CNT and H2 Production During CH4 Decomposition over Ni/CeZrO2. I. A Mechanistic Study


Abstract
1. Introduction
2. Experimental
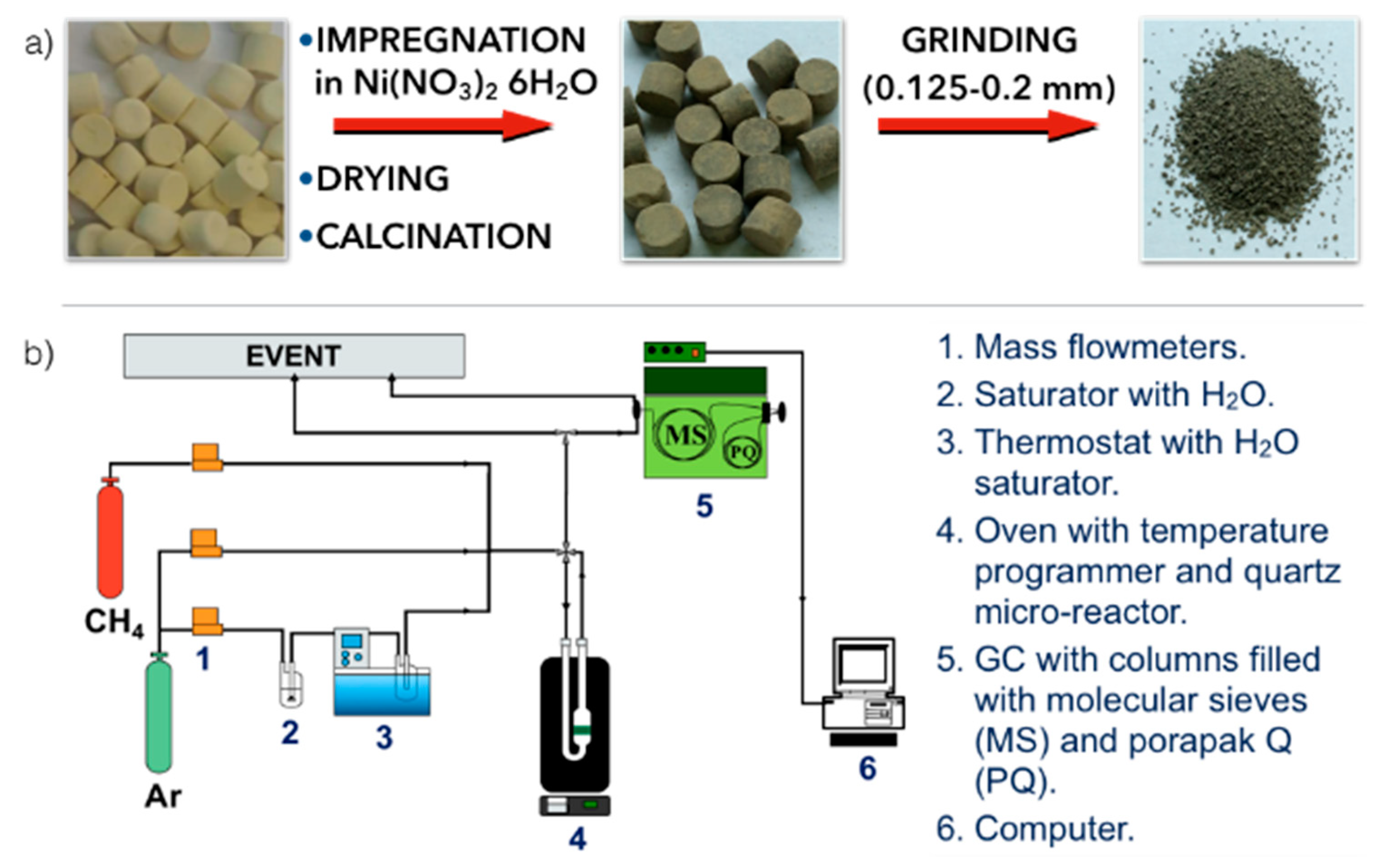
2.1. Preparation of the Formed Ni/CeZrO2 Catalyst
2.2. Tests of CH4 Decomposition and Catalyst Regeneration in H2O
2.3. Catalyst Characterization
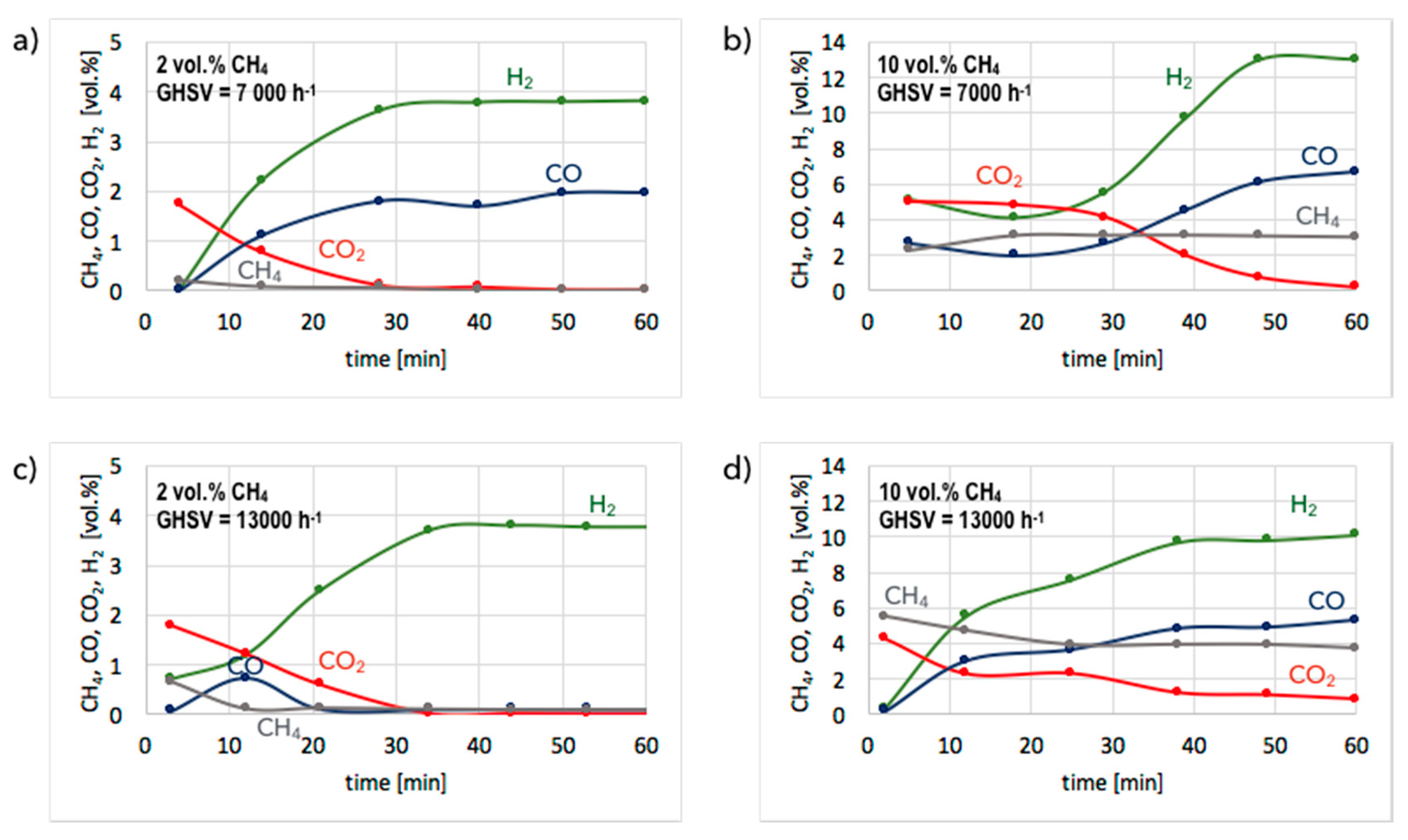
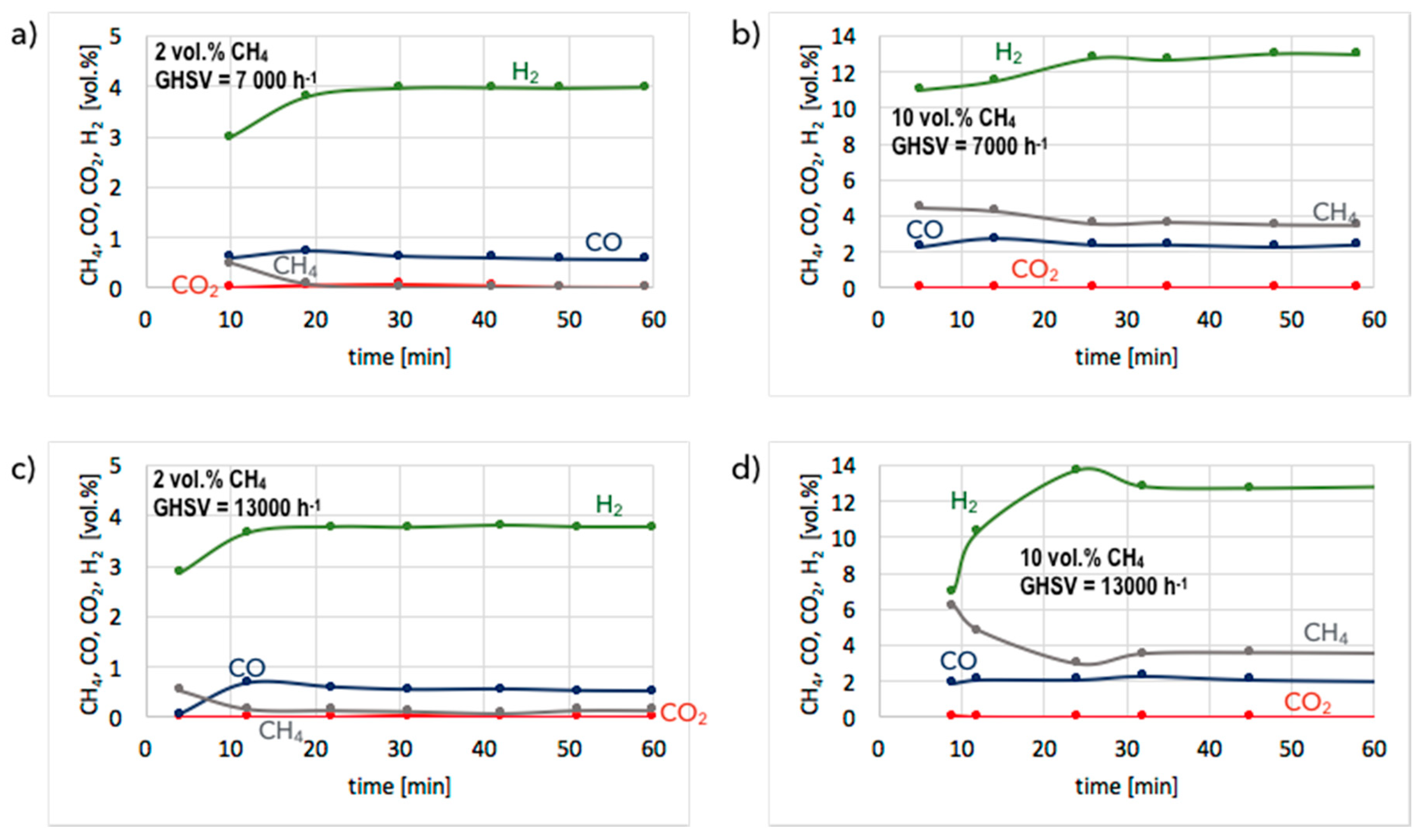
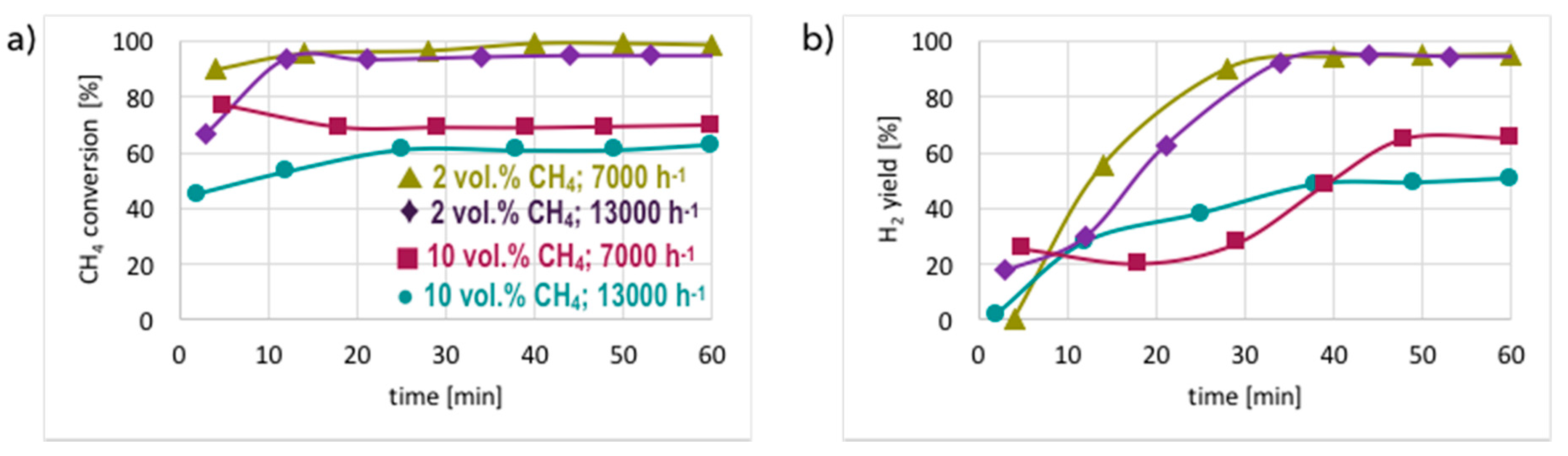
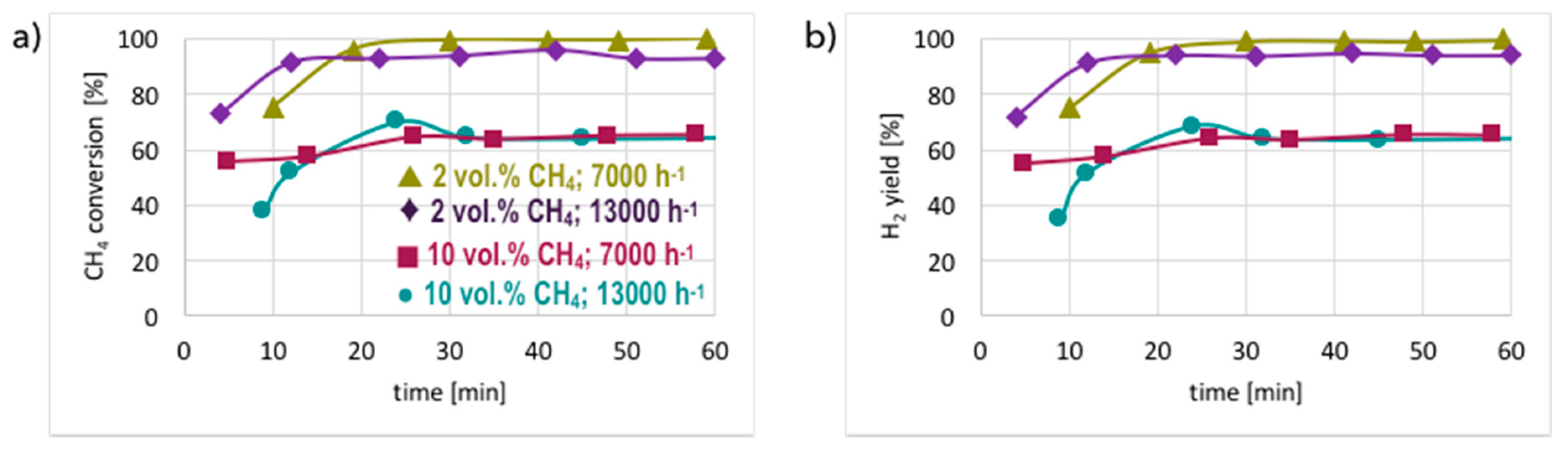
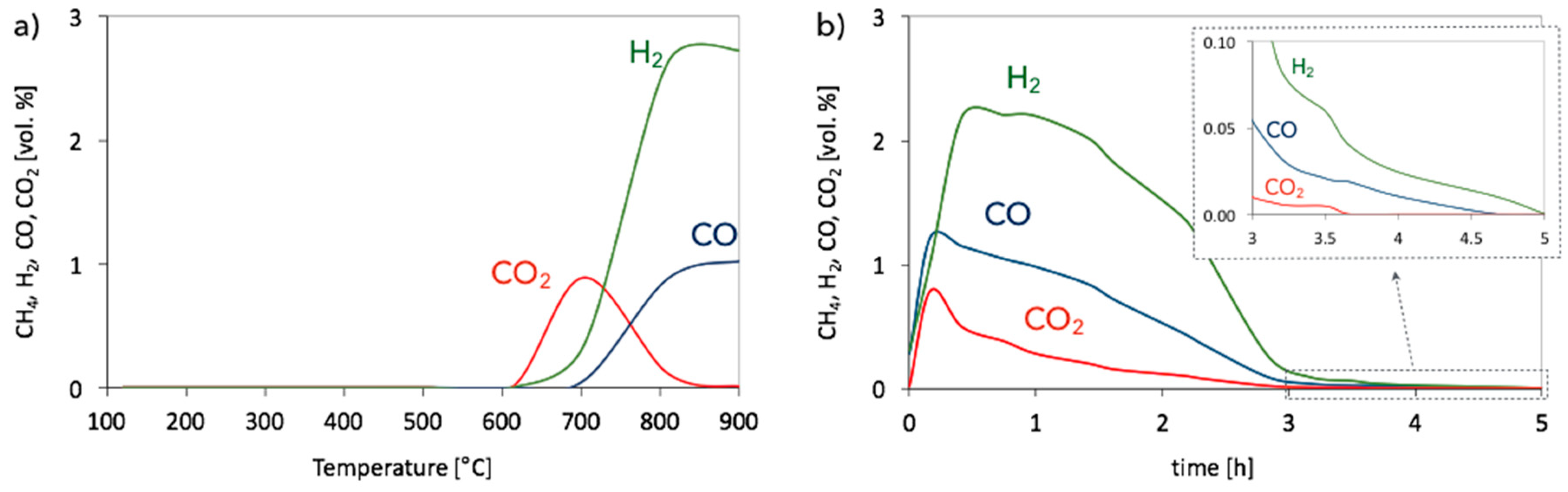
3. Results and Discussion
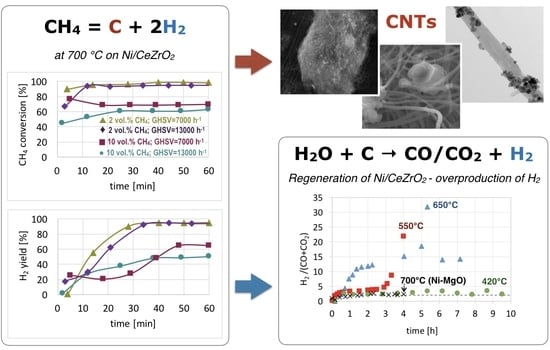
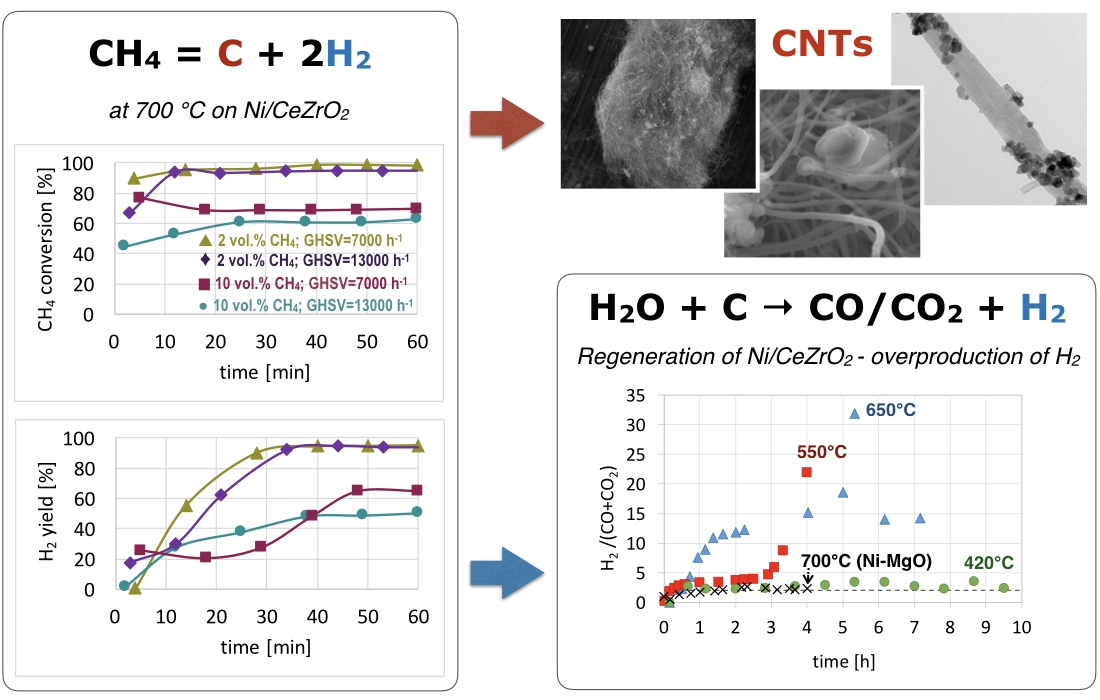
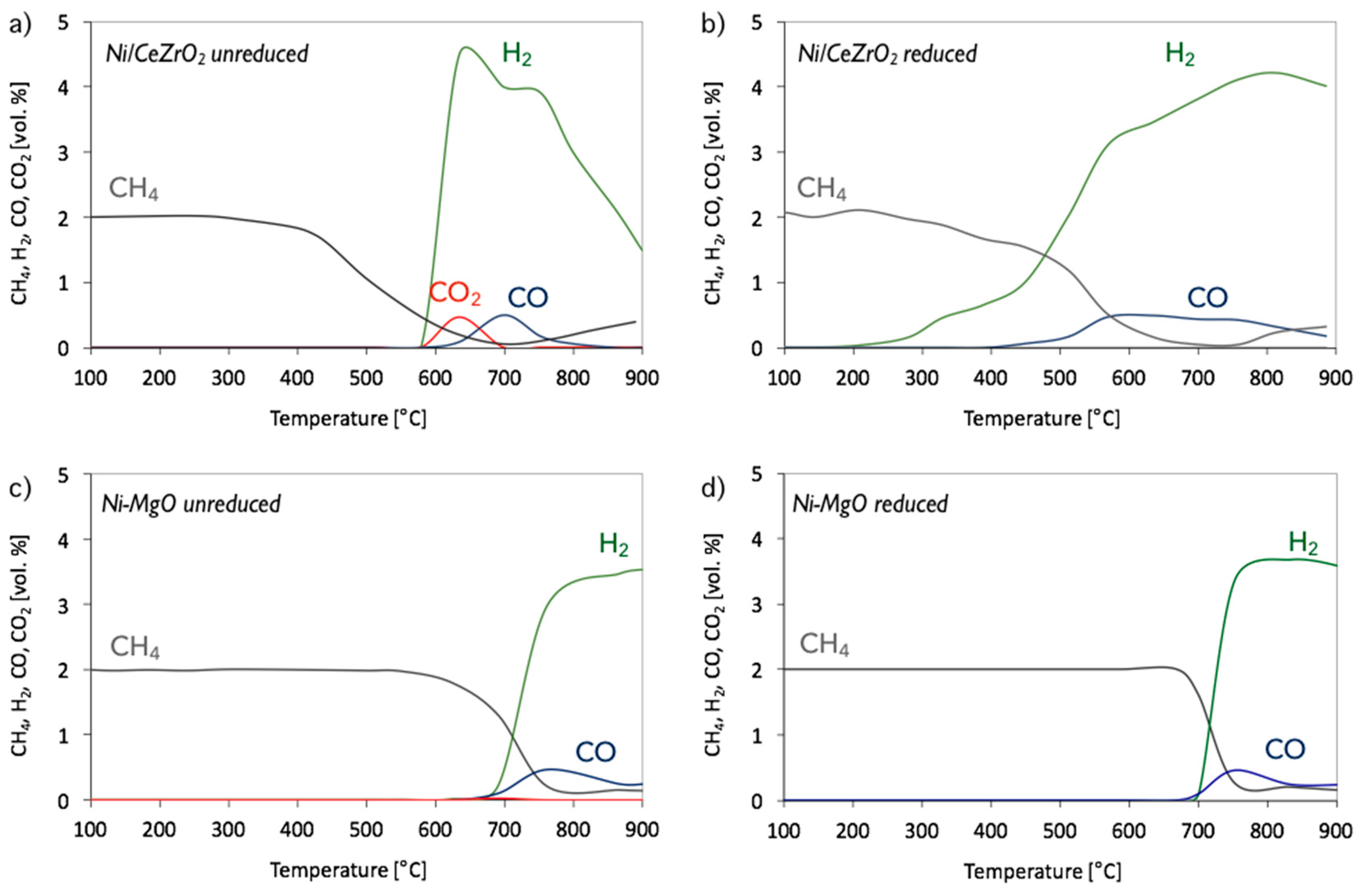
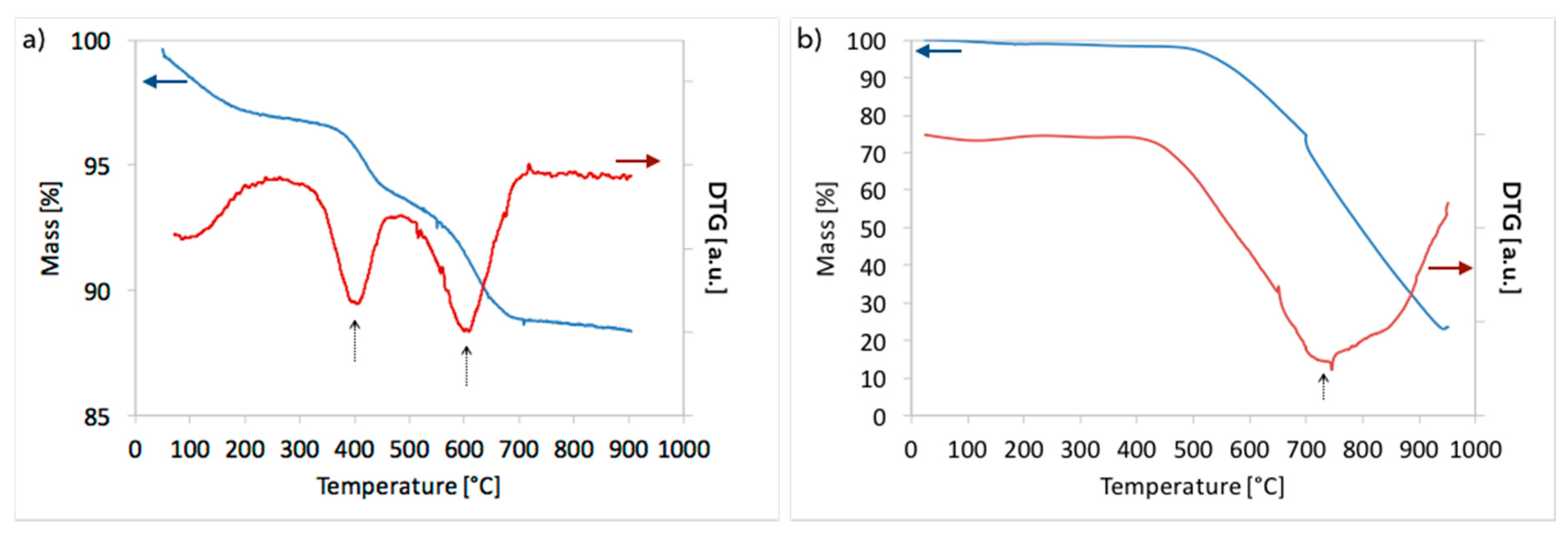
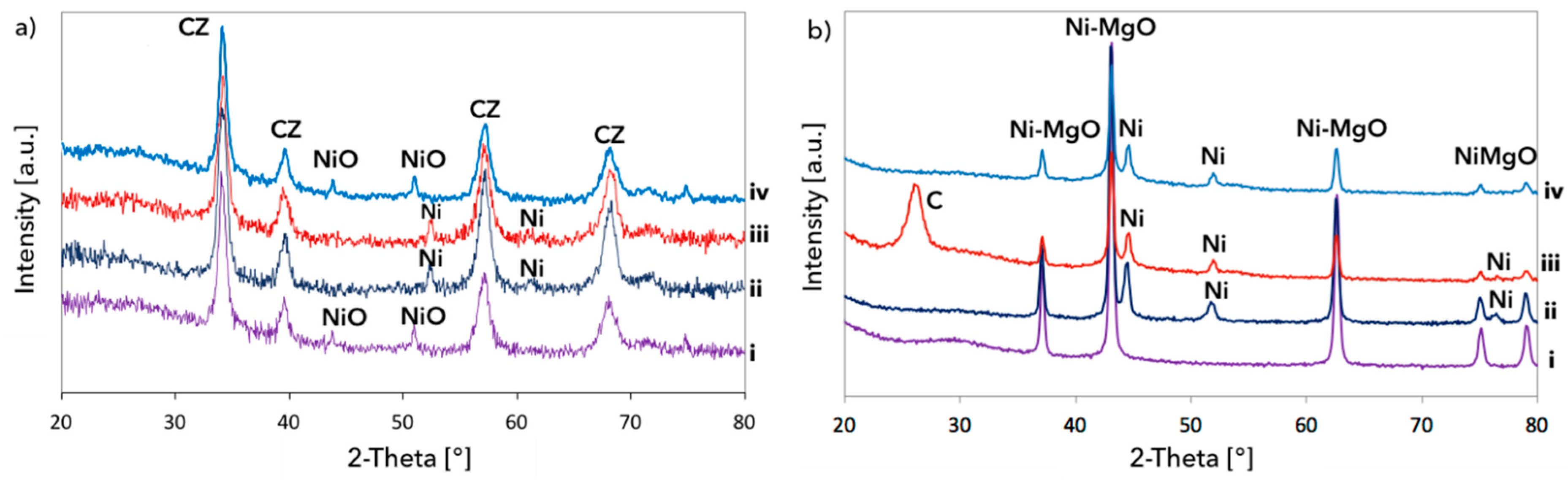
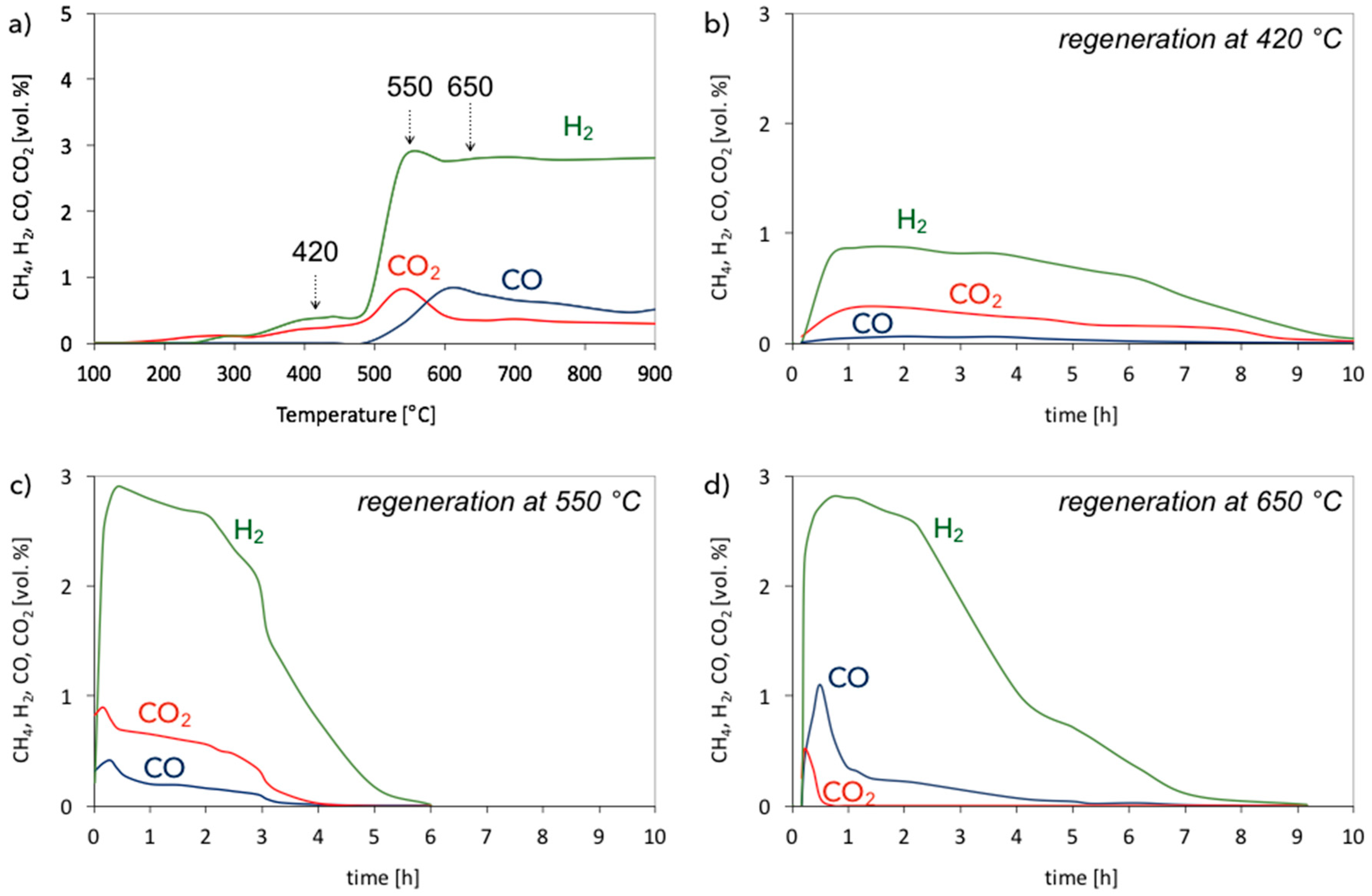
3.1. CH4 Decomposition to CNTs and H2
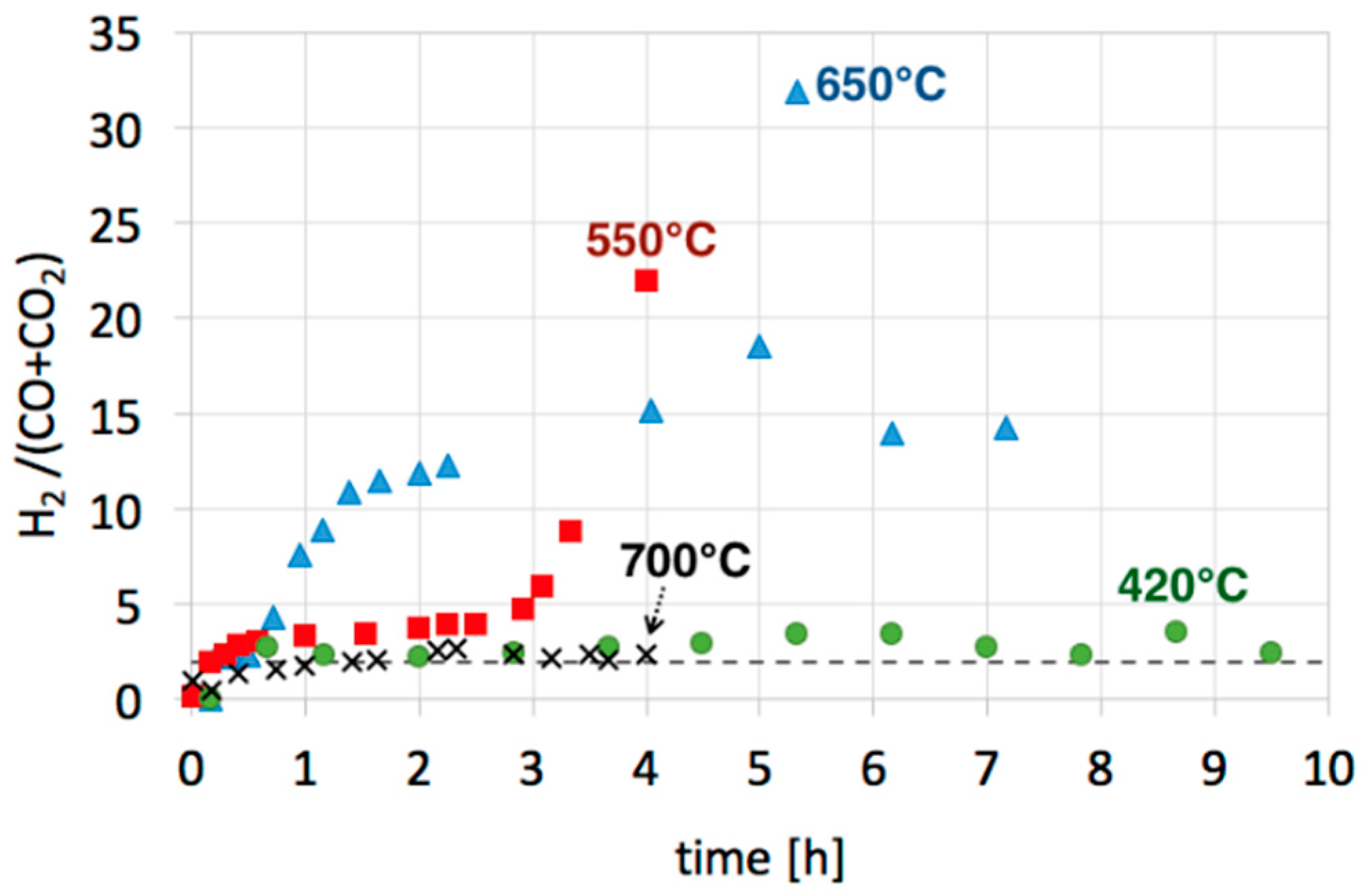
3.2. Catalyst Regeneration with H2O
4. Conclusions
Supplementary Materials
Funding
Acknowledgments
Conflicts of Interest
References
- Serrano, D.P.; Botas, J.A.; Guil-Lopez, R. H2 Production from Methane Pyrolysis Over Commercial Carbon Catalysts: Kinetic and Deactivation Study. Int. J. Hydrog. Energy 2009, 34, 4488–4494. [Google Scholar] [CrossRef]
- Zarębska, K.; Pernak-Misko, K. Zgazowanie węgla—perspektywa dla gospodarki wodorowej. Gosp. Sur. Min. 2007, 23, 243–255. [Google Scholar]
- Molenda, J. Fundamentalne znaczenie badań naukowych dla rozwoju gospodarki wodorowej. Polityka Energetyczna 2008, 11, 61–68. [Google Scholar]
- Rajeshwar, K. Hydrogen generation at irradiated oxide semiconductor–solution interfaces. J. Appl. Electrochem. 2007, 37, 765–787. [Google Scholar] [CrossRef]
- Gallo, A.; Marelli, M.; Psaro, R.; Gombac, V.; Montini, T.; Fornasiero, P.; Pievo, R.; Dal Santo, V. Bimetallic Au–Pt/TiO2 photocatalysts active under UV-A and simulated sunlight for H2 production from ethanol. Green Chem. 2012, 14, 330–333. [Google Scholar] [CrossRef]
- Laguna-Bercero, M.A. Recent advances in high temperature electrolysis using solid oxide fuel cells: A review. J. Power Sources 2012, 203, 4–16. [Google Scholar] [CrossRef]
- Millet, P.; Ngameni, R.; Grigoriev, S.A.; Mbemba, N.; Brisset, F.; Ranjbari, A.; Etievant, C. PEM water electrolyzers: From electrocatalysis to stack development. Int. J. Hydrog. Energy 2010, 35, 5043–5052. [Google Scholar] [CrossRef]
- Huczko, A. Nanorurki Węglowe. Czarne Diamenty XXI Wieku. BEL Studio, Poland. 2004. Available online: http://polona.pl/item/4535131 (accessed on 28 April 2018).
- Thompson, B.C.; Moulton, S.A.; Gilmore, K.J.; Higgins, M.J.; Whitten, P.G.; Wallach, G.G. Carbon nanotube biogels. Carbon 2009, 47, 1282–1291. [Google Scholar] [CrossRef]
- Kokai, F.; Takahashi, K.; Kasuya, D.; Ichihashi, T.; Yudasaka, M. Synthesis of single-wall carbon nanotubes by millisecond-pulsed CO2 laser vaporization at room temperature. Chem. Phys. Lett. 2000, 332, 449–454. [Google Scholar] [CrossRef]
- Zhao, X.; Ohkohchi, M.; Inoue, S.; Suzuki, T.; Kadoya, T.I.; Ando, Y. Large-scale purification of single-wall carbon nanotubes prepared by electric arc discharge. Diam. Relat. Mater. 2006, 15, 1098–1102. [Google Scholar] [CrossRef]
- Bacs, R.R.; Laurent, C.; Peigney, A.; Bacsa, W.S.; Vaugien, T.; Rousset, A. High specific surface area carbon nanotubes from catalytic chemical vapor deposition process. Chem. Phys. Lett. 2000, 323, 566–571. [Google Scholar] [CrossRef]
- Tran, K.Y.; Heinrichs, B.; Colomer, J.F.; Pirard, J.P.; Lambert, S. Carbon nanotubes synthesis by the ethylene chemical catalytic vapour deposition (CCVD) process on Fe, Co, and Fe–Co/Al2O3 sol–gel catalysts. Appl. Catal. A Gen. 2007, 318, 63–69. [Google Scholar] [CrossRef]
- Zhu., J.; Yudasaka, M.; Iijima, S. A catalytic chemical vapor deposition synthesis of double-walled carbon nanotubes over metal catalysts supported on a mesoporous material. Chem. Phys. Lett. 2003, 380, 496–502. [Google Scholar] [CrossRef]
- Vahlas, C.; Caussat, B.G.; Serp, P.; Angelopoulos, G.N. Principles and Applications of CVD Powder Technology. Mater. Sci. Eng. R Rep. 2006, 53, 1–72. [Google Scholar] [CrossRef]
- Nagaraju, N.; Fonseca, A.; Konya, Z.; Nagy, J.B. Alumina and silica supported metal catalysts for the production of carbon nanotubes. J. Mol. Cat. A Chem. 2002, 181, 57–62. [Google Scholar] [CrossRef]
- Qingwen, L.; Hao, Y.; Yan, C.; Jin, Z.; Zhongfan, L. A scalable CVD synthesis of high-purity single-walled carbon nanotubes with porous MgO as support material. J. Mater. Chem. 2002, 12, 1179–1183. [Google Scholar] [CrossRef]
- Park, J.B.; Choi, G.S.; Cho, Y.S.; Hong, S.Y.; Kim, D.; Choi, S.Y.; Lee, J.H.; Cho, K.I. Characterization of Fe-catalyzed carbon nanotubes grown by thermal chemical vapor deposition. J. Cryst. Growth 2002, 244, 211–217. [Google Scholar] [CrossRef]
- Emmenegger, C.; Bonard, J.M.; Mauron, P.; Sudan, P.; Lepora, A.; Grobety, B.; Zuttel, A.; Schlapbach, L. Synthesis of carbon nanotubes over Fe catalyst on aluminium and suggested growth mechanism. Carbon 2003, 41, 539–547. [Google Scholar] [CrossRef]
- Pinheiro, J.P.; Schouler, M.C.; Gadelle, P. Nanotubes and nanofilaments from carbon monoxide disproportionation over Co/MgO catalysts: I. Growth versus catalyst state. Carbon 2003, 41, 2949–2959. [Google Scholar] [CrossRef]
- Takenaka, S.; Kobayashi, S.; Ogihara, H.; Otsuka, K. Catalytic deuteration of cyclohexanone and allied reactions over platinum metals. J. Catal. 2003, 217, 79–87. [Google Scholar] [CrossRef]
- Flahaut, E.; Peigney, A.; Laurent, C.; Rousset, A. Synthesis of single-walled carbon nanotube–Co–MgO composite powders and extraction of the nanotubes. J. Mater. Chem. 2000, 10, 249–252. [Google Scholar] [CrossRef]
- Li, Y.L.; Kinloch, I.A.; Shaffer, M.S.P.; Geng, J.; Johnson, B.; Windle, A.H. Synthesis of single-walled carbon nanotubes by a fluidized-bed method. Chem. Phys. Lett. 2004, 384, 98–102. [Google Scholar] [CrossRef]
- Hao, Y.; Qunfeng, Z.; Fei, W.; Weizhong, Q.; Guohua, L. Agglomerated CNTs synthesized in a fluidized bed reactor: Agglomerate structure and formation mechanism. Carbon 2003, 41, 2855–2863. [Google Scholar] [CrossRef]
- Maghsoodi, S.; Khodadadi, A.; Mortazavi, Y. A novel continuous process for synthesis of carbon nanotubes using iron floating catalyst and MgO particles for CVD of methane in a fluidized bed reactor. Appl. Surf. Sci. 2010, 256, 2769–2774. [Google Scholar] [CrossRef]
- Wang, Y.; Wei, F.; Luo, G.; Yu, H.; Gu, G. The large-scale production of carbon nanotubes in a nano-agglomerate fluidized-bed reactor. Chem. Phys. Lett. 2002, 364, 568–572. [Google Scholar] [CrossRef]
- Corrias, M.; Caussat, B.; Ayral, A.; Durand, J.; Kihn, Y.; Alck, P.; Serp, P. Carbon nanotubes produced by fluidized bed catalytic CVD: First approach of the process. Chem. Eng. Sci. 2003, 58, 4475–4482. [Google Scholar] [CrossRef]
- Hsieh, C.T.; Lin, Y.T.; Chen, W.Y.; Wei, J.L. Parameter setting on growth of carbon nanotubes over transition metal/alumina catalysts in a fluidized bed reactor. Powder Technol. 2009, 192, 16–22. [Google Scholar] [CrossRef]
- Weizhong, Q.; Tang, L.; Zhanwen, W.; Fei, W.; Zhifei, L.; Guohua, L.; Yongdan, L. Production of hydrogen and carbon nanotubes from methane decomposition in a two-stage fluidized bed reactor. Appl. Catal. A Gen. 2004, 260, 223–228. [Google Scholar] [CrossRef]
- Wang, Y.; Wei, F.; Gu, G.; Yu, H. Agglomerated carbon nanotubes and its mass production in a fluidized-bed reactor. Phys. B 2002, 323, 327–329. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhao, M.Q.; Huang, J.Q.; Liu, Y.; Wang, Y.; Qian, W.Z.; Wei, F. Vertically aligned carbon nanotube arrays grown on a lamellar catalyst by fluidized bed catalytic chemical vapor deposition. Carbon 2009, 47, 2600–2610. [Google Scholar] [CrossRef]
- See, C.H.; Dunens, O.M.; MacKenzie, K.J.; Harris, A.T. Process Parameter Interaction Effects during Carbon Nanotube Synthesis in Fluidized Beds. Ind. Eng. Chem. Res. 2008, 47, 7686–7692. [Google Scholar] [CrossRef]
- Son, S.Y.; Lee, Y.; Won, S.; Lee, D.H.; Kim, S.D.; Sung, S.W. High-Quality Multiwalled Carbon Nanotubes from Catalytic Decomposition of Carboneous Materials in Gas–Solid Fluidized Beds. Ind. Eng. Chem. Res. 2008, 47, 2166–2175. [Google Scholar] [CrossRef]
- Liu, X.B.; Sun, H.; Chen, Y.; Lau, R.; Yang, Y.H. Preparation of large particle MCM-41 and investigation on its fluidization behavior and application in single-walled carbon nanotube production in a fluidized-bed reactor. Chem. Eng. J. 2008, 142, 331–336. [Google Scholar] [CrossRef]
- Global Carbon Nanotubes Market—Industry Beckons. Available online: http://www.nanowerk.com/spotlight/spotid=23118.php (accessed on 28 April 2018).
- Li, Y.; Fu, Q.; Flytzani-Stephanopoulos, M. Low-temperature water-gas shift reaction over Cu- and Ni-loaded cerium oxide catalysts. Appl. Catal. B Environ. 2000, 27, 179–191. [Google Scholar] [CrossRef]
- Diaz, E.; de Rias, B.; Lopez-Fonsees, R.; Ordonez, S. Characterization of ceria–zirconia mixed oxides as catalysts for the combustion of volatile organic compounds using inverse gas chromatography. J. Chromatogr. A 2006, 1116, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.R.; Kim, T.; Zhou, G.; Fornasiero, P.; Gorte, R.J. Evidence for Entropy Effects in the Reduction of Ceria–Zirconia Solutions. Chem. Mater. 2006, 18, 5363–5369. [Google Scholar] [CrossRef]
- Łamacz, A.; Krztoń, A.; Djéga-Mariadassou, G. Steam reforming of model gasification tars compounds on nickel based ceria-zirconia catalysts. Catal. Today 2011, 176, 347–351. [Google Scholar] [CrossRef]
- Łamacz, A.; Krztoń, A.; Musi, A.; Da Costa, P. Reforming of Model Gasification Tar Compounds. Catal. Lett. 2009, 128, 40–48. [Google Scholar] [CrossRef]
- Łamacz, A.; Krztoń, A. Hydrogen production by catalytic decomposition of selected hydrocarbons and H2O dissociation over CeZrO2 and Ni/CeZrO2. Int. J. Hydrog. Energy 2013, 38, 8772–8782. [Google Scholar] [CrossRef]
- Łamacz, A.; Matus, K.; Liszka, B.; Silvestre-Albero, J.; Lafjah, M.; Dintzer, T.; Janowska, I. The impact of synthesis method of CNT supported CeZrO2 and Ni-CeZrO2 on catalytic activity in WGS reaction. Catal. Today 2018, 301, 172–182. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | CH4 (vol.%) | GHSV (h−1) | △m (%) | C/Ni (mol/mol) |
|---|---|---|---|---|
| unreduced | 2 | 7000 | 2.1 | 1.1 |
| unreduced | 2 | 13,000 | 2.5 | 1.3 |
| unreduced | 10 | 7000 | 8.3 | 4.5 |
| unreduced | 10 | 13,000 | 7.9 | 4.2 |
| pre-reduced | 2 | 7000 | 3.6 | 1.9 |
| pre-reduced | 2 | 13,000 | 3.1 | 1.7 |
| pre-reduced | 10 | 7000 | 9.0 | 4.8 |
| pre-reduced | 10 | 13,000 | 8.8 | 4.7 |
| Ni/CeZrO2: | SSA (m2/g) | Pore Volume (cc/g) | Pore Size (nm) |
|---|---|---|---|
| fresh | 78 | 0.25 | 13.2 |
| pre-reduced in H2 | 74 | 0.25 | 13.1 |
| after CH4 decomposition * | 83 | 0.26 | 11.4 |
| regenerated in H2O ** | 95 | 0.28 | 13.1 |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Łamacz, A. CNT and H2 Production During CH4 Decomposition over Ni/CeZrO2. I. A Mechanistic Study. ChemEngineering 2019, 3, 26. https://doi.org/10.3390/chemengineering3010026
Łamacz A. CNT and H2 Production During CH4 Decomposition over Ni/CeZrO2. I. A Mechanistic Study. ChemEngineering. 2019; 3(1):26. https://doi.org/10.3390/chemengineering3010026
Chicago/Turabian StyleŁamacz, Agata. 2019. "CNT and H2 Production During CH4 Decomposition over Ni/CeZrO2. I. A Mechanistic Study" ChemEngineering 3, no. 1: 26. https://doi.org/10.3390/chemengineering3010026
APA StyleŁamacz, A. (2019). CNT and H2 Production During CH4 Decomposition over Ni/CeZrO2. I. A Mechanistic Study. ChemEngineering, 3(1), 26. https://doi.org/10.3390/chemengineering3010026