3.1. Carbon Deposition during Decomposition of CH4
The decomposition of CH4 is a few step dehydrogenation which requires the presence of reduced metal sites. Those Ni0 active sites were formed on both Ni/CeZrO2 and Ni/MgO catalysts at the beginning of each experiment as a result of the in-situ reduction by the flowing CH4. The mechanisms of CH4 decomposition on Ni/CeZrO2 and Ni/MgO were discussed in detail in the first part of this work.
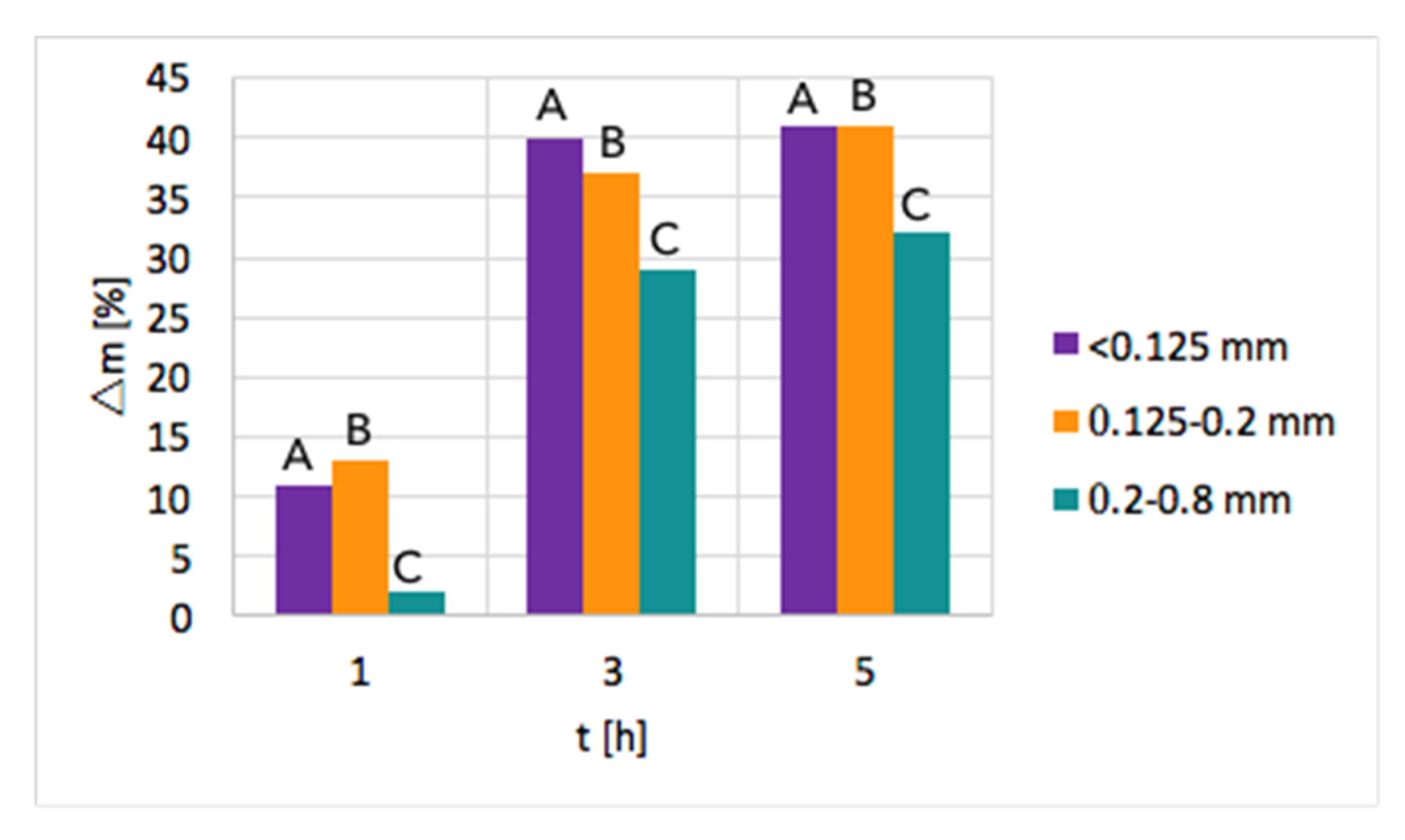
Ni/CeZrO2: The tests of CH4 decomposition were first carried out over three fractions of Ni/CeZrO
2, i.e., <0.125 mm, 0.125–0.2 mm, and 0.2–0.8 mm. The influence of time and particle size on the increase of sample mass due to carbon deposition is presented in
Figure 2. As can be seen, a significant mass increase was observed when the test duration was increased from 1–3 h. Further extension of the time of CH
4 decomposition resulted in a smaller mass gain. Moreover, the highest mass increase of ca. 40% was observed for fractions A (<0.125 mm) and B (0.125–0.2 mm) that have smaller particles; thus, the contact area with CH
4 molecules is the biggest. The lowest carbon deposition occurred for fraction C (0.2–0.8 mm).
After the tests, the catalyst bed was cooled down to 200 °C in flowing Ar to avoid oxygen entering the reactor, which at temperatures above 300 °C would cause oxidation of carbon species. After reaching 200 °C, the reactor was unsealed so that some air could enter and passivate the Ni3C that was formed on the catalyst during CH4 decomposition. Ignoring the passivation step resulted in a strongly exothermic oxidation of Ni3C during catalyst tipping at room temperature. Passivation worked well for fractions B and C, whereas it was challenging in the case of fraction A, whose fine grains allowed the highest carbon deposition. Lasting even 24 h, attempts to passivate the carbide in fraction A were unsuccessful, and the catalyst burned in contact with air during tipping. Owing to these problems and the low carbon deposition on fraction C, the next tests of methane decomposition were carried out over fraction B of Ni/CeZrO2, i.e., particles of 0.125–0.2 mm.
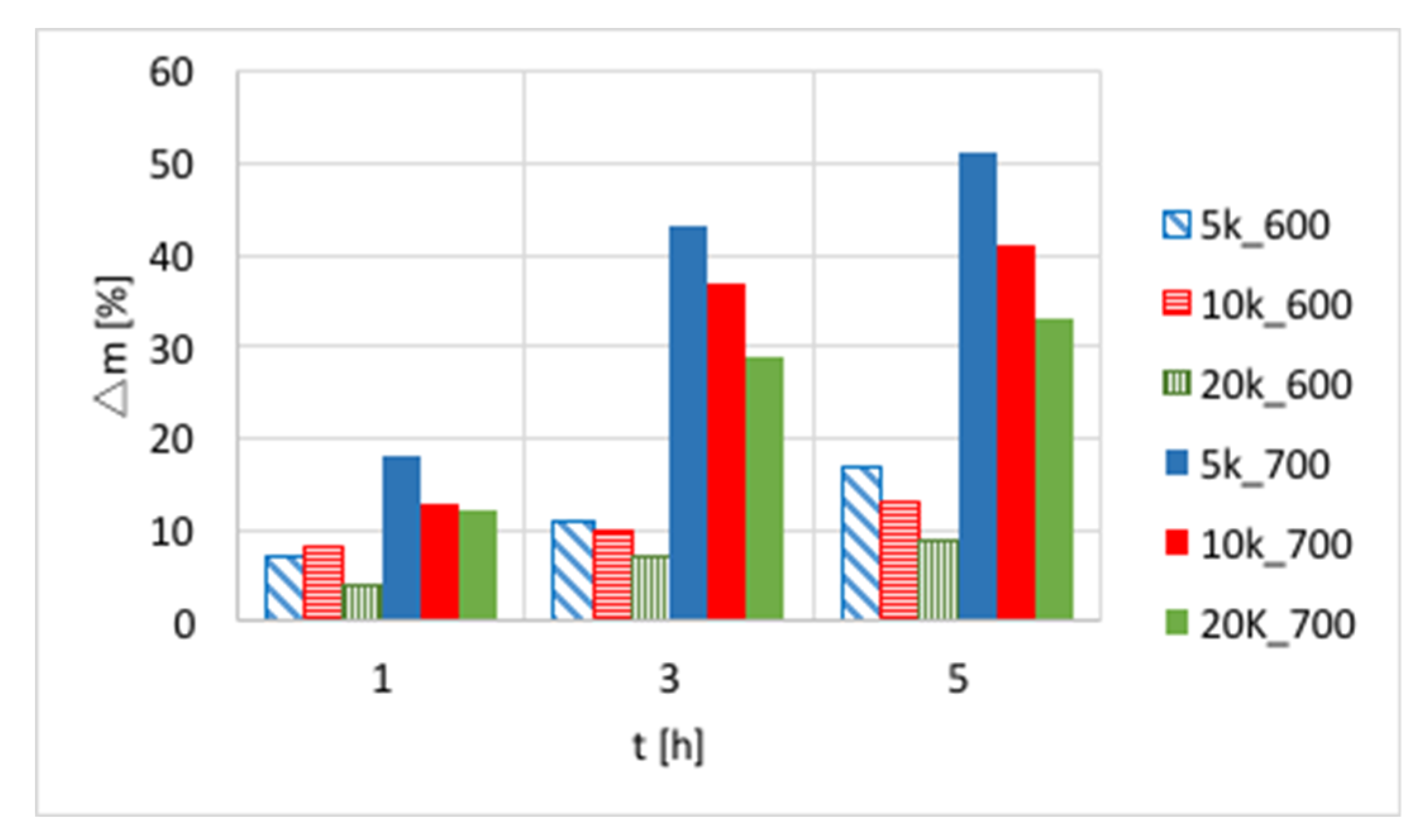
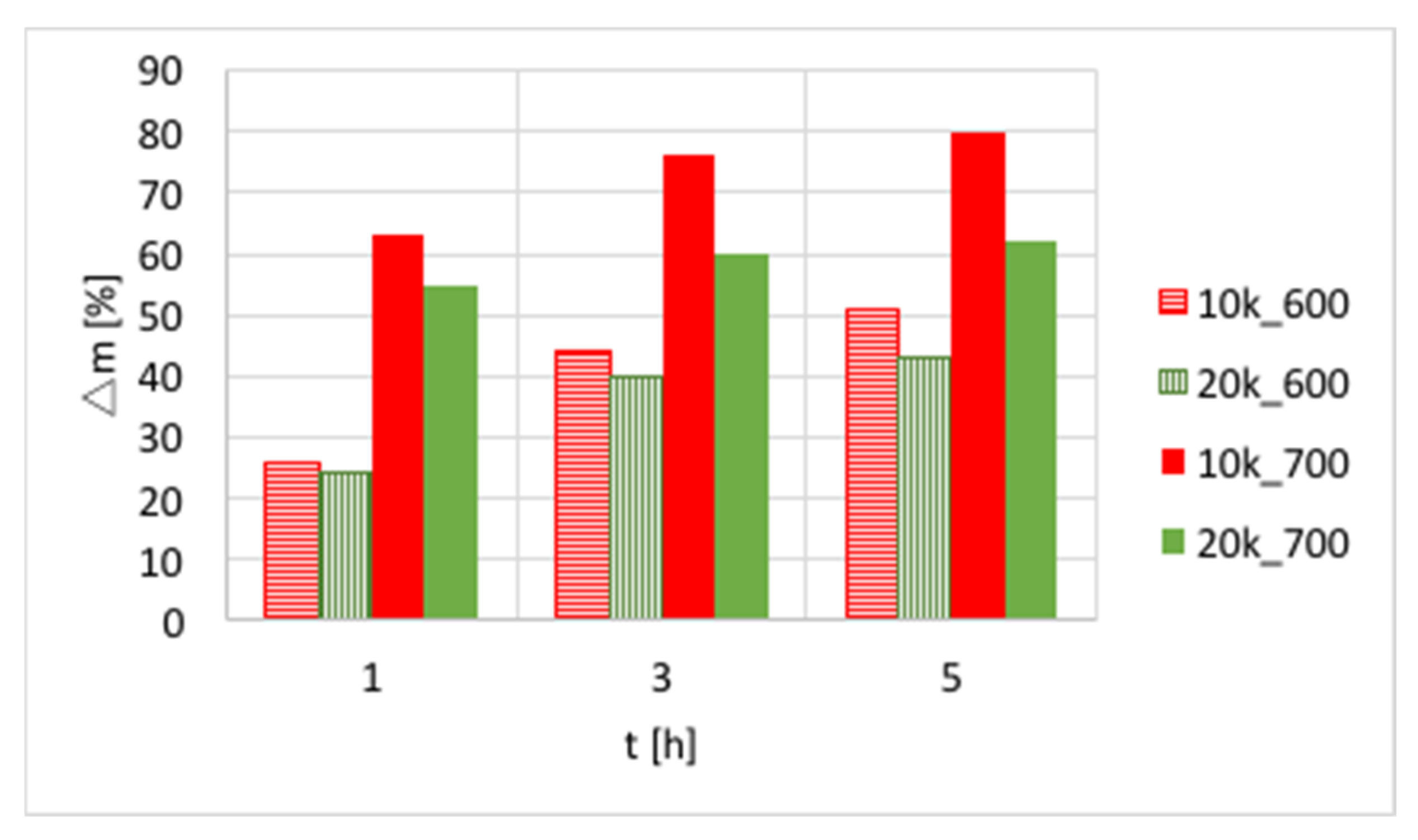
The influences of temperature (600 or 700 °C), test duration (1, 3, and 5 h) and GHSV (5000, 10,000 and 20,000 h
−1) on the increase of catalyst mass are presented in
Figure 3 and in
Table S1. Again, a significant increase of carbon deposition was noticed after test prolongation from 1–3 h, and a less important change was observed when it was extended to 5 h. The amount of carbon deposit on Ni/CeZrO
2 at 600 °C was significantly lower than at 700 °C. Moreover, the highest carbon deposition (51%) was observed for low GHSV values. However, low values of GHSV favor the formation of coke.
After the tests, the carbon deposit was separated from the catalyst. Unfortunately, complete separation of carbon deposit from the catalyst is impossible because there will always remain coke on the catalyst surface, whereas removed carbon fraction will contain small particles of the catalyst. The growth of CNTs on Ni particles often causes metal detachment from the support.
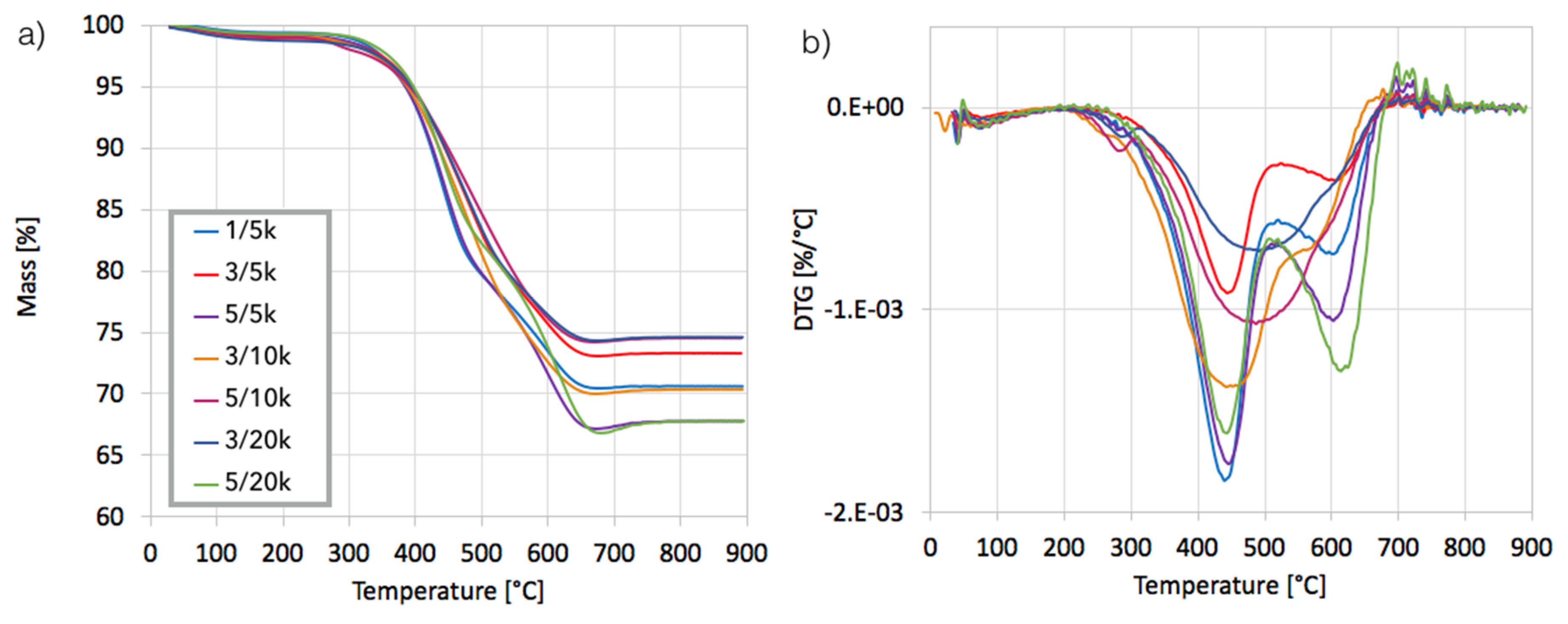
After separation, the catalyst samples were subjected to TGA/DTG in order to determine the amount and the nature of remaining carbon deposit. According to TG plots (
Figure 4a), Ni/CeZrO
2 still contained from 24–32% of carbon, both the amorphous, which was oxidized with a maximum at 410 °C, and the structural, which was oxidized with a maximum at 580 °C (
Figure 4b). It was found that the carbon deposit that was still present in the catalyst sample was composed in levels of 61–85% from the amorphous carbon and in 15–39% from the structural carbon. There was no relationship between the parameters of the CH
4 decomposition and the amount of particular carbon type in the sample.
The Ni/CeZrO
2 before and after CH
4 decomposition was characterized using SEM/EDS. SEM pictures of spent catalyst are presented in
Figure 5, whereas EDS analyses and mapping for Ni, Ce, Zr, and O for fresh and spent catalyst samples are shown in
Figures S2–S6.
Microscopic observations of spent Ni/CeZrO
2 catalyst proved the presence of randomly oriented and curved CNTs (
Figure 5a). Presented in
Figure 6, CNTs had outer/inner diameters of 64/6.3 (a) and 47/4.4 nm (b). The occurrence of CNTs in the catalyst sample after the separation process has also been noticed (
Figure 5b). During formation, CNTs blew up the catalyst grains from the inside and broke them into smaller particles. Some of the catalyst particles, especially those that had attached to CNTs, percolate to the carbon fraction, which can be observed in
Figure 5c,d. The EDS analysis (
Figure S5) proved the presence of Ce and Zr in that sample, whereas mapping showed a good distribution of Ni, Ce, and Zr (
Figure S6). Catalyst particles were not only covered loosely with CNTs or dispersed freely over CNTs, but as was observed by TEM (
Figure 6), some of them were also strongly attached to external CNT walls (similarly to Ni/CeZrO
2@CNT hybrid material obtained by deposition of Ni/CeZrO
2 on functionalized CNTs [
27]). The diameters of catalyst particles visible in
Figure 6 ranged from 7–19 nm. The process of CH
4 decomposition in a fluidized bed reactor leads to formation of such a hybrid material that can be further used as a catalyst, e.g., in WGS [
27] or DRM (
Figure S1) reactions. The content of Ni/CeZrO
2 in the carbon fraction was about 35 wt.%, which was determined by burning the sample in air at 800 °C. From SEM, TEM, and TGA observations, it is obvious, that it is impossible to obtain a pure CNT fraction as a result of CH
4 decomposition on Ni/CeZrO
2. However, the fraction containing CNTs and the catalyst can be subjected to further processing with acids in order to remove catalyst particles, it or can be used as a catalyst itself, e.g., in the WGS [
27] or DRM reaction.
Ni-MgO: Methane decomposition in a fluidized bed reactor was also performed over Ni-MgO (0.125–0.2 mm fraction). The increase of catalyst mass owing to carbon deposition at different T and GHSV is presented in
Figure 7 and
Table S2. Significantly higher carbon deposition was observed at 700 °C, whereas an increase in GHSV negatively influenced carbon build-up. Similar to Ni/CeZrO
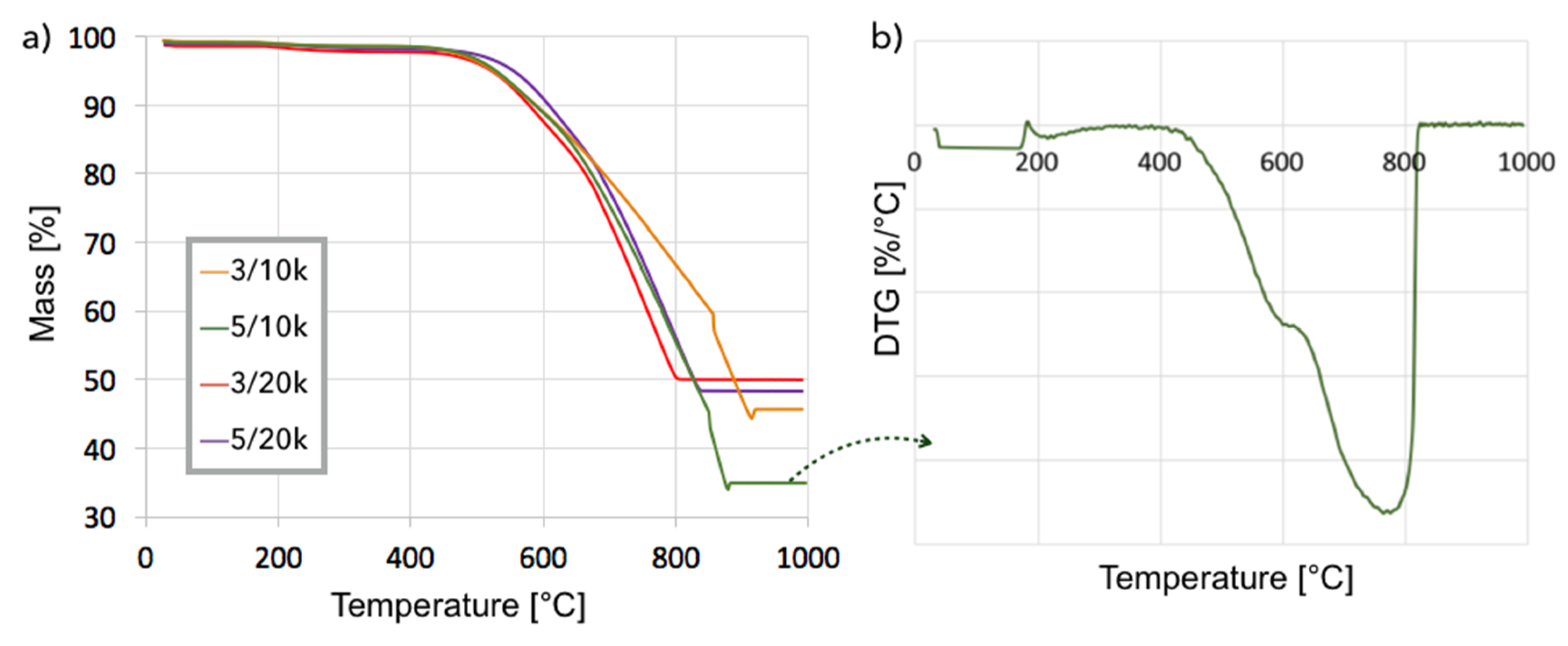
2, the fastest increase in carbon deposition was observed during the first 3 h of the test. The TGA analysis for samples obtained at 700 °C (
Figure 8a) showed that samples contained from 52–65% carbon, whereas the DTG plot for one of the samples (test: 5 h, GHSV = 10,000 h
−1) indicated that it contained mostly structural carbon (negative peak at 770 °C). After the separation of carbon deposits, the Ni-MgO was also subjected to TGA/DTG (
Figure 9), which revealed the presence of ca. 10% of carbon deposits, composed in 68% of the amorphous and in 32% of structural carbon. The separation process was significantly easier for Ni-MgO than for the Ni/CeZrO
2 catalyst.
3.2. H2 Production during Decomposition of CH4
Hydrogen formation during CH
4 decomposition in a fluidized bed reactor over Ni/CeZrO
2 (three granulations—fractions A, B, and C) and Ni-MgO catalysts was studied during tests carried out at 700 °C for 5 h and at GHSV = 10,000 h
−1. Neither CO
2 nor CO was detected in the outlet gas during tests in the fluidized bed reactor which indicates that the reduction of catalysts with CH
4 was very fast. Conversely, the formation of CO
2 and CO was observed during tests carried out in a micro-reactor [
28]. The composition of the gas at the outlet of fluidized bed reactor during tests of CH
4 decomposition over Ni/CeZrO
2 and Ni/MgO catalysts is presented in
Figure S7, whereas CH
4 conversions and H
2 yields are displayed in
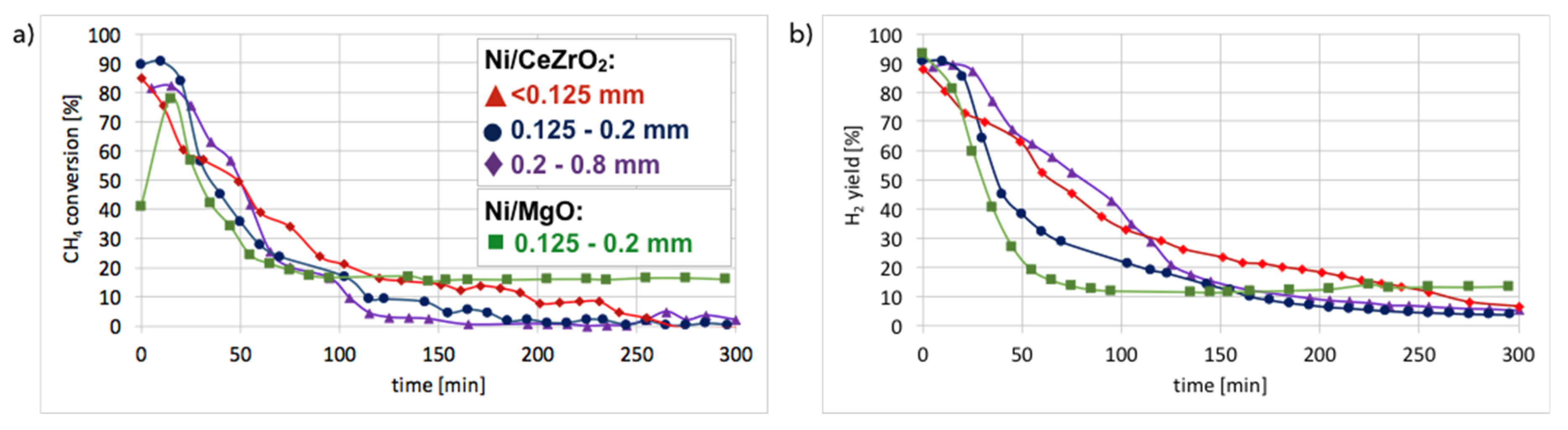
Figure 10. It can be observed that in all cases, CH
4 conversion and H
2 yield were the highest in the first 20 minutes of test and then decreased rapidly. The best performance was noticed for fraction A (particle size < 0.125 mm), for which the highest carbon deposition was also observed (
Figure 2).
The lowest CH
4 conversion and H
2 yield occurred for fraction C (0.2–0.8 mm), which also showed the lowest mass increase after the test. CH
4 conversion was higher for Ni-MgO, but the H
2 yield was higher for Ni/CeZrO
2. However, Ni-MgO showed a more stable ratio of H
2 yield to CH
4 conversion (YH
2/CCH
4), which oscillated around 0.8 throughout the experiment (
Figure S7). For Ni/CeZrO
2, this ratio was stable during the first 60 minutes and then varied depending on the catalyst granulation. The lower stability of Ni/CeZrO
2 (compared to Ni-MgO) is caused by very good red-ox properties of ceria-zirconia and high oxygen mobility in its lattice. The most stable YH
2/CCH
4 ratio was observed for fraction A of Ni/CeZrO
2 (<0.125 mm), whereas for fraction C (0.2–0.8 mm) H
2 yield was much higher than CH
4 conversion. The stoichiometric YH
2/CCH
4 ratio for CH
4 decomposition is 2, whereas its value on the Ni/CeZrO
2 catalyst was sometimes significantly surpassed. High H
2 yield compared to CH
4 conversion could be caused by the accumulation of CH
4 or products of its partial dehydrogenation (CH
x) on the catalyst surface earlier during the experiment, followed by higher H
2 release to the gas phase after some time. It seems that the bigger the catalyst grains, the more CH
4 (or CH
x) was trapped in the first hour of the experiment. The YH
2/CCH
4 < 2 may be explained by CH
x formation on the catalyst surface; thus, the H
2 release to the gas phase was lower.
It was calculated that 3.94 dm
3 of CH
4 was consumed and 7.79 dm
3 of H
2 was produced during that test over Ni/CeZrO
2 (0.125–0.2 mm), so the H
2/CH
4 ratio was 1.97 (very close to the stoichiometric H
2/CH
4 ratio of 2). Slightly different CH
4 consumption and H
2 production were noticed for Ni-MgO (respectively, 4.33 and 7.35 dm
3). The Ni-MgO showed higher carbon deposition than Ni/CeZrO
2 and more important CH
4 consumption, but it also contained more Ni.
Table S3 shows the number of CH
4 moles consumed and the number of H
2 moles produced over 1 Ni mol. It can be observed that the performance of Ni/CeZrO
2 is significantly better than Ni-MgO.
3.3. H2 Production during Catalyst Regeneration with H2O
Ni/CeZrO2: The Ni/CeZrO
2 catalyst after separation of the carbon deposit was regenerated in flowing 4.15 vol.% H
2O/Ar in temperature-programmed conditions in a micro-reactor (discussed in the supporting file,
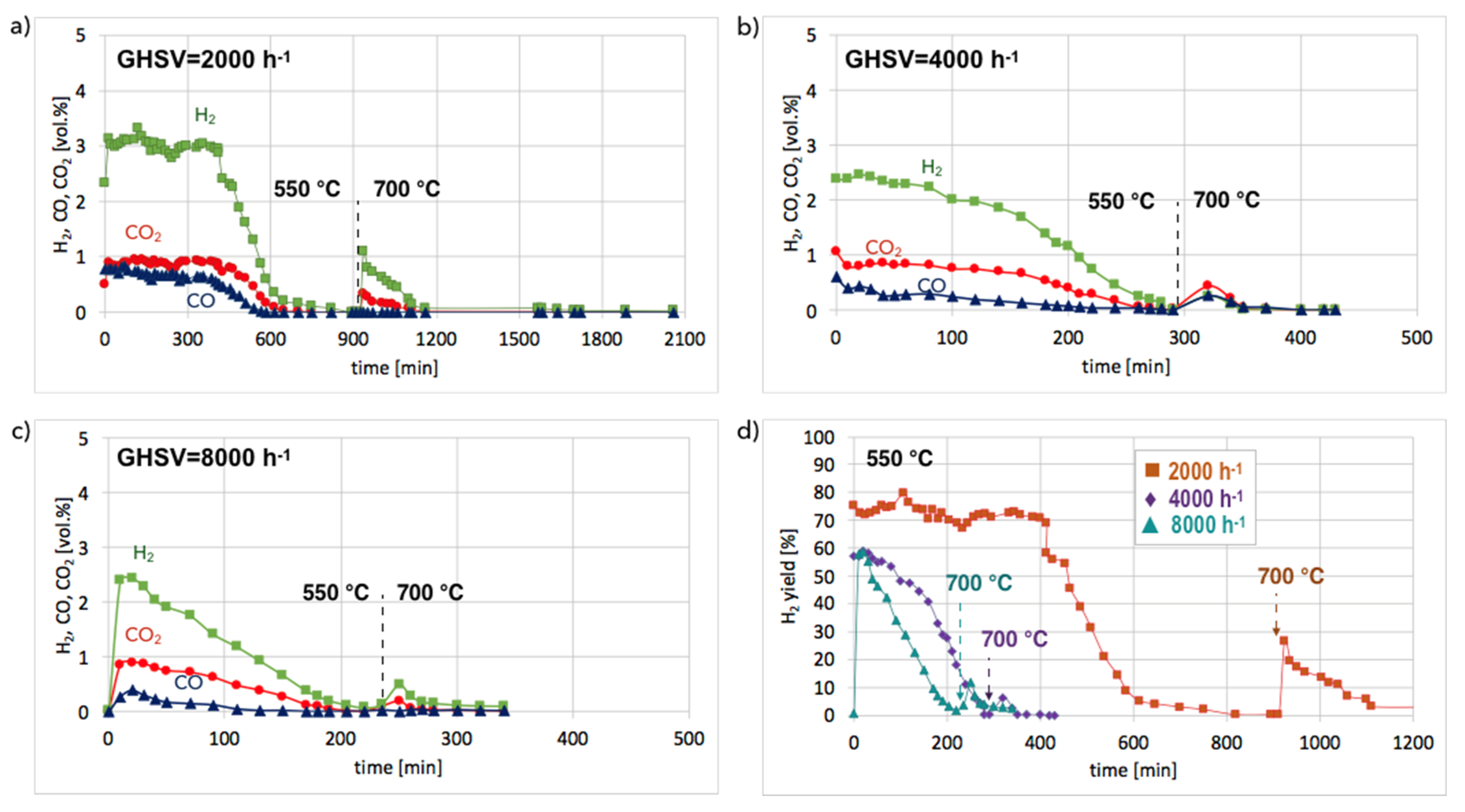
Figures S9 and S10) and in isothermal conditions in a fluidized bed reactor (
Figure 11). According to the results of tests carried out in temperature-programmed conditions, the lowest temperature for catalyst regeneration with the highest H
2 formation is ca. 550 °C. Hence, the Ni/CeZrO
2 after CH
4 decomposition (at 700 °C/3 h) and separation of carbon deposits was subjected to regeneration in flowing 4.15 vol.% H
2O/Ar at three GHSV values, i.e., 2000, 4000, and 8000 h
−1 at 550 °C. When CO and CO
2 stopped being formed, the temperature was increased from 550–700 °C in order to oxidize less reactive carbon deposits that could still be present on the catalyst surface. Regeneration of Ni/CeZrO
2 was the longest and occurred with the highest H
2 formation when the GHSV value was the lowest, i.e., 2000 h
−1. Formation of H
2, CO, and CO
2 were constant for the first 400 minutes (6 h 40 min) of the test. The increase in GHSV to 4000 and 8000 h
−1 resulted in shortening the time of H
2, CO, and CO
2 formation to ca. 1 hour and 20 minutes, respectively. It was also observed that CO formation decreased with the GHSV increase.
No CO formation was observed after 650, 260, and 205 minutes of the test run for GHSVs 2000, 4000, and 8000 h
−1, respectively, whereas H
2 was still being produced. It can be assumed that after the oxidation of carbon deposits (evidenced by CO and CO
2 desorption to the gas phase), H
2O dissociated on Ni
0 (oxidation of Ni to NiO) and oxygen vacancies in CeZrO
2 (oxidation of Ce
3+ to Ce
4+). At low GHSV values, those reactions occurred at a higher rate, and H
2 formation was longer than at GHSV = 4000 and 8000 h
−1. The increase of temperature to 700 °C resulted in an immediate increase in H
2, CO, and CO
2, which was due to oxidation of less reactive carbon deposits, i.e., CNTs, that were still present on the catalyst surface, as was proved by TGA. This phenomenon was more pronounced in the case of the test carried out at GHSV = 2000 h
−1, and the catalyst regeneration occurred for another 16 hours.
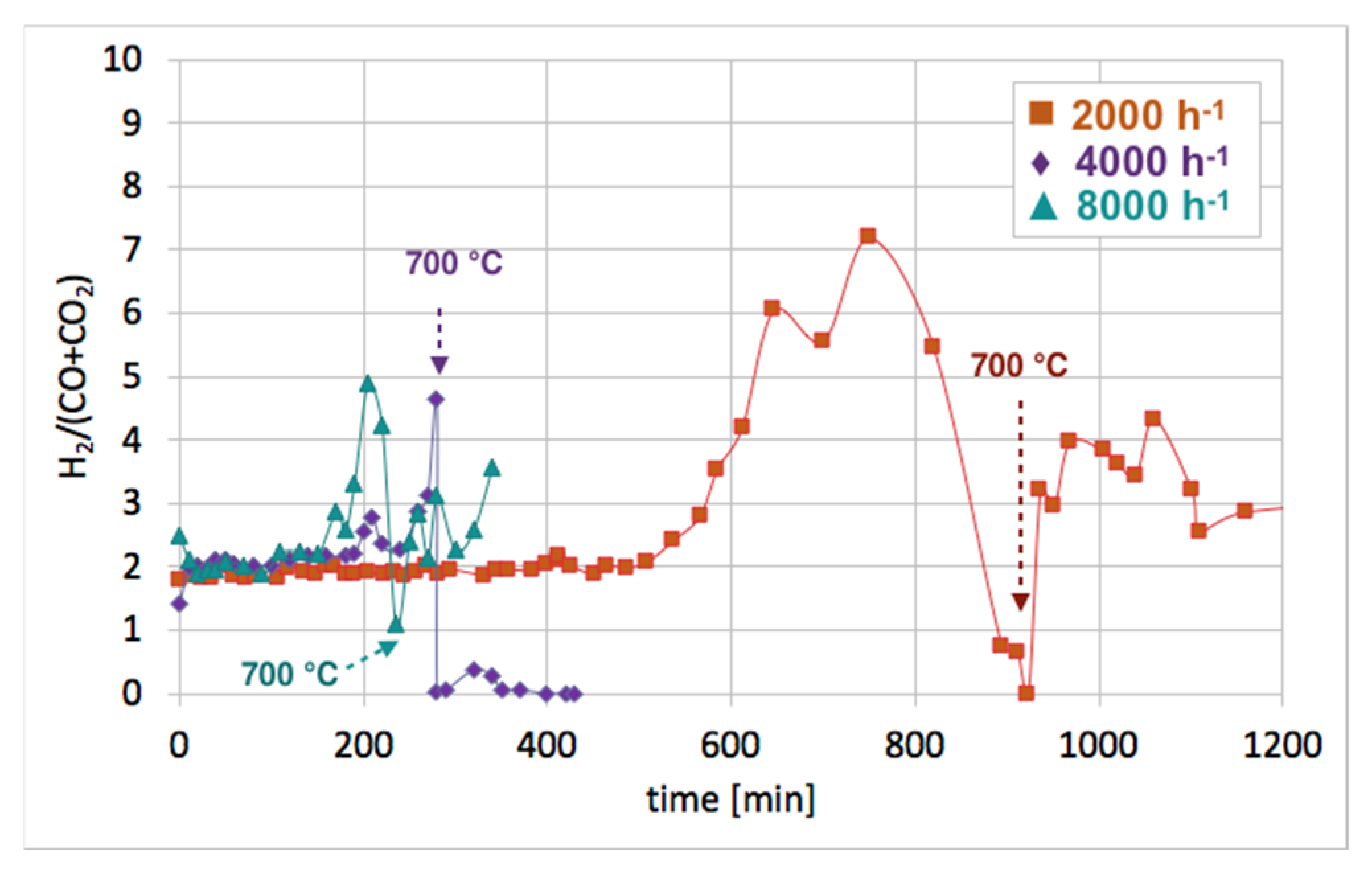
Figure 12 shows the influence of GHSV on the H
2/(CO + CO
2) ratio during catalyst regeneration. It can be observed that when regeneration was carried out at 550 °C and reactive carbon deposits were being oxidized, this ratio was stable and close to 2, i.e., the stoichiometric value of H
2/(CO + CO
2), assuming that all CO underwent oxidation to CO
2. The increase in this ratio above 2 speaks for H
2 overproduction, which can be assigned to H
2O dissociation on Ni
0 and/or Ce
3+. It was calculated that the H
2 formation was coming mainly from the oxidation of the carbon deposits into CO
2. The participation of this reaction in total H
2 production increased with GHSV. For all GHSVs, some part of H
2 was produced in reactions other than oxidation of carbon deposits (Equations (S5) and (S6)), i.e., in the H
2O dissociation on Ni
0 and/or Ce
3+ (Equation (S1)). However, the participation of H
2O dissociation on Ni
0 and Ce
3+ active sites decreased with increasing GHSV. For the test carried out at GHSV = 2000 h
−1, about 19% of H
2 was produced in that way (
Table 1). At a low value of GHSV, H
2 production increases significantly. As was shown in
Table 2, about 74 dm
3 of H
2 was produced during catalyst regeneration at GHSV = 2000 h
−1, whereas the increase of that value to 4000 and 8000 h
−1 resulted in a drastic decrease in H
2 formation to 3.8 and 1.4 dm
3, respectively.
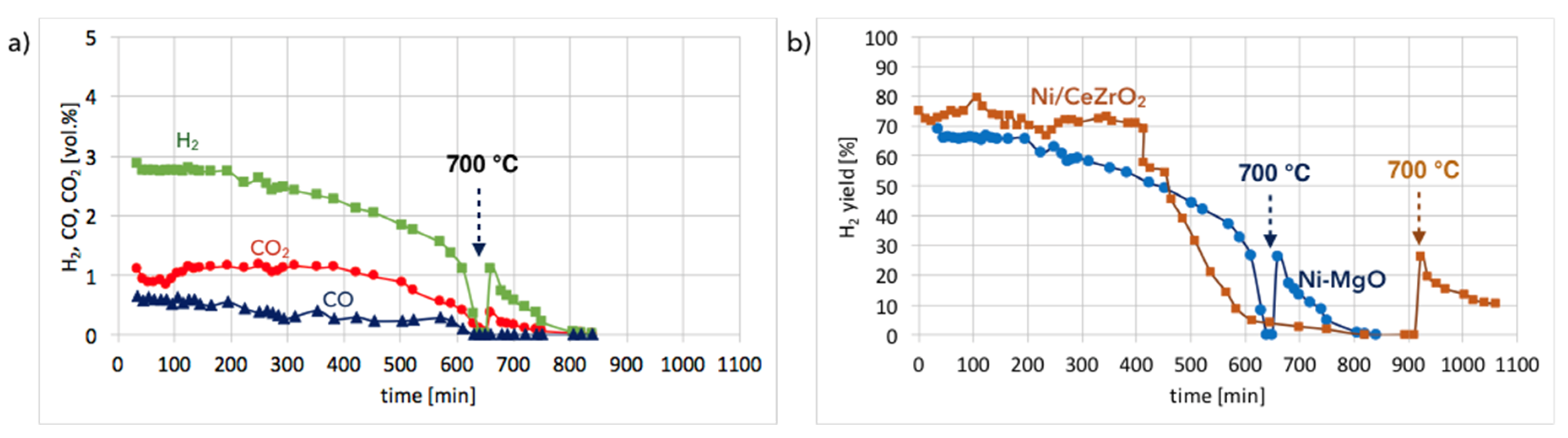
Ni-MgO:Figure 13a shows the gas composition on the reactor outlet during Ni-MgO regeneration. Stable H
2 formation was observed during the first 200 minutes of the test run at 550 °C. Next it decreased slowly. After 10 hours, a sharp H
2 decrease can be noticed. The temperature increase to 700 °C resulted in a rapid formation of H
2, CO, and CO
2, indicating that there was still structural carbon in the sample. Comparing H
2 formation during the regeneration of Ni/CeZrO
2 with Ni-MgO (
Figure 13b), it can be observed that this process took longer for Ni/CeZrO
2, which was due to the H
2O dissociation on Ni
0 and Ce
3+. In the case of Ni-MgO, only the oxidation of Ni to NiO can take place after the removal of carbon deposits with H
2O. However, the rate of that reaction is low at 550 °C. Within the first 200 minutes of the test run, the H
2/(CO + CO
2) ratio over Ni-MgO was only 1.7; thus, it was lower than for Ni/CeZrO
2, which was 2. It was calculated that during the regeneration of Ni-MgO, about 74% of H
2 was produced in the reaction of carbon oxidation to CO
2, while 21% of H
2 was produced in the reaction of CO oxidation. The remaining 5% was a result of H
2O dissociation on Ni
0 leading to its partial re-oxidation to NiO.
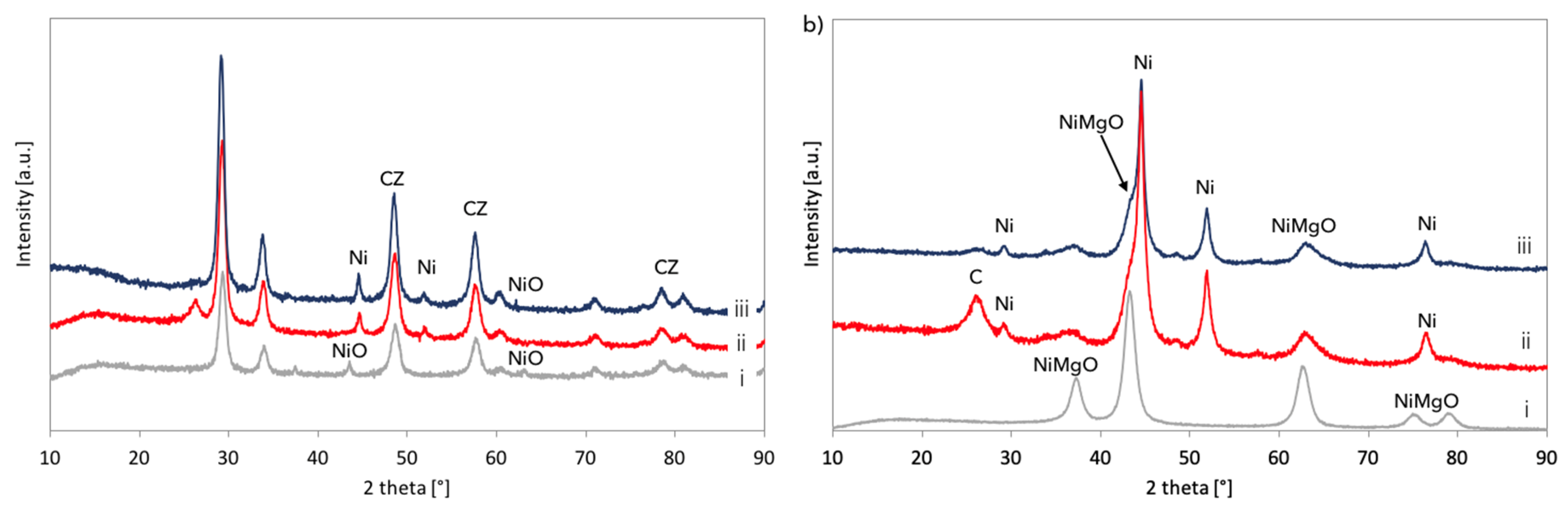
XRD analyses of fresh, spent, and regenerated Ni/CeZrO
2 and Ni-MgO are presented in
Figure 14. It was confirmed that both catalysts underwent a reduction in flowing CH
4, and carbon deposits were formed (diffractograms (ii)). According to the outlet gas composition, regeneration of Ni/CeZrO
2 and Ni-MgO was accomplished after over 34 and 14 hours, respectively. However, for both catalysts, a small intensity peak at ca. 26° indicating carbon presence is still observed on diffractograms (iii). Moreover, both catalysts were not re-oxidized after treatment with steam at 700 °C. Extended exposure of Ni/CeZrO
2 to steam at high temperatures caused some increase of Ni and CeZrO
2 crystallites size (
Table 3) that were calculated from the Scherrer equation for reflexions at 44.6° (for Ni) and 29.3° (for CeZrO
2). EDS analysis and mapping during SEM observations (
Figures S11 and S12) showed a carbon presence in the Ni/CeZrO
2 sample. As was measured by samples burning in air at 900 °C, the regenerated Ni/CeZrO
2 and Ni-MgO still contained 10 and 23% of carbon, respectively.
3.4. CH4 Decomposition and Catalyst Regeneration with H2O in Cycles
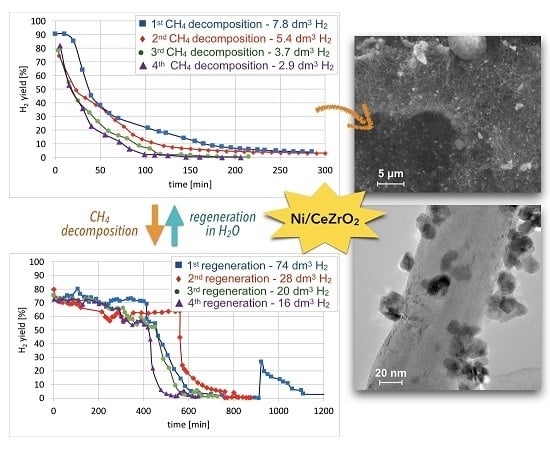
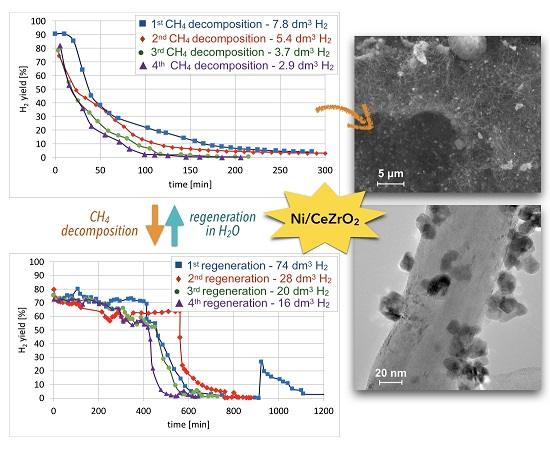
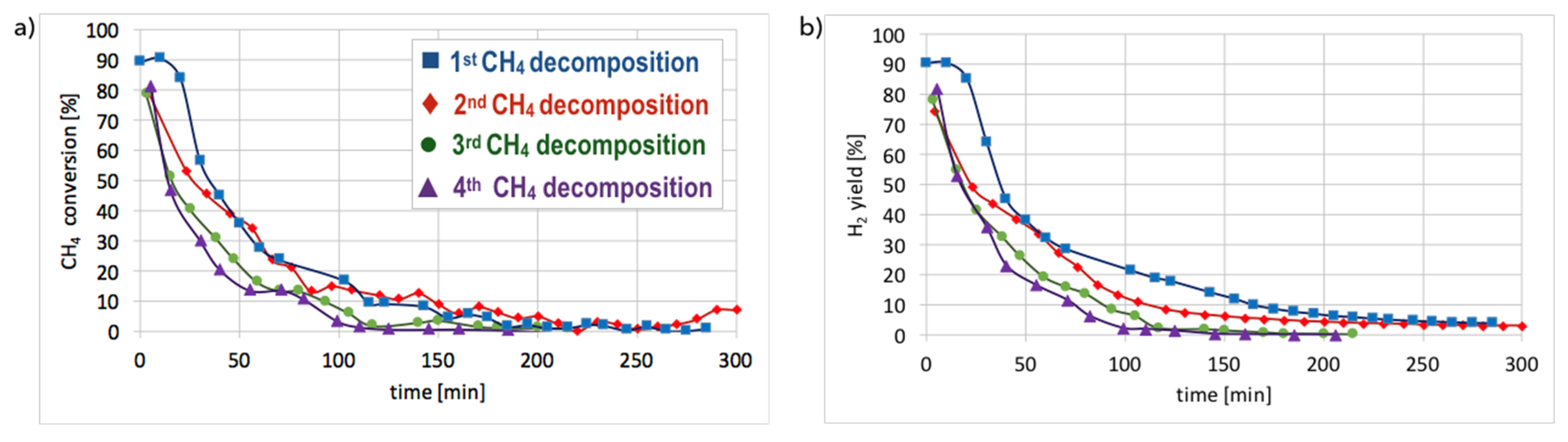
Cyclic tests of CH
4 decomposition (at 700 °C for 5 h, GHSV = 10,000 h
−1) followed by separation of carbon deposits and catalyst regeneration with H
2O (at 550 and 700 °C, GHSV = 2000 h
−1) were performed over Ni/CeZrO
2 (0.125–0.2 mm). Conversions of CH
4 and H
2 yields during tests are presented in
Figure 15. High H
2 yields and CH
4 conversions were observed at the beginning of each test, but the time of CH
4 decomposition with H
2 formation was shorter for each next test. Hence, the Ni/CeZrO
2 deactivated faster in consecutive cycles.
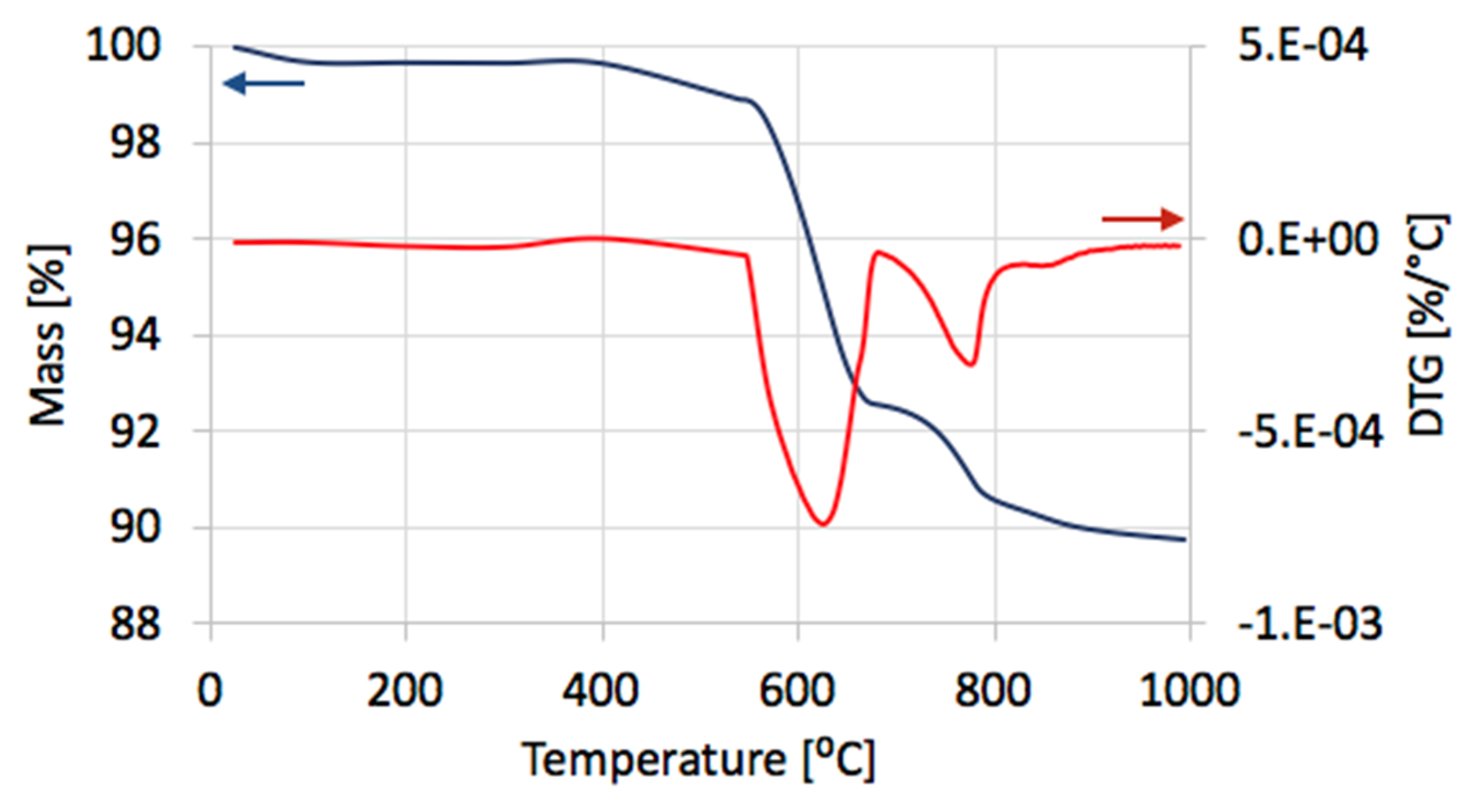
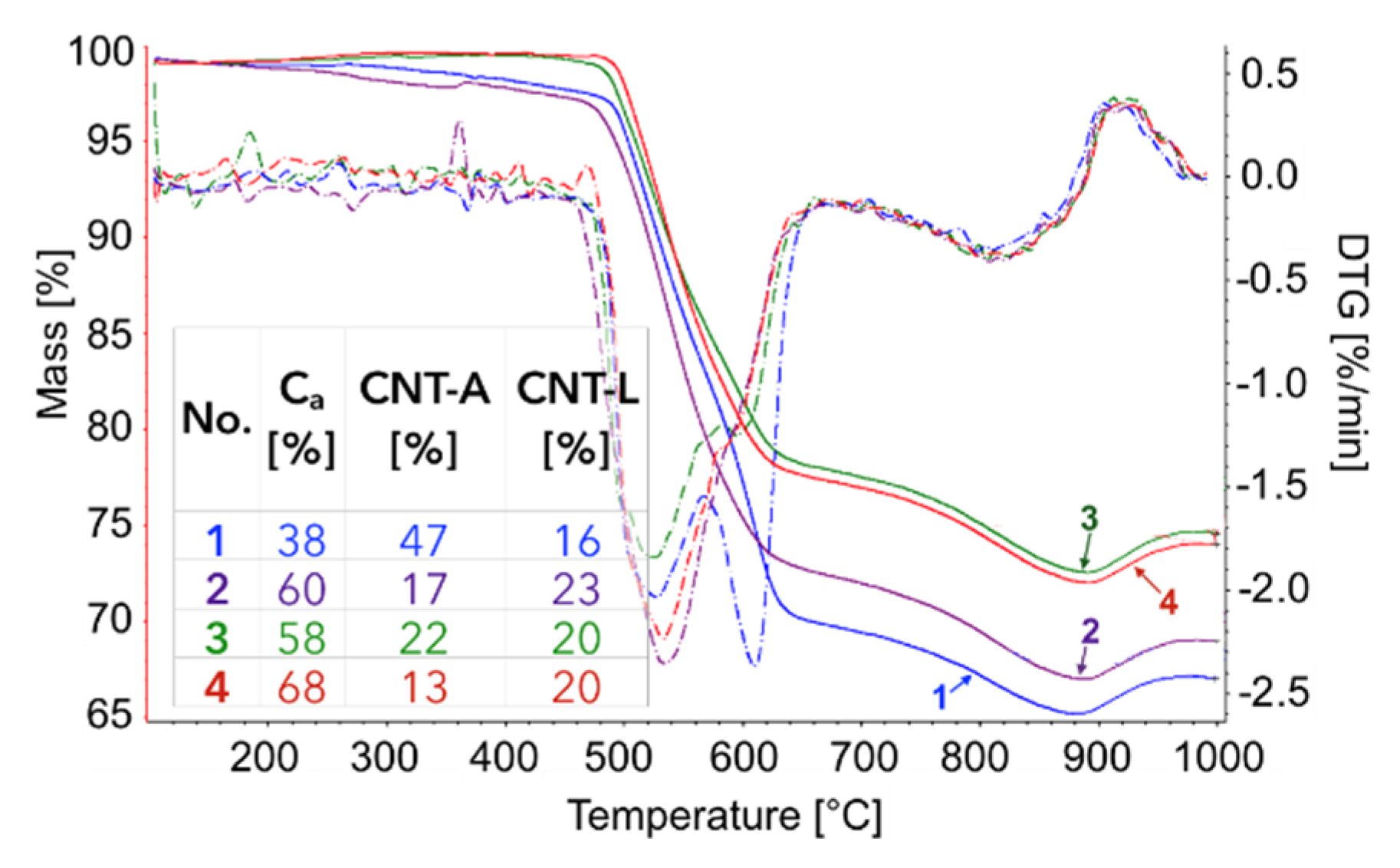
The TGA of the catalyst samples after each test (performed in flowing H
2O/Ar,
Figure 16) indicated that CH
4 decomposition resulted in the deposition of 33, 30, 27.5, and 28% of carbon (for the 1st, 2nd, 3rd, and 4th decomposition test, respectively). Carbon deposits were gasified with H
2O at ca. 550, 610, and 800 °C. Oxidation with H
2O of the most reactive carbon deposit, i.e., amorphous carbon (C
a), occurred at 550 °C, whereas oxidation of structural carbon was observed at 610 °C (for CNTs being in good contact with catalyst particles (CNT-A)) and at 800 °C (oxidation of CNTs being in loose contact with catalyst (CNT-L)). The most carbon deposits were formed during the 1st CH
4 decomposition, and this amount decreased with each next test. Moreover, the content of amorphous carbon in the sample increased with each next test. The decreasing formation of CNTs in each subsequent test could be caused by decreasing the amount of Ni sites accessible for CH
4 adsorption and dehydration. The reduction of the number of active Ni sites might be a consequence of (i) detachment of Ni particles from catalyst during CNT formation and their passing to carbon fraction, (ii) incomplete regeneration of the catalyst (described below in the text) and/or (iii) sintering of the catalyst leading to some coverage of Ni with CeZrO
2.
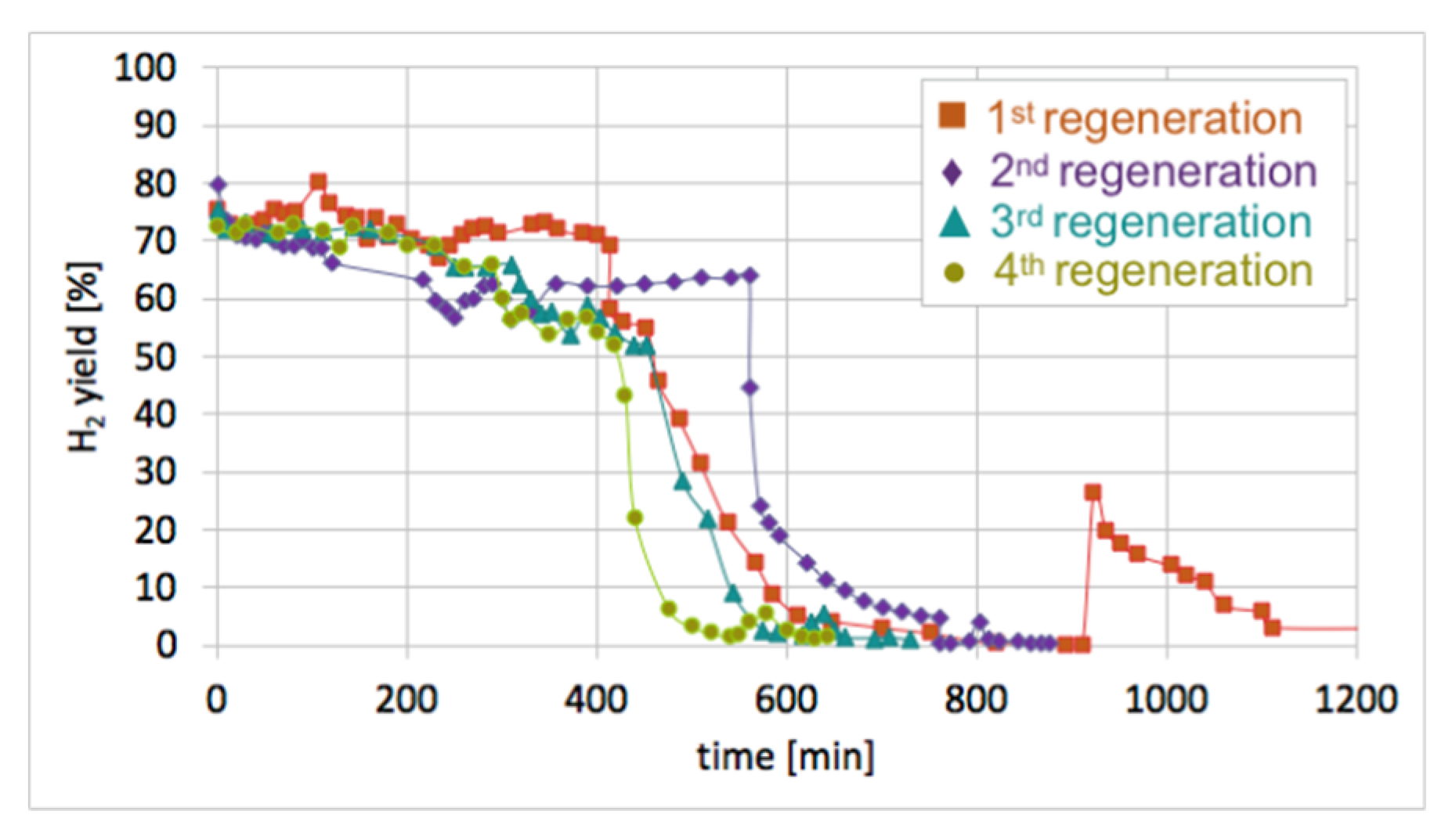
After each test of CH
4 decomposition, the carbon deposit was separated, and the catalyst was subjected to regeneration with H
2O at 550 and 700 °C. It can be seen in
Figure 17 that durations of the catalyst regeneration at 550 °C (i.e., until no more H
2, CO, and CO
2 were released to the gas phase (
Figure S13)), were 910, 790, 590, and 540 minutes for the first, second, third, and fourth regenerations, respectively. The shortening of regeneration time in each subsequent test can be explained by the decreasing amount of carbon deposits, as was demonstrated in
Figure 15 and
Figure 16.
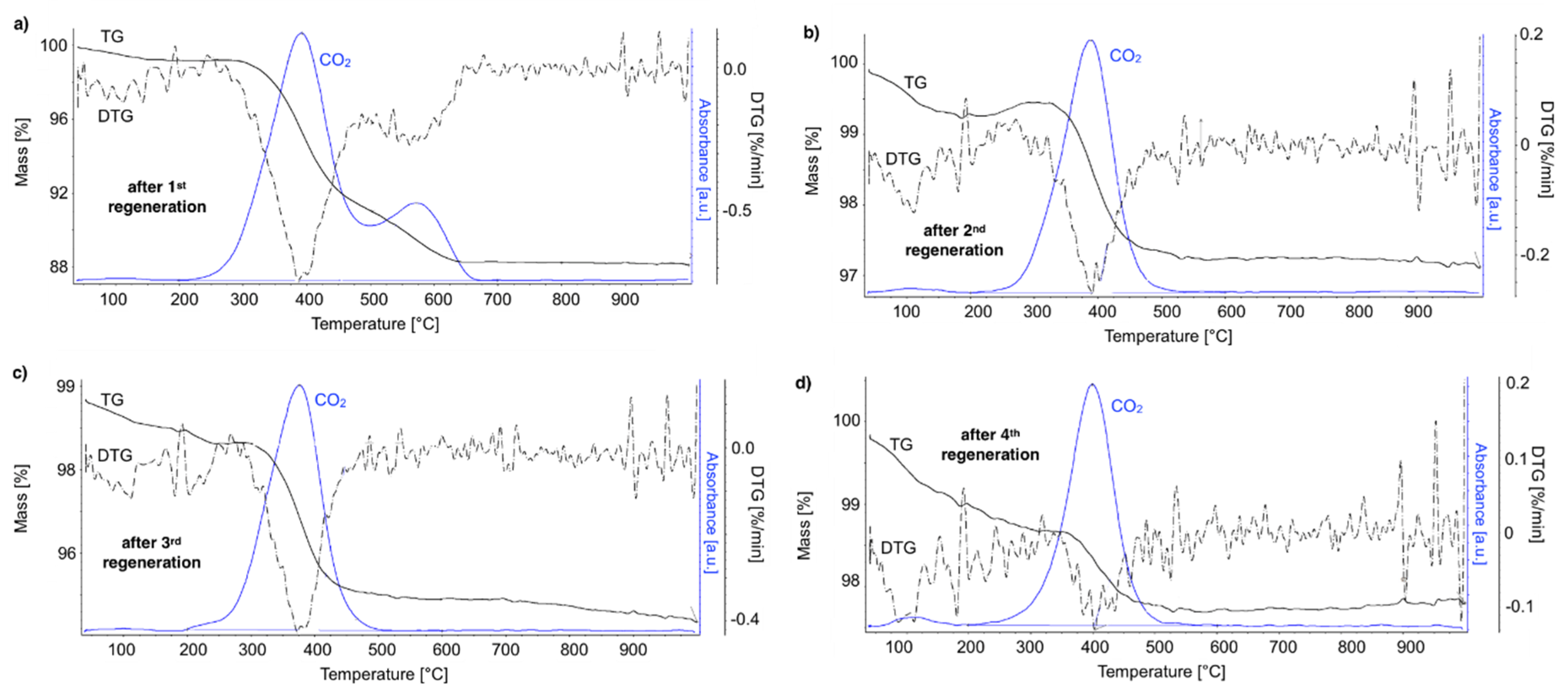
After regeneration, the catalyst samples were subjected to TGA/DTG (performed in air) with CO
2 detection using FTIR (
Figure 18).
Table 4 shows a mass decrease and CO
2 production during those analyses. TGA revealed that after the first regeneration with H
2O, the catalyst sample still contained 11.9% of the carbon deposits: The amorphous carbon (oxidized at ca. 400 °C) and CTNs (oxidized at ca. 570 °C). Oxidation of both types of carbon deposits resulted in the formation of CO
2 during TGA. After further regeneration tests, the catalyst samples contained only the amorphous carbon species. By these observations, one may conclude that catalyst regenerations in a fluidized bed reactor have not been accomplished. The lack of CO
2 and CO in the gas phase at the end of the regenerations carried out at 550 and 700 °C (
Figure S13) suggested that all carbon deposits were removed. However, the TGA revealed the presence of both the amorphous and structural carbon. This implies that some carbon deposits formed during CH
4 decomposition were resistant to oxidation with H
2O in a fluidized bed.
Sequential processes of CH
4 decomposition followed by catalyst regeneration with H
2O in a fluidized bed reactor were found to be less and less efficient in terms of CH
4 consumption and H
2 production (
Table 5). The number of CH
4 and H
2 moles consumed and produced on 1 Ni mole was decreasing in each next test of methane decomposition (
Table S3). The loss of active phase caused decreasing H
2 production during CH
4 decomposition tests. It determined the amount of H
2 produced during the regeneration step (the fewer carbon deposits in the catalyst, the less H
2 produced). Moreover, the increase in Ni and CeZrO
2 particle size could also contribute to a decrease in catalyst efficiency.






















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}