Pathological Mechanisms and Therapeutic Targets for Trigeminal Neuropathic Pain


Abstract
1. Introduction
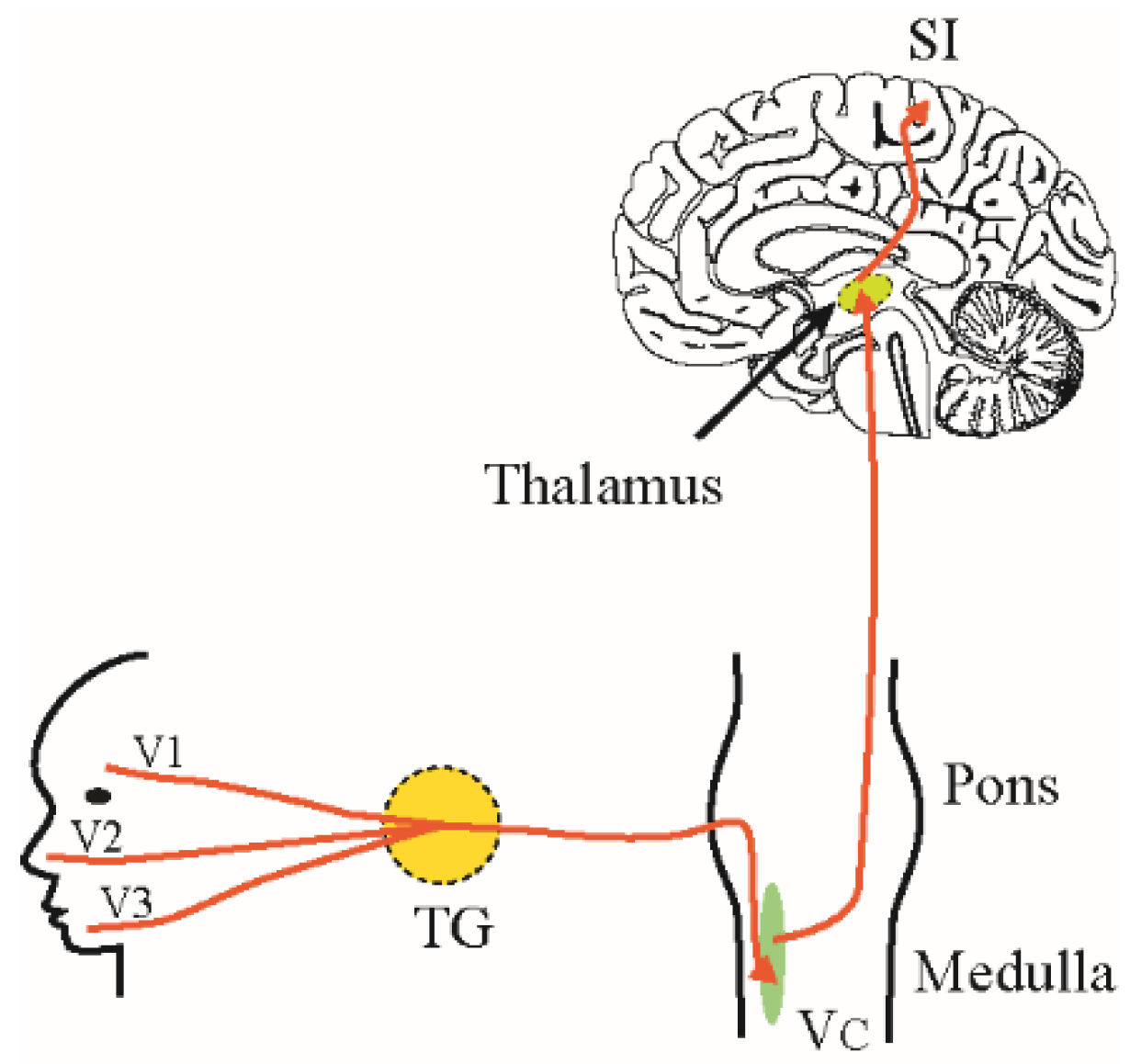
2. Trigeminal Pain Pathways
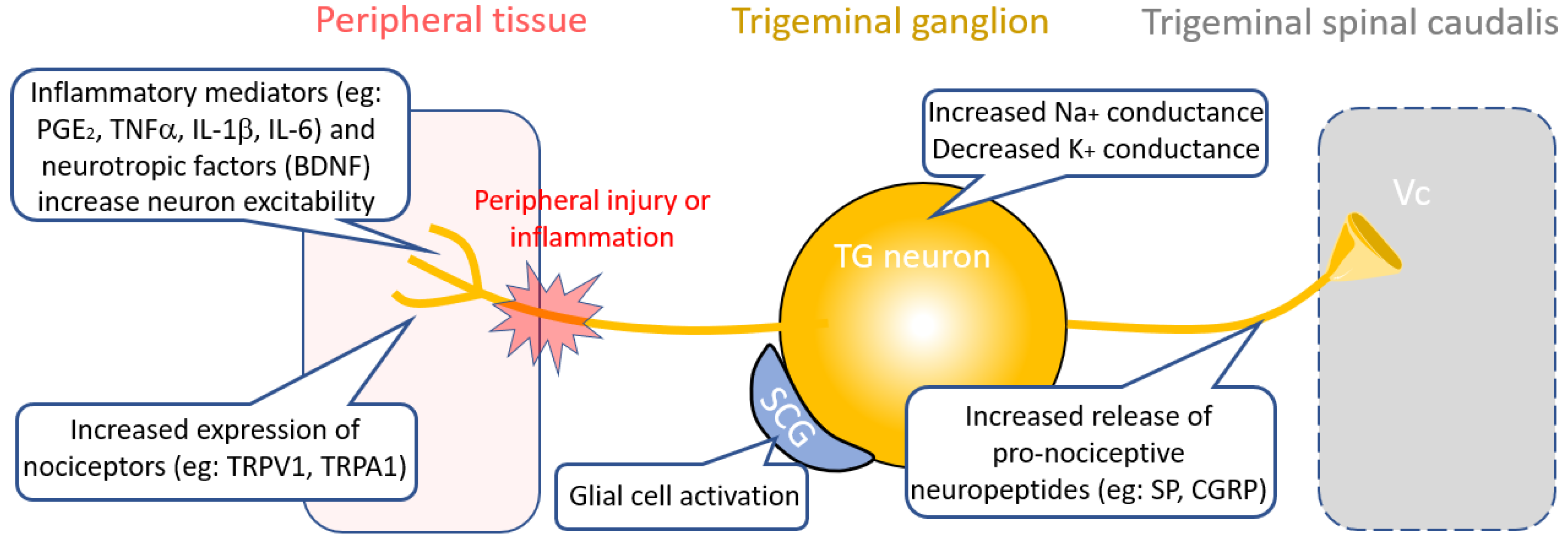
3. Therapies Targeting Peripheral Nociceptive Circuit Dysfunction
3.1. Inflammatory Targets
3.2. Neuropeptide Targets
3.3. Ion Channel Targets
3.3.1. TRP Channels
3.3.2. Sodium Channels
3.3.3. Potassium Channels and Non-Selective Cation Channels
3.3.4. Calcium Channels
3.4. Glial Targets
3.5. Cross-Excitation from Injured Nerve Fibers
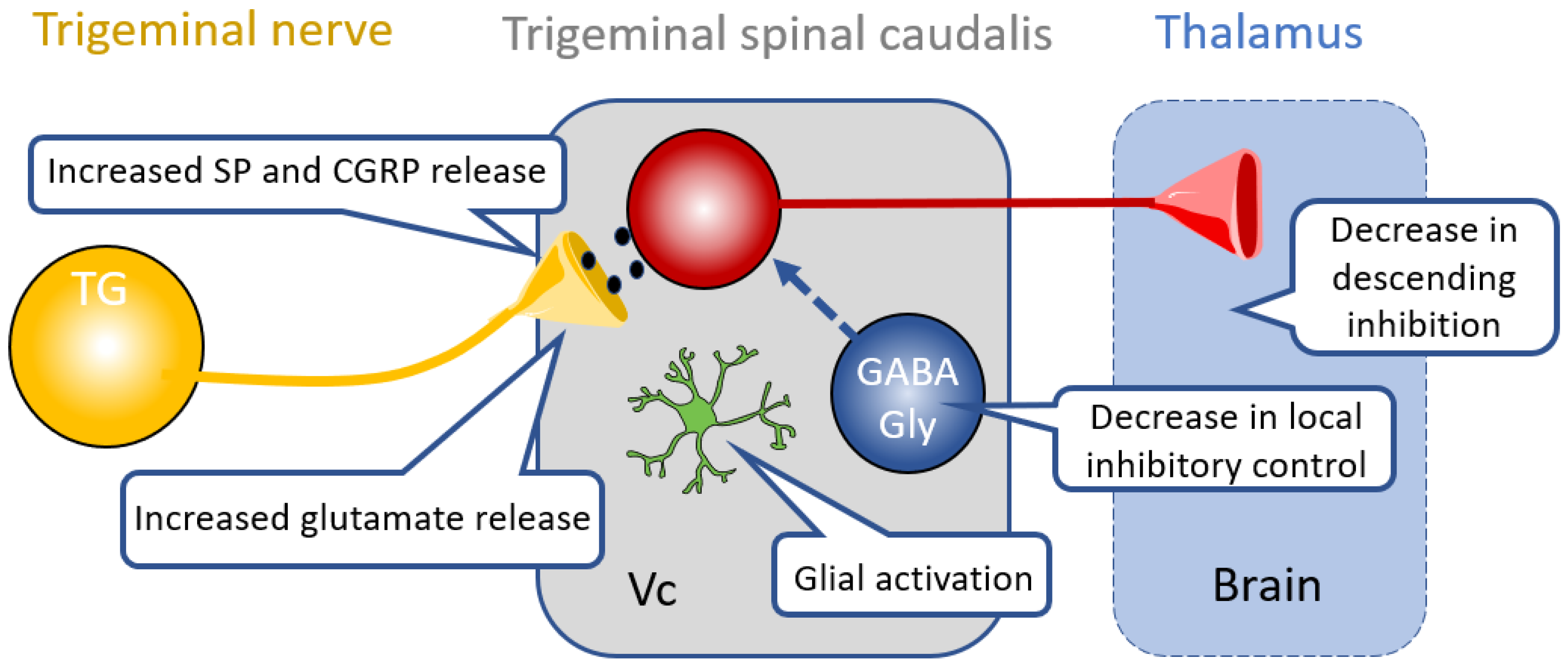
4. Therapies Targeting Central Nociceptive Circuit Dysfunction
4.1. Glia in the CNS
4.2. Inhibitory Neurotransmission in Central Pathways
4.3. Descending Modulation
5. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Tseng, W.-T.; Tsai, M.-L.; Iwata, K.; Yen, C.-T. Long-Term Changes in Trigeminal Ganglionic and Thalamic Neuronal Activities following Inferior Alveolar Nerve Transection in Behaving Rats. J. Neurosci. 2012, 32, 16051–16063. [Google Scholar] [CrossRef] [PubMed]
- Leal, P.R.L.; Barbier, C.; Hermier, M.; Souza, M.A.; Cristino-Filho, G.; Sindou, M. Atrophic changes in the trigeminal nerves of patients with trigeminal neuralgia due to neurovascular compression and their association with the severity of compression and clinical outcomes. J. Neurosurg. 2014, 120, 1484–1495. [Google Scholar] [CrossRef] [PubMed]
- Cruccu, G.; Biasiotta, A.; Di Rezze, S.; Fiorelli, M.; Galeotti, F.; Innocenti, P.; Mameli, S.; Millefiorini, E.; Truini, A. Trigeminal neuralgia and pain related to multiple sclerosis. Pain 2009, 143, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Gottfried, O.N.; Chin, S.; Davidson, H.C.; Couldwell, W.T. Trigeminal Amyloidoma: Case Report and Review of the Literature. Skull Base 2007, 17, 317–324. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wong, R.H.; Farhat, H.I. Charcot–Marie–Tooth and trigeminal neuralgia. Clin. Neurol. Neurosurg. 2013, 115, 2234–2235. [Google Scholar] [CrossRef] [PubMed]
- Behrman, S.; Knight, G. Herpes Simplex Associated with Trigeminal Neuralgia. Neurology 1954, 4, 525. [Google Scholar] [CrossRef] [PubMed]
- Gunduz, A.; Uygunoglu, U.; Uluduz, D.; Saip, S.; Siva, A.; Goksan, B.; Kiziltan, M.E. Reduced inhibition in brainstem circuits in classical trigeminal neuralgia. Eur. J. Pain 2019, 23, 142–149. [Google Scholar] [CrossRef]
- Costa, G.M.F.; Leite, C.M.d.A. Trigeminal neuralgia: Peripheral and central mechanisms. Rev. Dor 2015, 16, 297–301. [Google Scholar] [CrossRef]
- Matthews, B. Peripheral and Central Aspects of Trigeminal Nociceptive Systems. Philos. Trans. R. Soc. B Boil. Sci. 1985, 308, 313–324. [Google Scholar] [CrossRef]
- Okada, S.; Katagiri, A.; Saito, H.; Lee, J.; Ohara, K.; Iinuma, T.; Bereiter, D.A.; Iwata, K. Differential activation of ascending noxious pathways associated with trigeminal nerve injury. Pain 2019, 160, 1342–1360. [Google Scholar] [CrossRef]
- Wang, J.; Li, Z.H.; Feng, B.; Zhang, T.; Zhang, H.; Li, H.; Chen, T.; Cui, J.; Zang, W.D.; Li, Y.Q. Corticotrigeminal projections from the insular cortex to the trigeminal caudal subnucleus regulate orofacial pain after nerve injury via extracellular signal-regulated kinase activation in insular cortex neurons. Front. Cell. Neurosci. 2015, 9, 493. [Google Scholar] [CrossRef] [PubMed]
- Gojyo, F.; Sugiyo, S.; Kuroda, R.; Kawabata, A.; Varathan, V.; Shigenaga, Y.; Takemura, M. Effects of somatosensory cortical stimulation on expression of c-Fos in rat medullary dorsal horn in response to formalin-induced noxious stimulation. J. Neurosci. Res. 2002, 68, 479–488. [Google Scholar] [CrossRef] [PubMed]
- Chai, B.; Guo, W.; Wei, F.; Dubner, R.; Ren, K. Trigeminal-Rostral Ventromedial Medulla circuitry is involved in orofacial hyperalgesia contralateral to tissue injury. Mol. Pain 2012, 8, 78. [Google Scholar] [CrossRef] [PubMed]
- Shankland, W.E., 2nd. Factors that affect pain behavior. Cranio 2011, 29, 144–154. [Google Scholar] [CrossRef] [PubMed]
- Tenenbaum, H.; Mock, D.; Gordon, A.; Goklberg, M.; Grossi, M.; Locker, D.; Davis, K. Sensory and Affective Components of Orofacial Pain: Is it all in your Brain? Crit. Rev. Oral Boil. Med. 2001, 12, 455–468. [Google Scholar] [CrossRef]
- Bushnell, M.C.; Ceko, M.; Low, L.A. Cognitive and emotional control of pain and its disruption in chronic pain. Nat. Rev. Neurosci. 2013, 14, 502–511. [Google Scholar] [CrossRef]
- Basbaum, A.I.; Bautista, D.M.; Scherrer, G.; Julius, D. Cellular and Molecular Mechanisms of Pain. Cell 2009, 139, 267–284. [Google Scholar] [CrossRef] [PubMed]
- Ellis, A.; Bennett, D.L.H. Neuroinflammation and the generation of neuropathic pain. Br. J. Anaesth. 2013, 111, 26–37. [Google Scholar] [CrossRef]
- Takeda, M.; Takahashi, M.; Kitagawa, J.; Kanazawa, T.; Nasu, M.; Matsumoto, S. Brain-derived neurotrophic factor enhances the excitability of small-diameter trigeminal ganglion neurons projecting to the trigeminal nucleus interpolaris/caudalis transition zone following masseter muscle inflammation. Mol. Pain 2013, 9, 49. [Google Scholar] [CrossRef]
- Kiguchi, N.; Kobayashi, D.; Saika, F.; Matsuzaki, S.; Kishioka, S. Inhibition of peripheral macrophages by nicotinic acetylcholine receptor agonists suppresses spinal microglial activation and neuropathic pain in mice with peripheral nerve injury. J. Neuroinflamm. 2018, 15, 96. [Google Scholar] [CrossRef]
- Lee, S.; Shi, X.Q.; Fan, A.; West, B.; Zhang, J. Targeting macrophage and microglia activation with colony stimulating factor 1 receptor inhibitor is an effective strategy to treat injury-triggered neuropathic pain. Mol. Pain 2018, 14, 1744806918764979. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Yamamoto, S.; Zhang, J.; Kometani, M.; Tomiyama, D.; Kohno, K.; Tozaki-Saitoh, H.; Inoue, K.; Tsuda, M. Duloxetine inhibits microglial p2x4 receptor function and alleviates neuropathic pain after peripheral nerve injury. PLoS ONE 2016, 11, e0165189. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, J.L.; Cornelison, L.E.; Blankenship, B.A.; Durham, P.L. Vagus nerve stimulation inhibits trigeminal nociception in a rodent model of episodic migraine. Pain Rep. 2017, 2, 1. [Google Scholar] [CrossRef] [PubMed]
- Koopman, F.A.; Chavan, S.S.; Miljko, S.; Grazio, S.; Sokolovic, S.; Schuurman, P.R.; Mehta, A.D.; Levine, Y.A.; Faltys, M.; Zitnik, R.; et al. Vagus nerve stimulation inhibits cytokine production and attenuates disease severity in rheumatoid arthritis. Proc. Natl. Acad. Sci. USA 2016, 113, 8284–8289. [Google Scholar] [CrossRef] [PubMed]
- Iwatsuki, K.; Arai, T.; Ota, H.; Kato, S.; Natsume, T.; Kurimoto, S.; Yamamoto, M.; Hirata, H. Targeting Anti-Inflammatory Treatment Can Ameliorate Injury-Induced Neuropathic Pain. PLoS ONE 2013, 8, e57721. [Google Scholar] [CrossRef] [PubMed]
- Wolf, G.; Gabay, E.; Tal, M.; Yirmiya, R.; Shavit, Y. Genetic impairment of interleukin-1 signaling attenuates neuropathic pain, autotomy, and spontaneous ectopic neuronal activity, following nerve injury in mice. Pain 2006, 120, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Guptarak, J.; Wanchoo, S.; Durham-Lee, J.; Wu, Y.; Živadinović, D.; Paulucci-Holthauzen, A.; Nesic, O. Inhibition of IL-6 signaling: A novel therapeutic approach to treating spinal cord injury pain. Pain 2013, 154, 1115–1128. [Google Scholar] [CrossRef] [PubMed]
- Iwasa, T.; Afroz, S.; Inoue, M.; Arakaki, R.; Oshima, M.; Raju, R.; Waskitho, A.; Inoue, M.; Baba, O.; Matsuka, Y. IL-10 and CXCL2 in trigeminal ganglia in neuropathic pain. Neurosci. Lett. 2019, 703, 132–138. [Google Scholar] [CrossRef]
- Milligan, E.D.; Sloane, E.M.; Langer, S.J.; Cruz, P.E.; Chacur, M.; Spataro, L.; Wieseler-Frank, J.; Hammack, S.E.; Maier, S.F.; Flotte, T.R.; et al. Controlling neuropathic pain by adeno-associated virus driven production of the anti-inflammatory cytokine, interleukin-10. Mol. Pain 2005, 1, 9. [Google Scholar] [CrossRef]
- Goto, T.; Iwai, H.; Kuramoto, E.; Yamanaka, A. Neuropeptides and ATP signaling in the trigeminal ganglion. Jpn. Dent. Sci. Rev. 2017, 53, 117–124. [Google Scholar] [CrossRef]
- Goadsby, P.J.; Reuter, U.; Hallström, Y.; Broessner, G.; Bonner, J.H.; Zhang, F.; Sapra, S.; Picard, H.; Mikol, D.D.; Lenz, R.A. A Controlled Trial of Erenumab for Episodic Migraine. N. Engl. J. Med. 2017, 377, 2123–2132. [Google Scholar] [CrossRef] [PubMed]
- Holland, P.R.; Goadsby, P.J. Targeted CGRP Small Molecule Antagonists for Acute Migraine Therapy. Neurotherapeutics 2018, 15, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Traynor, K. FDA approves licensing of erenumab-aooe to prevent migraine. Am. J. Heal. Pharm. 2018, 75, 929–930. [Google Scholar] [CrossRef] [PubMed]
- Levinson, S.R.; Luo, S.; Henry, M.A. THE ROLE OF SODIUM CHANNELS IN CHRONIC PAIN. Muscle Nerve 2012, 46, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, K.; Takeda, M.; Tsuboi, Y.; Kondo, M.; Kitagawa, J.; Matsumoto, S.; Kobayashi, A.; Sessle, B.J.; Shinoda, M.; Iwata, K. Alteration of primary afferent activity following inferior alveolar nerve transection in rats. Mol. Pain 2010, 6, 9. [Google Scholar] [CrossRef]
- Takeda, M.; Tanimoto, T.; Ikeda, M.; Nasu, M.; Kadoi, J.; Yoshida, S.; Matsumoto, S. Enhanced excitability of rat trigeminal root ganglion neurons via decrease in A-type potassium currents following temporomandibular joint inflammation. Neuroscience 2006, 138, 621–630. [Google Scholar] [CrossRef]
- Kitagawa, J.; Takeda, M.; Suzuki, I.; Kadoi, J.; Tsuboi, Y.; Honda, K.; Matsumoto, S.; Nakagawa, H.; Tanabe, A.; Iwata, K. Mechanisms involved in modulation of trigeminal primary afferent activity in rats with peripheral mononeuropathy. Eur. J. Neurosci. 2006, 24, 1976–1986. [Google Scholar] [CrossRef]
- Yaksh, T.L. Calcium channels as therapeutic targets in neuropathic pain. J. Pain 2006, 7, S13–S30. [Google Scholar] [CrossRef]
- Brittain, J.M.; Duarte, D.B.; Wilson, S.M.; Zhu, W.; Ballard, C.; Johnson, P.L.; Liu, N.; Xiong, W.; Ripsch, M.S.; Wang, Y.; et al. Suppression of inflammatory and neuropathic pain by uncoupling crmp-2 from the presynaptic ca(2)(+) channel complex. Nat. Med. 2011, 17, 822–829. [Google Scholar] [CrossRef]
- Hingtgen, C.; Waite, K.; Vasko, M. Prostaglandins facilitate peptide release from rat sensory neurons by activating the adenosine 3’,5’-cyclic monophosphate transduction cascade. J. Neurosci. 1995, 15, 5411–5419. [Google Scholar] [CrossRef]
- DeMartini, C.; Greco, R.; Zanaboni, A.M.; Francesconi, O.; Nativi, C.; Tassorelli, C.; Deseure, K. Antagonism of Transient Receptor Potential Ankyrin Type-1 Channels as a Potential Target for the Treatment of Trigeminal Neuropathic Pain: Study in an Animal Model. Int. J. Mol. Sci. 2018, 19, 3320. [Google Scholar] [CrossRef]
- Park, C.K.; Kim, M.S.; Fang, Z.; Li, H.Y.; Jung, S.J.; Choi, S.Y.; Lee, S.J.; Park, K.; Kim, J.S.; Oh, S.B. Functional expression of thermo-transient receptor potential channels in dental primary afferent neurons: Implication for tooth pain. J. Biol. Chem. 2006, 281, 17304–17311. [Google Scholar] [CrossRef] [PubMed]
- Mickle, A.D.; Shepherd, A.J.; Mohapatra, D.P. Nociceptive TRP Channels: Sensory Detectors and Transducers in Multiple Pain Pathologies. Pharmaceuticals 2016, 9, 72. [Google Scholar] [CrossRef] [PubMed]
- Scholz, J.; Woolf, C.J. Can we conquer pain? Nat. Neurosci. 2002, 5, 1062–1067. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y.; Park, C.-K.; Cho, I.-H.; Jung, S.J.; Kim, J.S.; Oh, S.B. Differential Changes in TRPV1 expression after trigeminal sensory nerve injury. J. Pain 2008, 9, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Urano, H.; Ara, T.; Fujinami, Y.; Hiraoka, B.Y. Aberrant TRPV1 Expression in Heat Hyperalgesia Associated with Trigeminal Neuropathic Pain. Int. J. Med Sci. 2012, 9, 690–697. [Google Scholar] [CrossRef] [PubMed]
- Haas, E.T.; Rowland, K.; Gautam, M. Tooth injury increases expression of the cold sensitive TRP channel TRPA1 in trigeminal neurons. Arch. Oral Boil. 2011, 56, 1604–1609. [Google Scholar] [CrossRef] [PubMed]
- Michot, B.; Lee, C.S.; Gibbs, J.L. TRPM8 and TRPA1 do not contribute to dental pulp sensitivity to cold. Sci. Rep. 2018, 8, 13198. [Google Scholar] [CrossRef] [PubMed]
- Meents, J.E.; Hoffmann, J.; Chaplan, S.R.; Neeb, L.; Schuh-Hofer, S.; Wickenden, A.; Reuter, U. Two TRPV1 receptor antagonists are effective in two different experimental models of migraine. J. Headache Pain 2015, 16, 1668. [Google Scholar] [CrossRef]
- Guo, R.; Zhou, Y.; Long, H.; Shan, D.; Wen, J.; Hu, H.; Yang, H.; Wu, Z.; Lai, W. Transient receptor potential Vanilloid 1-based gene therapy alleviates orthodontic pain in rats. Int. J. Oral Sci. 2019, 11, 11. [Google Scholar] [CrossRef]
- Tanaka, B.S.; Zhao, P.; Dib-Hajj, F.B.; Morisset, V.; Tate, S.; Waxman, S.G.; Dib-Hajj, S.D. A Gain-of-Function Mutation in Nav1.6 in a Case of Trigeminal Neuralgia. Mol. Med. 2016, 22, 338–348. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, J.; Jablonski, A.; Persson, A.-K.; Hao, J.-X.; Kouya, P.F.; Wiesenfeld-Hallin, Z.; Xu, X.-J.; Fried, K. Behavioral changes and trigeminal ganglion sodium channel regulation in an orofacial neuropathic pain model. Pain 2005, 119, 82–94. [Google Scholar] [CrossRef] [PubMed]
- Al-Quliti, K.W. Update on neuropathic pain treatment for trigeminal neuralgia. Neurosciences 2015, 20, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Ambrósio, A.F.; Soares-Da-Silva, P.; Carvalho, C.M.; Carvalho, A.P. Mechanisms of Action of Carbamazepine and Its Derivatives, Oxcarbazepine, BIA 2-093, and BIA 2-024. Neurochem. Res. 2002, 27, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Granger, P.; Biton, B.; Faure, C.; Vige, X.; Depoortere, H.; Graham, D.; Langer, S.Z.; Scatton, B.; Avenet, P. Modulation of the gamma-aminobutyric acid type A receptor by the antiepileptic drugs carbamazepine and phenytoin. Mol. Pharmacol. 1995, 47, 1189–1196. [Google Scholar] [PubMed]
- Arai, Y.-C.P.; Hatakeyama, N.; Nishihara, M.; Ikeuchi, M.; Kurisuno, M.; Ikemoto, T. Intravenous lidocaine and magnesium for management of intractable trigeminal neuralgia: A case series of nine patients. J. Anesth. 2013, 27, 960–962. [Google Scholar] [CrossRef]
- Liu, C.Y.; Lu, Z.Y.; Li, N.; Yu, L.H.; Zhao, Y.F.; Ma, B. The role of large-conductance, calcium-activated potassium channels in a rat model of trigeminal neuropathic pain. Cephalalgia 2015, 35, 16–35. [Google Scholar] [CrossRef]
- Contet, C.; Goulding, S.P.; Kuljis, D.A.; Barth, A.L. BK Channels in the Central Nervous System. Int. Rev. Neurobiol. 2016, 128, 281–342. [Google Scholar]
- Gangadharan, V.; Kuner, R. Pain hypersensitivity mechanisms at a glance. Dis. Model. Mech. 2013, 6, 889–895. [Google Scholar] [CrossRef]
- Yuan, M.; Zhou, H.; Xiao, Z.; Wang, W.; Li, X.; Chen, S.; Yin, X.; Xu, L. Efficacy and Safety of Gabapentin vs. Carbamazepine in the Treatment of Trigeminal Neuralgia: A Meta-Analysis. Pain Pract. 2016, 16, 1083–1091. [Google Scholar] [CrossRef]
- Obermann, M.; Yoon, M.S.; Sensen, K.; Maschke, M.; Diener, H.C.; Katsarava, Z. Efficacy of pregabalin in the treatment of trigeminal neuralgia. Cephalalgia 2008, 28, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Serpell, M.G. Gabapentin in neuropathic pain syndromes: A randomised, double-blind, placebo-controlled trial. Pain 2002, 99, 557–566. [Google Scholar] [CrossRef]
- Fink, K.; Meder, W.; Dooley, D.J.; Gothert, M. Inhibition of neuronal ca(2+) influx by gabapentin and subsequent reduction of neurotransmitter release from rat neocortical slices. Br. J. Pharm. 2000, 130, 900–906. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.P.; Angelotti, T.; Fauman, E. Pharmacology and mechanism of action of pregabalin: The calcium channel α2–δ (alpha2–delta) subunit as a target for antiepileptic drug discovery. Epilepsy Res. 2007, 73, 137–150. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R. Therapeutic use of botulinum toxin in pain treatment. Neuronal Signal. 2018, NS20180058. [Google Scholar] [CrossRef]
- Liu, J.; Xu, Y.Y.; Zhang, Q.L.; Luo, W.F. Efficacy and Safety of Botulinum Toxin Type A in Treating Patients of Advanced Age with Idiopathic Trigeminal Neuralgia. Pain Res. Manag. 2018, 2018, 5p. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.Z.; Unno, S.; Ando, H.; Masuda, Y.; Kitagawa, J. Neuron–Glia Crosstalk and Neuropathic Pain: Involvement in the Modulation of Motor Activity in the Orofacial Region. Int. J. Mol. Sci. 2017, 18, 2051. [Google Scholar] [CrossRef] [PubMed]
- Donegan, M.; Kernisant, M.; Cua, C.; Jasmin, L.; Ohara, P.T. Satellite glial cell proliferation in the trigeminal ganglia after chronic constriction injury of the infraorbital nerve. Glia 2013, 61, 2000–2008. [Google Scholar] [CrossRef]
- Goto, T.; Oh, S.B.; Takeda, M.; Shinoda, M.; Sato, T.; Gunjikake, K.K.; Iwata, K. Recent advances in basic research on the trigeminal ganglion. J. Physiol. Sci. 2016, 66, 381–386. [Google Scholar] [CrossRef]
- Ohara, P.T.; Vit, J.-P.; Bhargava, A.; Jasmin, L. Evidence for a role of connexin 43 in trigeminal pain using RNA interference in vivo. J. Neurophysiol. 2008, 100, 3064–3073. [Google Scholar] [CrossRef]
- Kaji, K.; Shinoda, M.; Honda, K.; Unno, S.; Shimizu, N.; Iwata, K. Connexin 43 contributes to ectopic orofacial pain following inferior alveolar nerve injury. Mol. Pain 2016, 12, 1744806916633704. [Google Scholar] [CrossRef] [PubMed]
- Afroz, S.; Arakaki, R.; Iwasa, T.; Oshima, M.; Hosoki, M.; Inoue, M.; Baba, O.; Okayama, Y.; Matsuka, Y. CGRP Induces Differential Regulation of Cytokines from Satellite Glial Cells in Trigeminal Ganglia and Orofacial Nociception. Int. J. Mol. Sci. 2019, 20, 711. [Google Scholar] [CrossRef] [PubMed]
- Magni, G.; Merli, D.; Verderio, C.; Abbracchio, M.P.; Ceruti, S. P2Y2receptor antagonists as anti-allodynic agents in acute and sub-chronic trigeminal sensitization: Role of satellite glial cells. Glia 2015, 63, 1256–1269. [Google Scholar] [CrossRef] [PubMed]
- Vit, J.-P.; Ohara, P.T.; Bhargava, A.; Kelley, K.; Jasmin, L. Silencing the Kir4.1 potassium channel subunit in satellite glial cells of the rat trigeminal ganglion results in pain-like behavior in the absence of nerve injury. J. Neurosci. 2008, 28, 4161–4171. [Google Scholar] [CrossRef]
- Vit, J.P.; Ohara, P.T.; Sundberg, C.; Rubi, B.; Maechler, P.; Liu, C.; Puntel, M.; Lowenstein, P.; Castro, M.; Jasmin, L. Adenovector gad65 gene delivery into the rat trigeminal ganglion produces orofacial analgesia. Mol. Pain 2009, 5, 42. [Google Scholar] [CrossRef] [PubMed]
- Tsuboi, Y.; Takeda, M.; Tanimoto, T.; Ikeda, M.; Matsumoto, S.; Kitagawa, J.; Teramoto, K.; Simizu, K.; Yamazaki, Y.; Shima, A.; et al. Alteration of the second branch of the trigeminal nerve activity following inferior alveolar nerve transection in rats. Pain 2004, 111, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Devor, M.; Govrin-Lippmann, R.; Rappaport, Z.H. Mechanism of trigeminal neuralgia: An ultrastructural analysis of trigeminal root specimens obtained during microvascular decompression surgery. J. Neurosurg. 2002, 96, 532–543. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Chang, L.; Sorkin, L.S.; Chaplan, S.R. Hyperpolarization-Activated, Cation-Nonselective, Cyclic Nucleotide-Modulated Channel Blockade Alleviates Mechanical Allodynia and Suppresses Ectopic Discharge in Spinal Nerve Ligated Rats. J. Pain 2005, 6, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Okada-Ogawa, A.; Suzuki, I.; Sessle, B.J.; Chiang, C.-Y.; Salter, M.W.; Dostrovsky, J.O.; Tsuboi, Y.; Kondo, M.; Kitagawa, J.; Kobayashi, A.; et al. Astroglia in Medullary Dorsal Horn (Trigeminal Spinal Subnucleus Caudalis) Are Involved in Trigeminal Neuropathic Pain Mechanisms. J. Neurosci. 2009, 29, 11161–11171. [Google Scholar] [CrossRef]
- Piao, Z.G.; Cho, I.-H.; Park, C.K.; Hong, J.P.; Choi, S.-Y.; Lee, S.J.; Lee, S.; Park, K.; Kim, J.S.; Oh, S.B. Activation of glia and microglial p38 MAPK in medullary dorsal horn contributes to tactile hypersensitivity following trigeminal sensory nerve injury. Pain 2006, 121, 219–231. [Google Scholar] [CrossRef] [PubMed]
- Tonkin, R.S.; Bowles, C.; Perera, C.J.; Keating, B.A.; Makker, P.G.S.; Duffy, S.S.; Lees, J.G.; Tran, C.; Don, A.S.; Fath, T.; et al. Attenuation of mechanical pain hypersensitivity by treatment with peptide5, a connexin-43 mimetic peptide, involves inhibition of nlrp3 inflammasome in nerve-injured mice. Exp. Neurol. 2018, 300, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Cao, Y.; Chiang, C.-Y.; Dostrovsky, J.O.; Sessle, B.J. The gap junction blocker carbenoxolone attenuates nociceptive behavior and medullary dorsal horn central sensitization induced by partial infraorbital nerve transection in rats. Pain 2014, 155, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.-Y.; Wang, J.; Xie, Y.-F.; Zhang, S.; Hu, J.W.; Dostrovsky, J.O.; Sessle, B.J. Astroglial Glutamate Glutamine Shuttle Is Involved in Central Sensitization of Nociceptive Neurons in Rat Medullary Dorsal Horn. J. Neurosci. 2007, 27, 9068–9076. [Google Scholar] [CrossRef]
- Imlach, W.L.; Bhola, R.F.; Mohammadi, S.A.; Christie, M.J. Glycinergic dysfunction in a subpopulation of dorsal horn interneurons in a rat model of neuropathic pain. Sci. Rep. 2016, 6, 37104. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Dong, H.; Gao, Y.; Gong, Y.; Ren, Y.; Gu, N.; Zhou, S.; Xia, N.; Sun, Y.-Y.; Ji, R.-R.; et al. A feed-forward spinal cord glycinergic neural circuit gates mechanical allodynia. J. Clin. Investig. 2013, 123, 4050–4062. [Google Scholar] [CrossRef] [PubMed]
- Eto, K.; Ishibashi, H.; Yoshimura, T.; Watanabe, M.; Miyamoto, A.; Ikenaka, K.; Moorhouse, A.J.; Nabekura, J. Enhanced GABAergic Activity in the Mouse Primary Somatosensory Cortex Is Insufficient to Alleviate Chronic Pain Behavior with Reduced Expression of Neuronal Potassium–Chloride Cotransporter. J. Neurosci. 2012, 32, 16552–16559. [Google Scholar] [CrossRef] [PubMed]
- Takeda, M.; Tanimoto, T.; Matsumoto, S. Change in mechanical receptive field properties induced by GABAA receptor activation in the trigeminal spinal nucleus caudalis neurons in rats. Exp. Brain Res. 2000, 134, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Iwata, K.; Imai, T.; Tsuboi, Y.; Tashiro, A.; Ogawa, A.; Morimoto, T.; Masuda, Y.; Tachibana, Y.; Hu, J. Alteration of Medullary Dorsal Horn Neuronal Activity Following Inferior Alveolar Nerve Transection in Rats. J. Neurophysiol. 2001, 86, 2868–2877. [Google Scholar] [CrossRef] [PubMed]
- Fujita, S.; Yamamoto, K.; Kobayashi, M. Trigeminal nerve transection-induced neuroplastic changes in the somatosensory and insular cortices in a rat ectopic pain model. eNeuro 2019, 6. [Google Scholar] [CrossRef]
- Zhang, S.; Chiang, C.; Xie, Y.-F.; Park, S.; Lu, Y.; Hu, J.; Dostrovsky, J.; Sessle, B. Central sensitization in thalamic nociceptive neurons induced by mustard oil application to rat molar tooth pulp. Neurosciences 2006, 142, 833–842. [Google Scholar] [CrossRef]
- Vasović, D.; Divović, B.; Treven, M.; Knutson, D.E.; Steudle, F.; Scholze, P.; Obradović, A.; Fabjan, J.; Brković, B.; Sieghart, W.; et al. Trigeminal neuropathic pain development and maintenance in rats are suppressed by a positive modulator of α6 GABA A receptors. Eur. J. Pain 2019, 23, 973–984. [Google Scholar] [CrossRef] [PubMed]
- Fan, P.-C.; Lai, T.-H.; Hor, C.C.; Lee, M.T.; Huang, P.; Sieghart, W.; Ernst, M.; Knutson, D.E.; Cook, J.; Chiou, L.-C. The α6 subunit-containing GABAA receptor: A novel drug target for inhibition of trigeminal activation. Neuropharmacology 2018, 140, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Park, Y.H.; Yang, K.Y.; Ju, J.S.; Bae, Y.C.; Han, S.K.; Ahn, D.K. Participation of central gabaa receptors in the trigeminal processing of mechanical allodynia in rats. Korean J. Physiol. Pharmacol. 2017, 21, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, T.; Marsala, M.; Sakabe, T.; Yaksh, T.L. Characterization of spinal amino acid release and touch-evoked allodynia produced by spinal glycine or GABA(A) receptor antagonist. Neurosciences 2000, 95, 781–786. [Google Scholar] [CrossRef]
- John, J.M.; Kaneko, M.; Hammond, D.L. Intrathecal bicuculline does not increase formalin-induced inflammation. Brain Res. 1998, 794, 320–324. [Google Scholar] [CrossRef]
- Price, T.J.; Cervero, F.; Koninck, Y.D. Role of Cation-Chloride-Cotransporters (CCC) in Pain and Hyperalgesia. Curr. Top. Med. Chem. 2005, 5, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Okada-Ogawa, A.; Nakaya, Y.; Imamura, Y.; Kobayashi, M.; Shinoda, M.; Kita, K.; Sessle, B.J.; Iwata, K. Involvement of medullary GABAergic system in extraterritorial neuropathic pain mechanisms associated with inferior alveolar nerve transection. Exp. Neurol. 2015, 267, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Idänpään-Heikkilä, J.J.; Guilbaud, G. Pharmacological studies on a rat model of trigeminal neuropathic pain: Baclofen, but not carbamazepine, morphine or tricyclic antidepressants, attenuates the allodynia-like behaviour. Pain 1999, 79, 281–290. [Google Scholar] [CrossRef]
- Reis, G.M.L.; Duarte, I.D.G. Baclofen, an agonist at peripheral GABAB receptors, induces antinociception via activation of TEA-sensitive potassium channels. Br. J. Pharmacol. 2006, 149, 733–739. [Google Scholar] [CrossRef] [PubMed]
- Ng, C.H.; Ong, W.Y. Increased expression of gamma-aminobutyric acid transporters GAT-1 and GAT-3 in the spinal trigeminal nucleus after facial carrageenan injections. Pain 2001, 92, 29–40. [Google Scholar] [CrossRef]
- Yadav, R.; Yan, X.; Maixner, D.W.; Gao, M.; Weng, H.-R. Blocking the GABA transporter GAT-1 ameliorates spinal GABAergic disinhibition and neuropathic pain induced by paclitaxel. J. Neurochem. 2015, 133, 857–869. [Google Scholar] [CrossRef] [PubMed]
- Zeilhofer, H.U.; Studler, B.; Arabadzisz, D.; Schweizer, C.; Ahmadi, S.; Layh, B.; Bosl, M.R.; Fritschy, J.M. Glycinergic neurons expressing enhanced green fluorescent protein in bacterial artificial chromosome transgenic mice. J. Comp. Neurol. 2005, 482, 123–141. [Google Scholar] [CrossRef] [PubMed]
- Cioffi, C.L. Modulation of glycine-mediated spinal neurotransmission for the treatment of chronic pain. J. Med. Chem. 2018, 61, 2652–2679. [Google Scholar] [CrossRef] [PubMed]
- Miraucourt, L.S.; Dallel, R.; Voisin, D.L. Glycine Inhibitory Dysfunction Turns Touch into Pain through PKCgamma Interneurons. PLoS ONE 2007, 2, e1116. [Google Scholar] [CrossRef] [PubMed]
- Imlach, W.L. New approaches to target glycinergic neurotransmission for the treatment of chronic pain. Pharmacol. Res. 2017, 116, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Chu, Y.; Han, L.; Li, M.; Li, Z.; LaVinka, P.C.; Sun, S.; Tang, Z.; Park, K.; Caterina, M.J.; et al. Central terminal sensitization of TRPV1 by descending serotonergic facilitation modulates chronic pain. Neuron 2014, 81, 873–887. [Google Scholar] [CrossRef]
- Okubo, M.; Castro, A.; Guo, W.; Zou, S.; Ren, K.; Wei, F.; Keller, A.; Dubner, R. Transition to persistent orofacial pain after nerve injury Involves supraspinal serotonin mechanisms. J. Neurosci. 2013, 33, 5152–5161. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Tang, Y.; Shu, H.; Tatum, D.; Bai, Q.; Crawford, J.; Xing, Y.; Lobo, M.K.; Bellinger, L.; Kramer, P.; et al. Dopamine receptor D2, but not D1, mediates descending dopaminergic pathway–produced analgesic effect in a trigeminal neuropathic pain mouse model. Pain 2019, 160, 334–344. [Google Scholar] [CrossRef]
- Castro, A.; Raver, C.; Li, Y.; Uddin, O.; Rubin, D.; Ji, Y.; Masri, R.; Keller, A. Cortical Regulation of Nociception of the Trigeminal Nucleus Caudalis. J. Neurosci. 2017, 37, 11431–11440. [Google Scholar] [CrossRef]
- Dostrovsky, J.O.; Shah, Y.; Gray, B.G. Descending inhibitory influences from periaqueductal gray, nucleus raphe magnus, and adjacent reticular formation. II. Effects on medullary dorsal horn nociceptive and nonnociceptive neurons. J. Neurophysiol. 1983, 49, 948–960. [Google Scholar] [CrossRef]
- Heinricher, M.M.; Tavares, I.; Leith, J.L.; Lumb, B.M. Descending control of nociception: Specificity, recruitment and plasticity. Brain Res. Rev. 2009, 60, 214–225. [Google Scholar] [CrossRef] [PubMed]
- Aicher, S.A.; Hermes, S.M.; Whittier, K.L.; Hegarty, D.M. Descending projections from the rostral ventromedial medulla (rvm) to trigeminal and spinal dorsal horns are morphologically and neurochemically distinct. J. Chem. Neuroanat. 2012, 43, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Zilliox, L.A. Neuropathic pain. Continuum 2017, 23, 512–532. [Google Scholar] [CrossRef] [PubMed]
- Hiroki, T.; Suto, T.; Saito, S.; Obata, H. Repeated administration of amitriptyline in neuropathic pain: Modulation of the noradrenergic descending inhibitory system. Anesth. Analg. 2017, 125, 1281–1288. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.; Sinclair, A.J.; Davies, B.; Matharu, M. Neurostimulation in the treatment of primary headaches. Pract. Neurol. 2016, 16, 362–375. [Google Scholar] [CrossRef] [PubMed]
- Osenbach, R. Neurostimulation for the Treatment of Intractable Facial Pain. Pain Med. 2006, 7, S126–S136. [Google Scholar] [CrossRef][Green Version]




{kind=link}
{kind=link}
{kind=link}
| Clinical Condition or Animal Model | Therapeutic Target | Therapeutic Agent | Effect on Symptoms, or Behavioral Outcome | Reference |
|---|---|---|---|---|
| Peripheral Nociceptive Pathways | ||||
| Infraorbital nerve constriction model, rat | IL-10 gene expression in glial cells | Viral vector (AAV) encoding IL-10 | Suppresses mechanical allodynia and thermal hyperalgesia | Iwasa et al., 2019; Milligan et al., 2005 [28,29] |
| Phase III clinical trial for migraine | CGRP receptor (antagonist) | Erenumab | Anti-migraine | Goadsby et al., 2017; Traynor K, 2018 [31,33] |
| Phase III clinical trial for migraine | CGRP receptor (antagonist) | Gepants (Ubrogepant, Rimegepant) | Anti-migraine | Holland PR, 2018 [32] |
| α-CGRP intra-TG injection model, rat | Glial cell (inhibitor) | Minocycline | Reduces thermal hyperalgesia | Afroz et al., 2019 [72] |
| TMJ inflammation induced by CFA injection, rat | P2Y2 receptor (antagonist) | AR-C118925 | Reduces mechanical allodynia | Magni et al., 2015 [73] |
| Inferior alveolar nerve transection (IANX) model, rat | Cx43 gap junctions | Gap27 (Cx43 blocking peptide) | Attenuates mechanical hypersensitivity | Kaji et al., 2016 [71] |
| Orofacial formalin model, rat | GAD65 gene expression (in SGC) | Viral vector (AAV) encoding GAD | Blocks pain behaviour (orofacial rub) | Vit et al., 2009 [75] |
| Clinical study of idiopathic trigeminal neuralgia in elderly (≥ 80 years) or adult > 60 years old patient groups | Acetylcholine release (inhibitor) | Botulinum Toxin Type A | Relief of trigeminal pain symptoms | Jing et al., 2018 [66] |
| Central Nociceptive Pathways | ||||
| Clinical case studies in 9 patients with intractable trigeminal neuralgia | NaV channels (antagonist) | Intravenous magnesium and lidocaine | Relief of trigeminal pain symptoms | Arai et al., 2013 [56] |
| Partial infraorbital nerve transection model, rat | Gap junction (blocker) | Carbenoxolone | Reduces facial mechanical hypersensitivity and central sensitization | Wang et al., 2014 [82] |
| Allyl isothiocyanate tooth pulp inflammation model, rat | Astroglial enzyme glutamine synthetase (inhibitor) | Methionine sulfoximine | Reduces central sensitization | Chiang et al., 2007 [83] |
| Chronic constriction injury of the infraorbital nerve, rat | α6 GABAAR | DK-I-56-1 | Reduces mechanical hypersensitivity | Vasovic et al., 2019 [91] |
| Subcutaneous injection of IL-1β, rat | GABAAR (antagonist) | Bicuculline | Allodynia in naïve rat and anti-allodynic effect in IL-1β injected rat | Kim et al., 2017 [93] |
| Inferior alveolar nerve transection (IANX) model, rat | GABAAR (agonist) | Muscimol | Decreases mechanical evoked response | Okada-ogawa et al., 2015 [97] |
| Chronic inferior alveolar nerve constriction, rat | GABABR (agonist) | Baclofen | Reduces mechanical allodynia like behaviour | Idanpaan-Heikkila et al., 1999; Reis et al., 2006 [98,99] |
| Primary headache, Intractable facial pain, in clinical use | Vagus nerve, cortex (transcranial stimulation). Occipital nerve, ventral tegmental area (invasive stimulation) | Neurostimulat-ion through transcranial magnetic stimulation or invasive brain stimulation | Reduction in headache symptoms, reduced facial pain | Miller et al., 2016 [115] Osenbach, 2006 [116] |
| Ion Channel Targets | ||||
| Chronic constriction injury of the infraorbital nerve, rat | TRPA1 (antagonist) | ADM_12 | Reduction of mechanical allodynia | Demartini et al., 2018 [41] |
| Orthodontic pain model, rat | TRPV1 receptors | Lentivirus delivery of shRNA for TRPV1 | Reduces pain from tooth movement | Guo et al., 2019 [50] |
| Phase III clinical trial for trigeminal neuralgia, human | Voltage dependent calcium channel (inhibitor) | Gabapentin | Reduces neuropathic pain and trigeminal neuralgia pain | Yuan et al., 2016; Serpell et al., 2002 [60,62] |
| Phase III clinical trial for trigeminal neuralgia, human | Voltage dependent calcium channels (inhibitor) | Pregabalin | Reduces trigeminal neuralgia related pain | Obermann et al., 2008 [61] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bista, P.; Imlach, W.L. Pathological Mechanisms and Therapeutic Targets for Trigeminal Neuropathic Pain. Medicines 2019, 6, 91. https://doi.org/10.3390/medicines6030091
Bista P, Imlach WL. Pathological Mechanisms and Therapeutic Targets for Trigeminal Neuropathic Pain. Medicines. 2019; 6(3):91. https://doi.org/10.3390/medicines6030091
Chicago/Turabian StyleBista, Pawan, and Wendy L. Imlach. 2019. "Pathological Mechanisms and Therapeutic Targets for Trigeminal Neuropathic Pain" Medicines 6, no. 3: 91. https://doi.org/10.3390/medicines6030091
APA StyleBista, P., & Imlach, W. L. (2019). Pathological Mechanisms and Therapeutic Targets for Trigeminal Neuropathic Pain. Medicines, 6(3), 91. https://doi.org/10.3390/medicines6030091