pH close to the intended pH. A partial explanation for
Kd differences comes from the inaccuracy of said pH balancing: the pH
eq values are within about 0.1 units. It can be shown that for trivalent metal to hydrogen exchange, which has the slope of approximately 3 in the pH
eq versus log
Kd plot, a rise of 0.1 pH units effectively doubles the
Kd. In short, if the
Kd is 500 L∙kg
−1 at pH
eq 0.90, it is expected to be approx. 1000 L∙kg
−1 at pH
eq 1.00. This inaccuracy is, however, not enough to explain all the reported results. Especially in the case of the dilute salts, pH was repeatedly determined to be within a few hundredths of a unit, and the
Kd differences and trends are clear. For low Na concentrations, the
Kd for Eu(III) steadily increases as salt concentration is increased from 0.001 mol∙L
−1 until 0.2 mol∙L
−1. As the concentration is increased further, possible competition takes place as
Kd starts to decrease, albeit at this point there is a slight drop in pH, which to some extent could explain lower
Kd, as explained earlier. For stronger salts however, the effect of competition is more easily seen as the
Kd of Eu(III) is halved when Na concentration is increased from 1 to 4 mol∙L
−1 in equal pH
eq. Since Eu is present in trace concentrations and Na or Sr is abundant, it is clear that the zirconium phosphate ion exchanger is greatly selective for trivalent metals over mono and divalent cations. The relative uncertainties for the values presented in
Table 2 are from 1% to 2% for NaCl, from 15% to 17% for SrCl
2 and from 2% to 14% for NaNO
3. The relative uncertainties for the values presented in
Figure 1 are between 1% and 15%.
3.3. Effect of Eu(III) Concentration on Am(III) Uptake
Effect of varying Eu and Am molarities on Eu and Am sorption was investigated. Binary solutions of Eu and Am in nitric acid (pH 1.5) with 0.1 M NaNO3 were created containing a set amount of carrier-free Am-241 tracer along with varying amounts of Eu, in the form of Eu-152 carrier-free tracer or inactive Eu diluted from Eu nitrate standard solution. Results show that in binary solutions with a vast excess of Eu, which has the higher sorption, Am sorption decreases as the amount of competing Eu increases (
Table 3). However, unexpected findings are seen elsewhere: when Eu is present in a lesser degree (molarities 1:39 to 1:1), Am sorption is greater than in Am solution without any Eu. A significant increase in the concentration of coexisting cations could open up the structure for higher Am sorption, but evidently that is it not the case here, since both the Eu and Am are present only in trace amounts and since Na is also present, in a far larger—and constant—amount.
Separation factors were approximately 100 in the molar ratio region they were determined in. This region is relevant to the Eu:Am molar ratios calculated from data from the literature [
20] of nuclide inventories for PWR used nuclear fuel. Two PWR fuels, greatly differing by their burn-up and decay time, were taken as example cases for calculations. Their total Eu:Am ratios were 1:13 for 72 GWd∙MTU
−1 burn-up with 8 years of decay and 1:147 for 38.6 GWd∙MTU
−1 burn-up with 22 years of decay, including all presented isotopes for both elements. Similarly, total Ln(III)/An(III) ratios were 1:18 for 72 GWd∙MTU
−1 with 8 years and 1:58 for 38.6 GWd∙MTU
−1 with 22 years, including all presented isotopes for every lanthanide and for the actinides Am, Cm and Np.
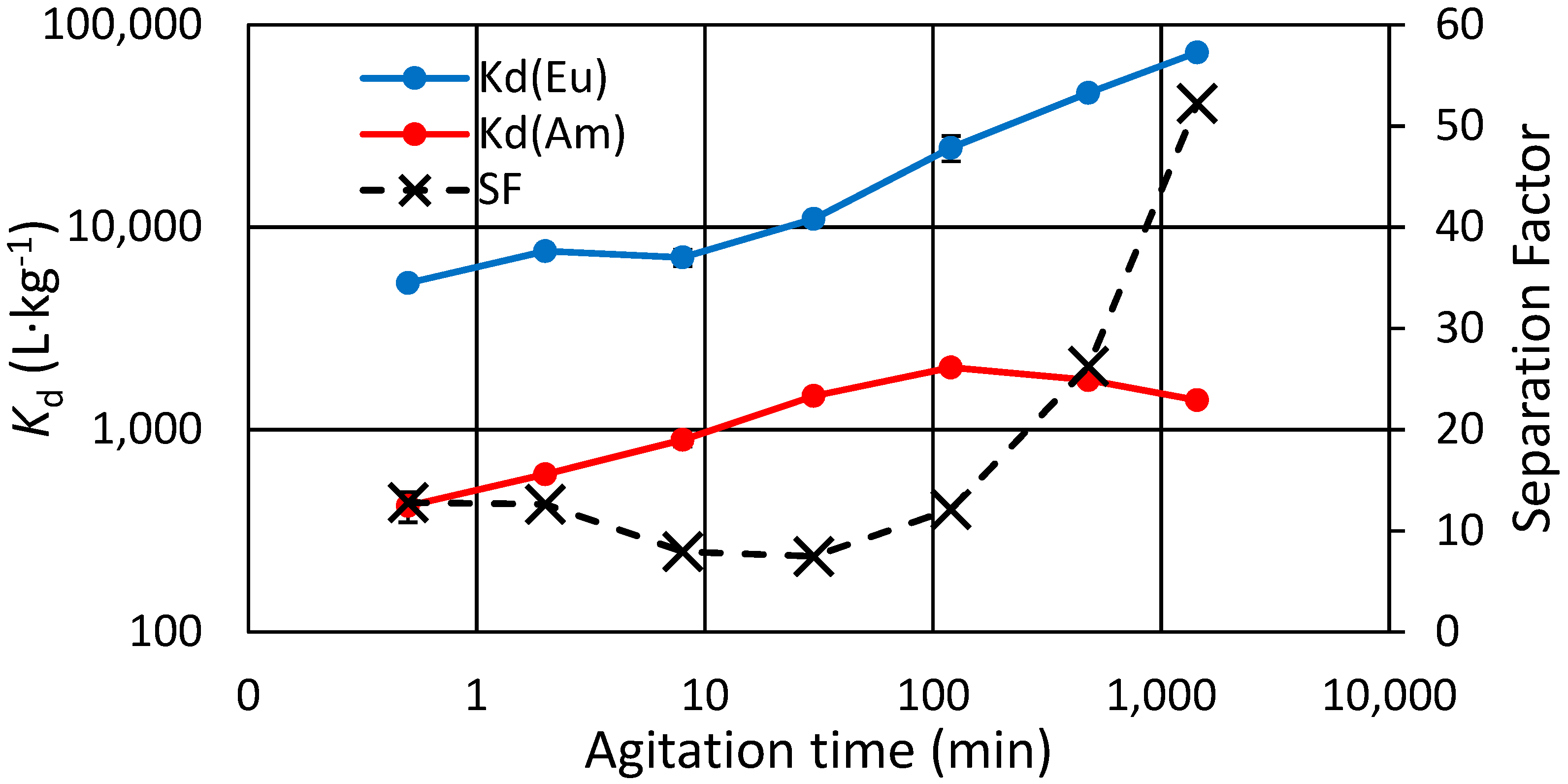
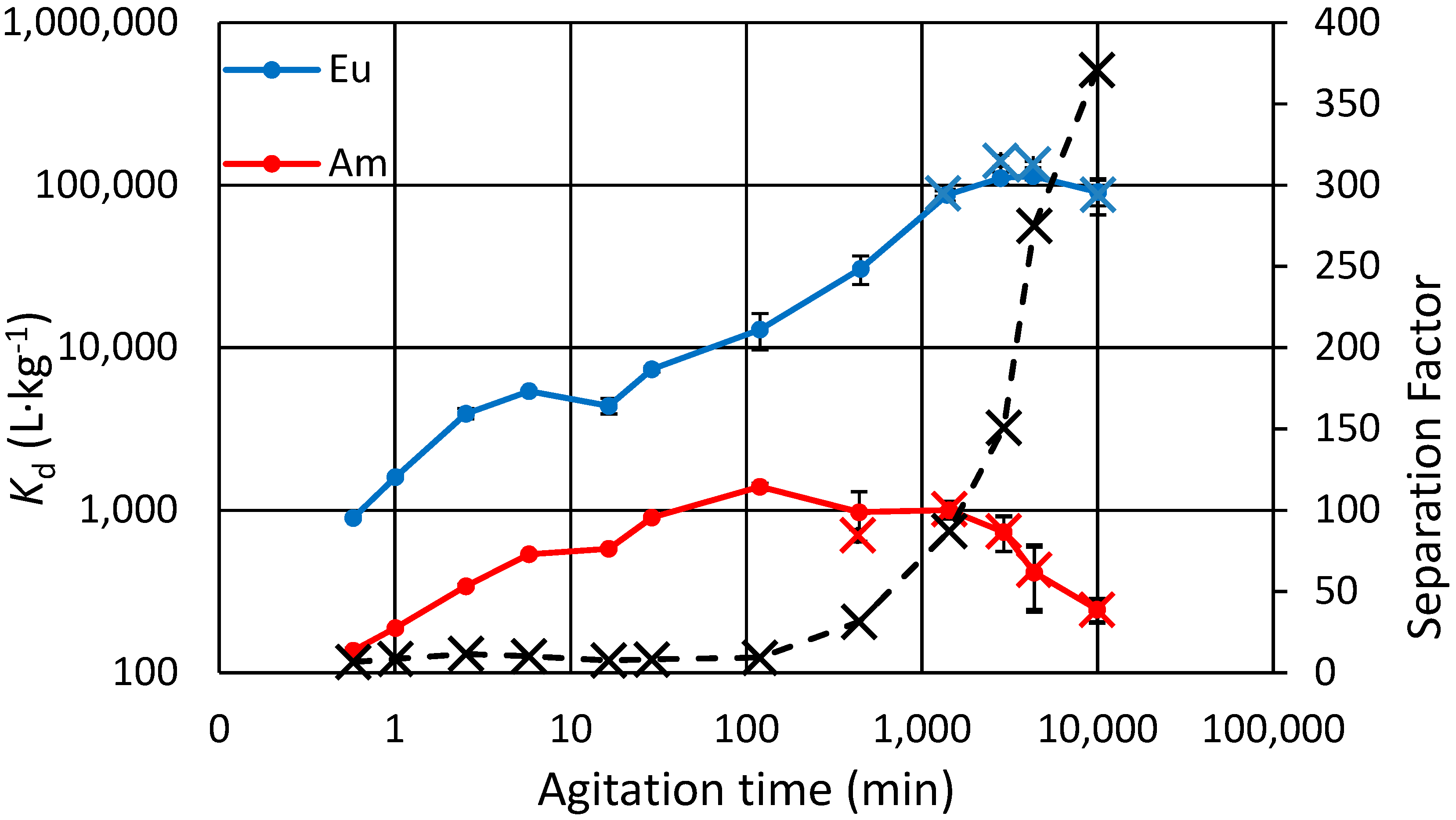
3.4. Kinetic Experiments, Binary and Separate Solutions
Eu(III) and Am(III) trace ion exchange kinetics were studied both in binary solution (
Figure 2) and in separate solutions (
Figure 3). Note that the term “separation factor” used here is not exactly the separation factor that is conventionally derived from equilibrium state distribution values, but rather, it is just the ratio of Eu and Am distribution at a specific time point. In the binary experiment it also accounts for possible competition since the solution has both analytes. It is seen that in the binary experiment the Am concentration in the exchanger starts to lower after only 2 h, as that of Eu steadily increases. The effect of possible Eu-Am competition can be ruled out based on the other kinetics experiment done with separate Eu and Am solutions, where similar behavior for Am is shown after the same 2 h (
Figure 3). The total experiment time there is longer, and after 24 h we note that the
Kd for Eu settles at its maximum while that for Am continues to decrease.
Previously we have proven stability of α-zirconium phosphate for both highly crystalline [
21] and highly amorphous [
22] material for similar metals: no lowering of the
Kd for Nd(III) nor Dy(III) were shown during the full duration of the kinetics experiment (three days). In those experiments the analyte concentrations were always 1 mmol∙L
−1, instead of trace levels used here, and also there was no Na present. It took 15 to 24 h for Nd and Dy to reach the stable plateau with maximum sorption, comparable to the 24 h taken for Eu to reach its maximum as reported here. The physical stability and the possibility of self-milling were considered: if particle size decreases enough due to the self-milling of the particles in the rotating sample during the prolonged experiment, a fraction of the analytes, along with a fraction of the smallest milled particles would go through the filtering procedure. Finally, that fraction would be wrongly attributed in the radiometric determination as “analytes in the solution”. This was tested by more demanding, centrifugal filtering of the samples before measurement. However, no significant difference between the 0.2 µm-filtered and the further filtered results are seen, proving the physical stability of the particles for the duration of the experiment, one week.
What is left to account for the decrease in Am uptake, is the slow kinetics of the major cation present: Na. In both experiments, Na is present in great abundance compared to the trace analytes. It is possible that Na is in these conditions favored against Am, but not against Eu. For the kinetic experiments, the materials were initially in H-form, not in Na-form. Thus, we would then expect to first see the increase of Am exchange with faster kinetics, but finally the replacement of some of the Am by Na with slower kinetics. Capacity was also considered: in the conditions of the experiment, 0.05 meq∙g−1 Na and Am combined are present, which is one sixth of the dynamic breakthrough capacity of the exchanger as reported later in the text (0.3 meq∙g−1).
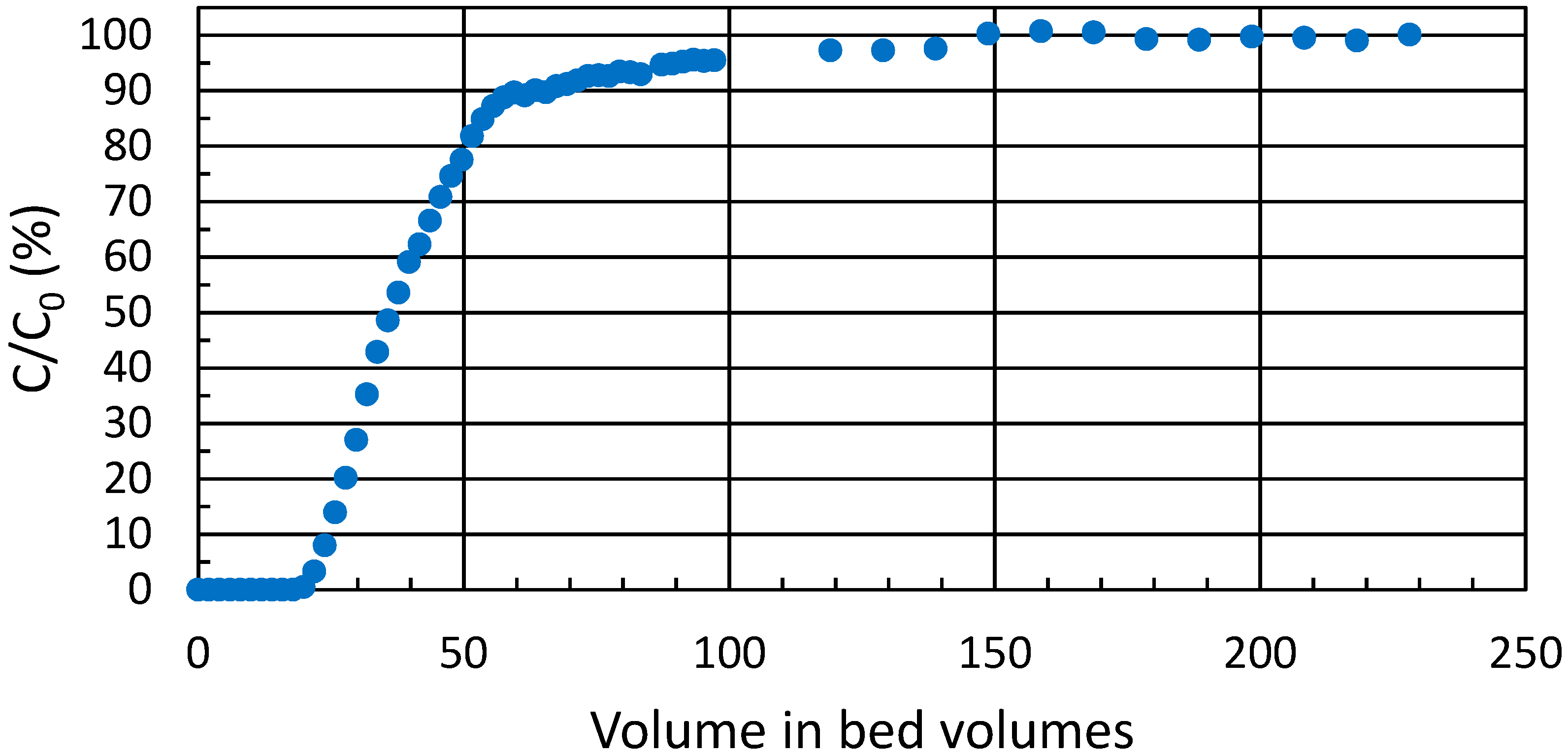
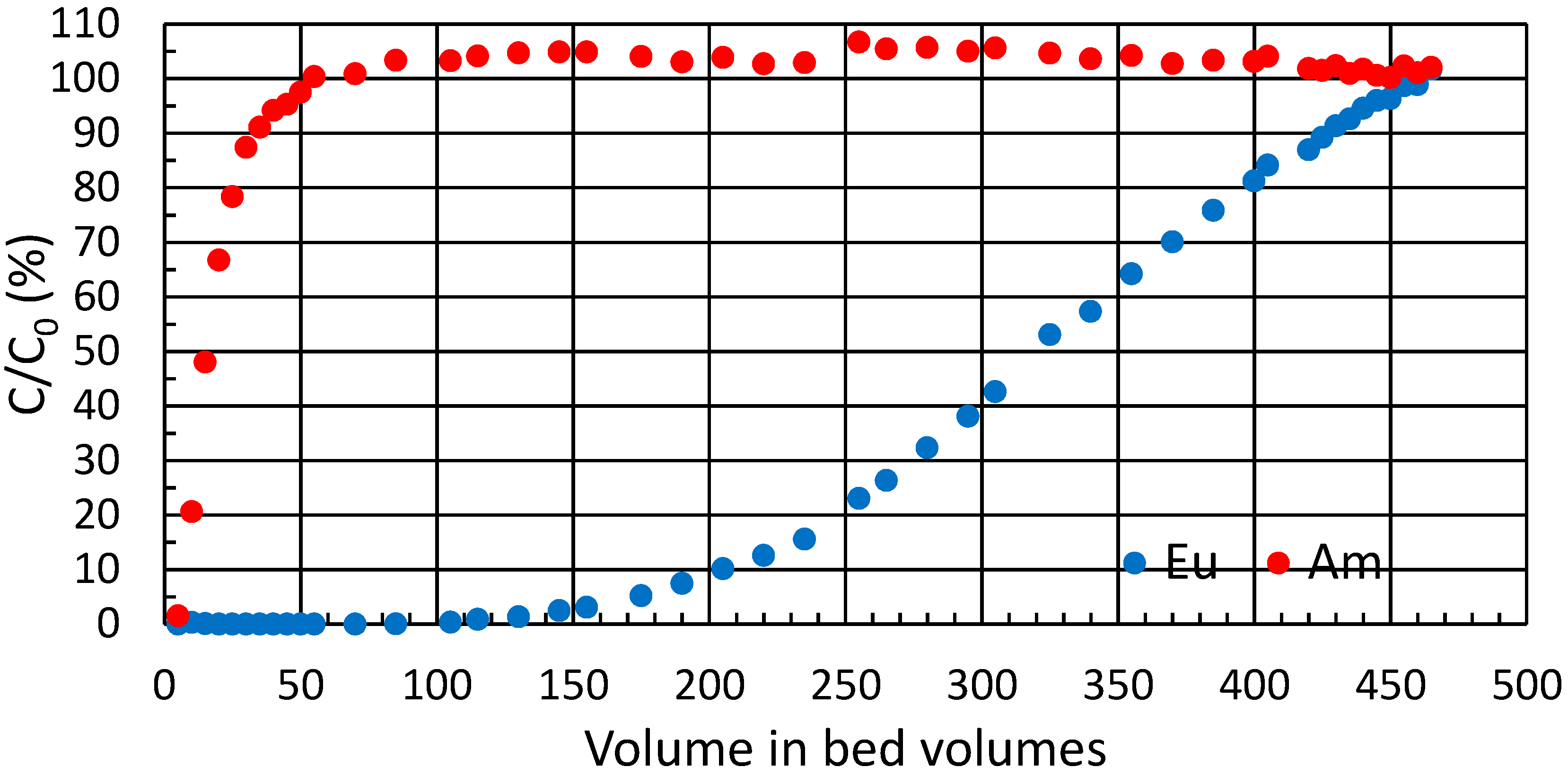
3.5. Breakthrough Experiment
The practical ion exchange capacity of ZrP for the trivalent metal ions of interest in column use was determined from a breakthrough experiment in high
Kd conditions (
Figure 4). During the experiment, a total of 0.10 mmol∙g
−1 of Eu(III) was taken up by the exchanger; or 0.30 meq∙g
−1. Theoretical capacity of crystalline α-zirconium phosphate is 6.6 meq∙g
−1 [
23] whereas amorphous zirconium phosphate can exceed this [
18,
22]. For the semi-crystalline zirconium phosphate batch used throughout the text, the experimental total capacity by titration has been earlier determined to be 7 meq∙g
−1 [
18]. Accordingly, approximately 4% of the ion exchange sites were available for Eu(III). This highlights the small proportion of the more readily available sites at surfaces and edges, because in the bulk interlayer space itself, when hydrogen is exchanged for a trivalent metal, the layers are pulled closer due to the high charge density of the metal, which in turn hinders the exchange of subsequent cations to the surrounding space.
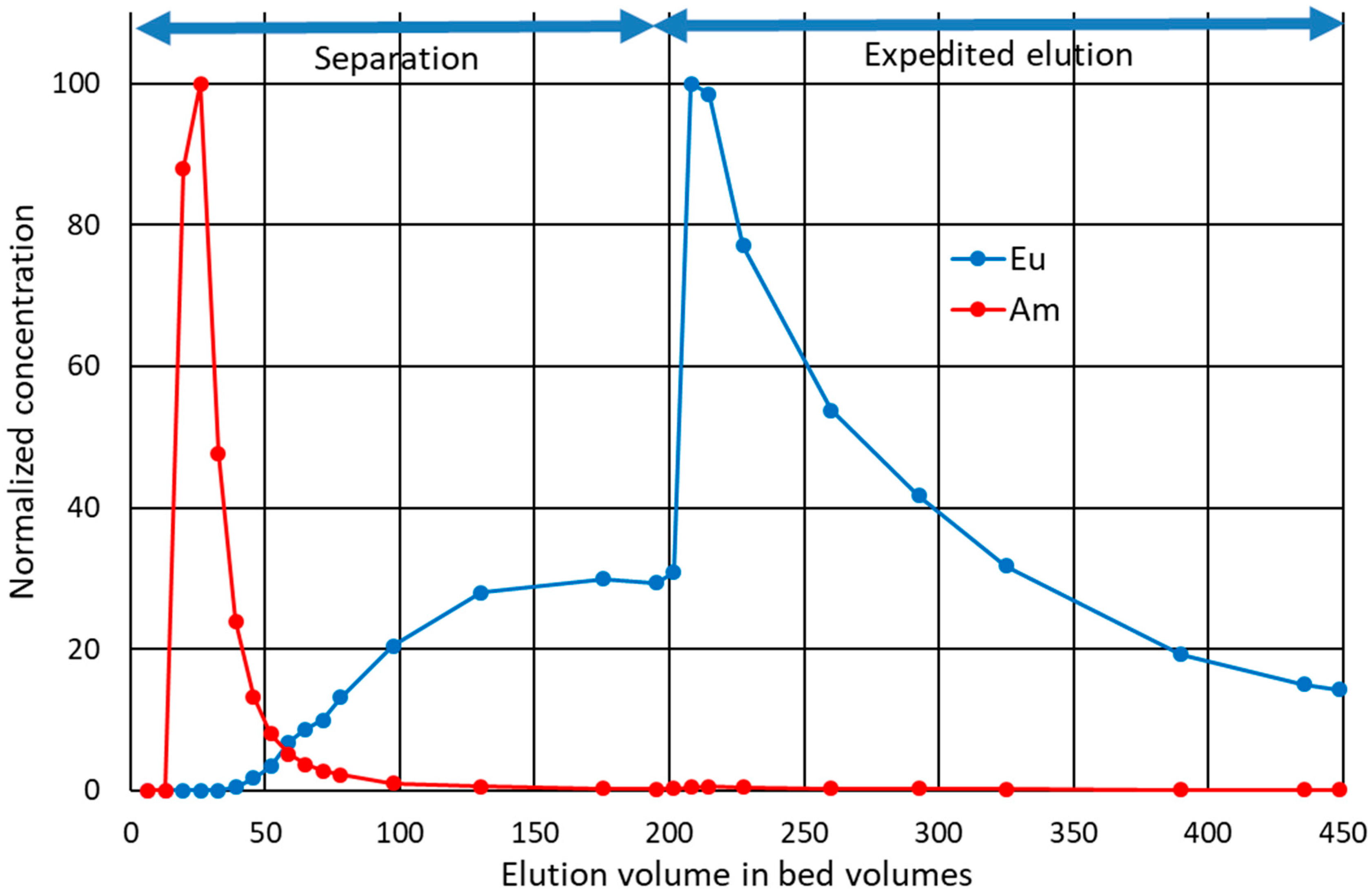
3.6. Load-Elution Column Separation Experiment
Selective elution of the less-bound Am was tested in a low-pressure, top-down, column-separation experiment. The loading was done in conditions in which both analytes have high retention, that is, high
Kd based on the batch experiments. The analytes were concentrated in the very top of the column. Selective elution was then achieved with moderate speed in conditions where Am had low retention and Eu had high retention. Results show that most of Am was quickly eluted in the first selected conditions (
Figure 5). Eu concentrations did not reach measurable quantities in the first 40 bed volumes, but started to slowly but steadily increase thereafter until reaching a plateau before 200 bed volumes. Eluent was then strengthened to enhance the elution of Eu and to see if more Am would elute with a stronger acid. The process for Eu was slow even in 1 M HNO
3. When changing the composition of the eluent, a minuscule but detectable amount of Am was also eluted. Approx. 88% of the total loaded Am was eluted during the first 130 bed volumes, and 93% during the whole experiment. Thus 7% of Am could not be eluted with selected final conditions or would be eluted extremely slowly and with low purity if the separation conditions or the expedited elution conditions were used continuously. From this 93%, dubbed the
available fraction, 82% was eluted with 99.999% molar purity in the first 39 bed volumes combined, or alternatively, 95% with 99.7% molar purity in the first 130 bed volumes combined. Most overlap between the analytes occurred between 40 and 130 bed volumes. In the combined eluate of the corresponding fractions, 13% of the available Am overlaps with Eu, and molar Am purity is 97%.
3.7. Constant Feed Column Separation Experiment
The separation capability of ZrP for an equimolar Am-Eu mixture was tested in a constant feed low-pressure column experiment. Based on preliminary trace concentration batch experiments, such conditions were selected where Am retention was low to negligible while Eu retention was as high as possible in comparison: 0.1 M HNO
3 with 0.1 M NaNO
3, where
Kd(Eu) was 2800 L∙kg
−1 and
Kd(Am) was 7 L∙kg
−1. In the selected conditions, the Am concentrations in the eluate quickly reached feed levels, before any significant amount of Eu was eluted (
Figure 6). Up to 330 L∙kg
−1 of the equimolar mixture was treated with molar Am purity above 99.5% in the cumulative volume of eluate, up to 630 L∙kg
−1 above 95 mol %, or up to 800 L∙kg
−1 above 90 mol %.
4. Conclusions
It has been reported earlier that zirconium phosphates show promise for actinide and lanthanide separation: they are highly selective towards trivalent metal ions from both groups, while clearly preferring the studied model lanthanide Eu(III) over the actinide Am(III) in select conditions, in low pH (0–3) [
14,
15,
17,
18]. The separation capabilities of a semi-crystalline α-zirconium phosphate were then further investigated and reported here. The order of preference, Eu(III) over Am(III), is favorable for the possible applications, since Am(III) is easily eluted from the material for further use, e.g., fuel fabrication, while the lanthanides can be kept in the solid separation material, convenient for their final disposal.
The separation factors for Eu:Am reported here, commonly approx. 100 and up to 400, are high given the physicochemical similarities between Eu(III) and Am(III). It was demonstrated that the concentration of Eu affects the uptake of Am even in the case of trace concentrations, where more than enough ion exchange sites are present. Remarkably, when dilute Eu was present, Am had higher uptake than in the equal solution on its own. Similarly, in another experiment, with dilute salt added into the system, Eu uptake was increased. The positive effect of Na
+ presence can be explained by the opening of ZrP layers when H
+ is exchanged to the larger Na
+ of the same charge. However, a similar effect of Sr
2+ presence for Eu
3+, or Eu
3+ presence for Am
3+ should not take place if considered in this manner, since the higher charge density pulls the layers closer and should make it harder for subsequent ions to be exchanged close by. Such effects were seen, so there must be another mechanism supporting the ‘co-exchange’. We have previously discussed [
17] that a portion of Eu(III) and Am(III) is exchanged in the form of M(NO
3)
2+. If it was the case such large species found their way to the interlayer space, it would help explain the co-exchange. However, it is more likely that such large species would only occupy the most readily available ion exchange sites, at the outer surfaces or edges of the layers. More importantly, the opposite happens when a larger concentration of Eu is present. Am uptake is lowered, which supports their separation.
The separation of Eu(III) and Am(III) was demonstrated in both a load-elution type column experiment (I), and in a constant feed column experiment (II):
(I) In higher pH, both elements were readily taken by the exchanger, with no leakage detected during the loading. Through elution in selected conditions, most of the less-favored Am was separated before any Eu started to elute. We found that 7% of Am could not be eluted in any of the select conditions, meaning an additional sorption mechanism is present, such as inclusion of some of the analytes into the ZrP structure. The majority (82% of the available 93%) of Am could be purified from Eu-Am mixture with extremely high 99.999% molar purity, while even more (95% of the available 93%) at a lower purity of 99.7 mol %. Thus, the achievable recovery depends on the requirements of an application.
(II) It was shown through the equimolar constant feed separation experiment that up to 330 L∙kg−1 of the solution could be treated with Am purity still above 99.5 mol % in the eluate. Alternatively, up to 630 L∙kg−1 could be treated with Am purity above 95 mol %, or up to 800 L∙kg−1 with Am purity above 90 mol %, again depending on the requirements of the application.
Throughout this study, some critical open questions were answered regarding the development of these materials for the purpose of dynamic separation applications in complex aqueous solutions. Promising numbers were achieved on the dynamic conditions, although the capacity needs to be improved. Work remains to be done, as the studied solutions have so far been rather simple and synthetic. The investigated Eu:Am ratios were in the range of a best estimate in PWR fuel, but the absolute levels remain an open question and is tied to the type of application in question (e.g., the PUREX raffinate, or a stream from an intermediate step during such reprocessing). In the future, the ongoing study will continue towards hot tests with non-synthetic solutions or elaborate simulated solutions. There, discussion will continue on feasibility of the material for the separation in real conditions, and further research effort will be focused especially on improving the dynamic separation and capacity of the material.
Through the separation experiments in columns, and the preliminary investigations, we have further demonstrated the capability of ion exchange for this difficult task of actinide-lanthanide separation in the fields of UNF processing and P&T. The study and use of ion exchange should not be limited to this specific separation case, as its solid phase and the possibility to work in simple mineral acids would offer desired features, such as one alternative or supportive to the current solvent extraction separation schemes. Ion exchangers can be deliberately engineered for very specific tasks, and are by their nature readily suitable for column use, e.g., on-line in industrial processes.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}