
Molecular Mechanisms of Environmental Metal Neurotoxicity: A Focus on the Interactions of Metals with Synapse Structure and Function




{kind=link}
{kind=link}
Abstract
1. Introduction
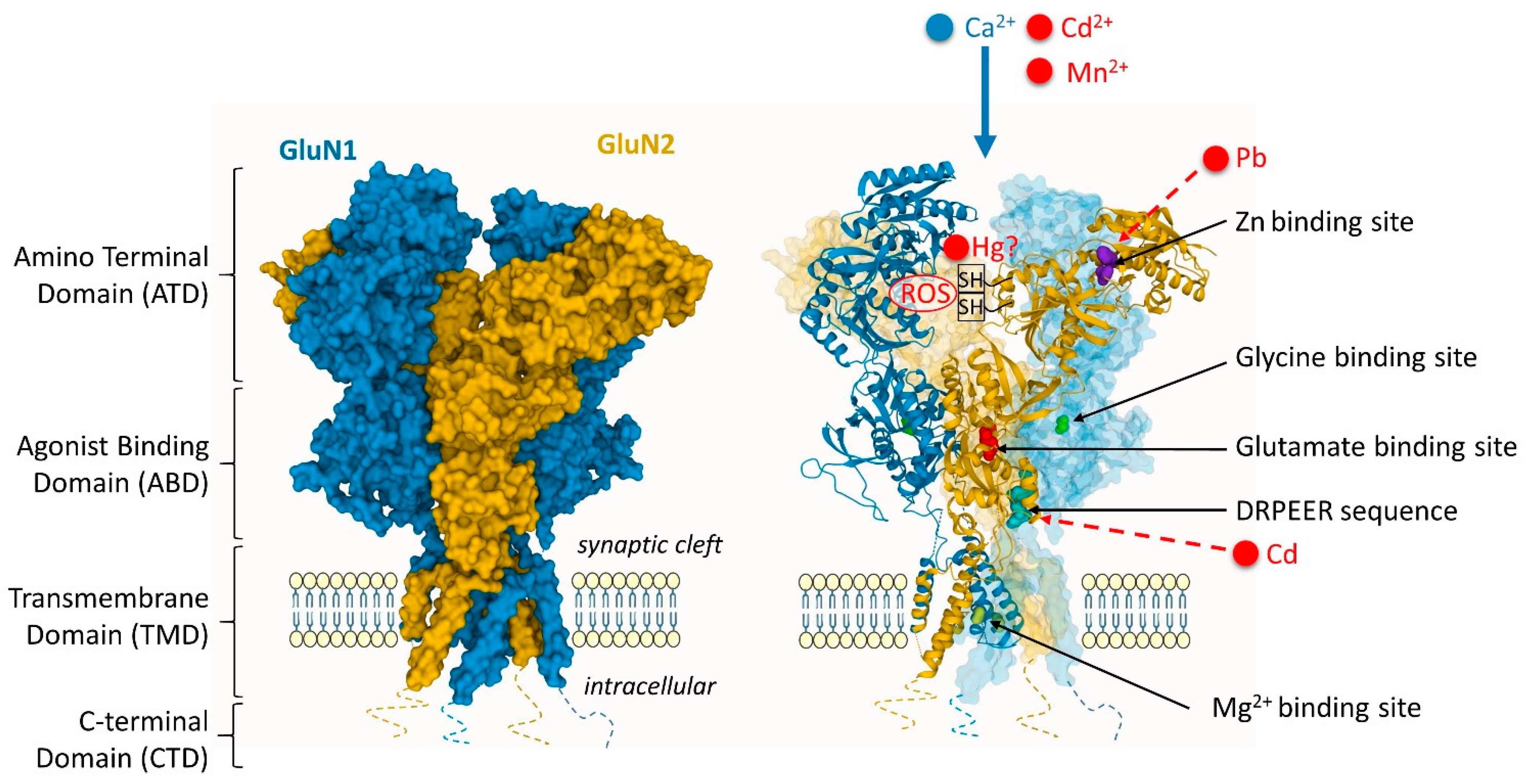
2. Metal Interactions with Neurotransmitter Receptors
2.1. Arsenic
2.2. Cadmium
2.3. Lead
2.4. Manganese
2.5. Mercury
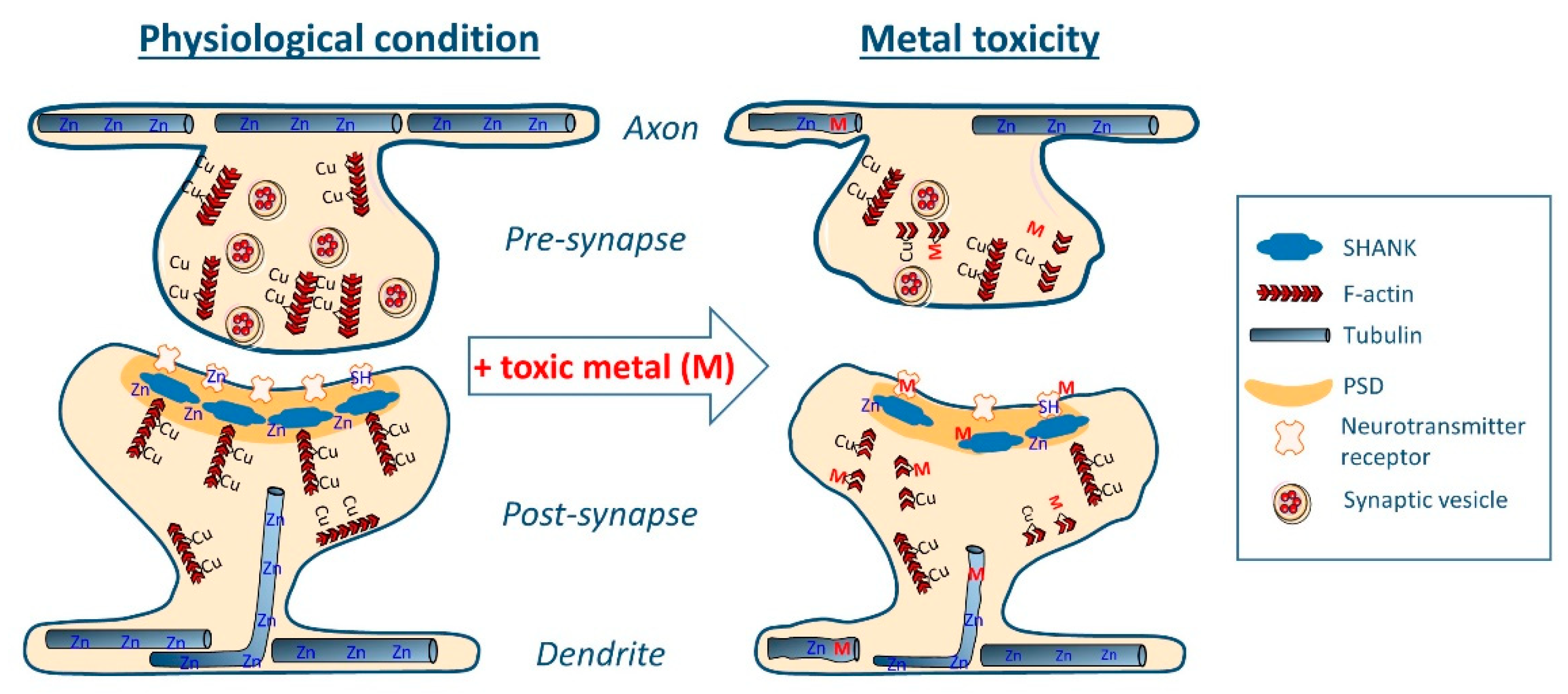
3. Metal-Interactions with Proteins of the Synaptic Structure
3.1. Arsenic
3.2. Cadmium
3.3. Lead
3.4. Manganese
3.5. Mercury
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- World Health Organization (WHO). Ten Chemicals of Public Health Concern. 2020. Available online: https://www.who.int/news-room/photo-story/photo-story-detail/10-chemicals-of-public-health-concern (accessed on 21 July 2021).
- ATSDR (Agency for Toxic Substances and Disease Registry). ATSDR’s Substance Priority List. 2019. Available online: https://www.atsdr.cdc.gov/spl/#2019spl (accessed on 21 July 2021).
- ATSDR (Agency for Toxic Substances and Disease Registry). ATDSR’s Toxicological Profiles. Available online: https://www.atsdr.cdc.gov/toxprofiledocs/index.html (accessed on 21 July 2021).
- Miah, M.R.; Ijomone, O.M.; Okoh, C.O.; Ijomone, O.K.; Akingbade, G.T.; Ke, T.; Krum, B.; da Cunha Martins, A., Jr.; Akinyemi, A.; Aranoff, N.; et al. The effects of manganese overexposure on brain health. Neurochem. Int. 2020, 135, 104688. [Google Scholar] [CrossRef] [PubMed]
- WHO. Arsenic—World Health Organizaiton. Fact Sheets. 2018. Available online: https://www.who.int/news-room/fact-sheets/detail/arsenic (accessed on 16 August 2021).
- Modabbernia, A.; Velthorst, E.; Reichenberg, A. Environmental risk factors for autism: An evidence-based review of systematic reviews and meta-analyses. Mol. Autism 2017, 8, 1–16. [Google Scholar] [CrossRef]
- Chandravanshi, L.; Shiv, K.; Kumar, S. Developmental toxicity of cadmium in infants and children: A review. Environ. Anal. Health Toxicol. 2021, 36, e2021003. [Google Scholar] [CrossRef] [PubMed]
- Weiss, B. Lead, Manganese, and Methylmercury as Risk Factors for Neurobehavioral Impairment in Advanced Age. Int. J. Alzheimer’s Dis. 2011, 2011, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ijomone, O.M.; Ifenatuoha, C.W.; Aluko, O.M.; Ijomone, O.K.; Aschner, M. The aging brain: Impact of heavy metal neurotoxicity. Crit. Rev. Toxicol. 2020, 50, 801–814. [Google Scholar] [CrossRef]
- Monnet-Tschudi, F.; Zurich, M.-G.; Boschat, C.; Corbaz, A.; Honegger, P. Involvement of Environmental Mercury and Lead in the Etiology of Neurodegenerative Diseases. Rev. Environ. Health 2006, 21, 105–117. [Google Scholar] [CrossRef]
- Charlet, L.; Chapron, Y.; Faller, P.; Kirsch, R.; Stone, A.T.; Baveye, P.C. Neurodegenerative diseases and exposure to the environmental metals Mn, Pb, and Hg. Coord. Chem. Rev. 2012, 256, 2147–2163. [Google Scholar] [CrossRef]
- Schofield, K. The Metal Neurotoxins: An Important Role in Current Human Neural Epidemics? Int. J. Environ. Res. Public Health 2017, 14, 1511. [Google Scholar] [CrossRef]
- Siblerud, R.; Mutter, J.; Moore, E.; Naumann, J.; Walach, H. A Hypothesis and Evidence That Mercury May be an Etiological Factor in Alzheimer’s Disease. Int. J. Environ. Res. Public Health 2019, 16, 5152. [Google Scholar] [CrossRef]
- Bakulski, K.M.; Seo, Y.A.; Hickman, R.C.; Brandt, D.; Vadari, H.S.; Hu, H.; Park, S.K. Heavy Metals Exposure and Alzheimer’s Disease and Related Dementias. J. Alzheimer Dis. 2020, 76, 1215–1242. [Google Scholar] [CrossRef]
- Raj, K.; Kaur, P.; Gupta, G.; Singh, S. Metals associated neurodegeneration in Parkinson’s disease: Insight to physiological, pathological mechanisms and management. Neurosci. Lett. 2021, 753, 135873. [Google Scholar] [CrossRef]
- Rahman, A.; Hannan, A.; Uddin, J.; Rahman, S.; Rashid, M.; Kim, B. Exposure to Environmental Arsenic and Emerging Risk of Alzheimer’s Disease: Perspective Mechanisms, Management Strategy, and Future Directions. Toxics 2021, 9, 188. [Google Scholar] [CrossRef]
- Skogheim, T.S.; Weyde, K.V.F.; Engel, S.M.; Aase, H.; Surén, P.; Øie, M.G.; Biele, G.; Reichborn-Kjennerud, T.; Caspersen, I.H.; Hornig, M.; et al. Metal and essential element concentrations during pregnancy and associations with autism spectrum disorder and attention-deficit/hyperactivity disorder in children. Environ. Int. 2021, 152, 106468. [Google Scholar] [CrossRef] [PubMed]
- Arora, M.; Reichenberg, A.; Willfors, C.; Austin, C.; Gennings, C.; Berggren, S.; Lichtenstein, P.; Anckarsäter, H.; Tammimies, K.; Bölte, S. Fetal and postnatal metal dysregulation in autism. Nat. Commun. 2017, 8, 15493. [Google Scholar] [CrossRef] [PubMed]
- Min, J.-Y.; Min, K.-B. Blood cadmium levels and Alzheimer’s disease mortality risk in older US adults. Environ. Health 2016, 15, 69. [Google Scholar] [CrossRef]
- Xu, L.; Zhang, W.; Liu, X.; Zhang, C.; Wang, P.; Zhao, X. Circulatory Levels of Toxic Metals (Aluminum, Cadmium, Mercury, Lead) in Patients with Alzheimer’s Disease: A Quantitative Meta-Analysis and Systematic Review. J. Alzheimer’s Dis. 2018, 62, 361–372. [Google Scholar] [CrossRef]
- Wu, X.; Cobbina, S.J.; Mao, G.; Xu, H.; Zhang, Z.; Yang, L. A review of toxicity and mechanisms of individual and mixtures of heavy metals in the environment. Environ. Sci. Pollut. Res. 2016, 23, 8244–8259. [Google Scholar] [CrossRef]
- Garza-Lombo, C.; Pappa, A.; Panayiotidis, M.I.; Gonsebatt, M.E.; Franco, R. Arsenic-induced neurotoxicity: A mechanistic appraisal. JBIC J. Biol. Inorg. Chem. 2019, 24, 1305–1316. [Google Scholar] [CrossRef]
- Wang, B.; Du, Y. Cadmium and Its Neurotoxic Effects. Oxidative Med. Cell. Longev. 2013, 2013, 898034. [Google Scholar] [CrossRef]
- Pacini, A.; Branca, J.J.V.; Morucci, G. Cadmium-induced neurotoxicity: Still much ado. Neural Regen. Res. 2018, 13, 1879–1882. [Google Scholar] [CrossRef]
- Sadiq, S.; Ghazala, Z.; Chowdhury, A.; Büsselberg, D. Metal Toxicity at the Synapse: Presynaptic, Postsynaptic, and Long-Term Effects. J. Toxicol. 2012, 2012, 132671. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, C. Interaction of metal ions with neurotransmitter receptors and potential role in neurodiseases. BioMetals 2014, 27, 1097–1113. [Google Scholar] [CrossRef] [PubMed]
- Penzes, P.; Cahill, M.; Jones, A.K.; VanLeeuwen, J.-E.; Woolfrey, K.M. Dendritic spine pathology in neuropsychiatric disorders. Nat. Neurosci. 2011, 14, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Wong, G.T.-H.; Chang, R.C.-C.; Law, A.C.-K. A breach in the scaffold: The possible role of cytoskeleton dysfunction in the pathogenesis of major depression. Ageing Res. Rev. 2013, 12, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Kilinc, D. The Emerging Role of Mechanics in Synapse Formation and Plasticity. Front. Cell. Neurosci. 2018, 12, 483. [Google Scholar] [CrossRef]
- Zieger, H.L.; Choquet, D. Nanoscale synapse organization and dysfunction in neurodevelopmental disorders. Neurobiol. Dis. 2021, 158, 105453. [Google Scholar] [CrossRef]
- Paoletti, P.; Bellone, C.; Zhou, Q. NMDA receptor subunit diversity: Impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci. 2013, 14, 383–400. [Google Scholar] [CrossRef]
- Karakas, E.; Furukawa, H. Crystal structure of a heterotetrameric NMDA receptor ion channel. Science 2014, 344, 992–997. [Google Scholar] [CrossRef] [PubMed]
- Huo, T.-G.; Li, W.-K.; Zhang, Y.-H.; Yuan, J.; Gao, L.-Y.; Yuan, Y.; Yang, H.-L.; Jiang, H.; Sun, G.-F. Excitotoxicity Induced by Realgar in the Rat Hippocampus: The Involvement of Learning Memory Injury, Dysfunction of Glutamate Metabolism and NMDA Receptors. Mol. Neurobiol. 2015, 51, 980–994. [Google Scholar] [CrossRef]
- Zhao, F.; Wang, Z.; Liao, Y.; Wang, G.; Jin, Y. Alterations of NMDA and AMPA receptors and their signaling apparatus in the hippocampus of mouse offspring induced by developmental arsenite exposure. J. Toxicol. Sci. 2019, 44, 777–788. [Google Scholar] [CrossRef]
- Luo, J.-H.; Qiu, Z.-Q.; Zhang, L.; Shu, W.-Q. Arsenite exposure altered the expression of NMDA receptor and postsynaptic signaling proteins in rat hippocampus. Toxicol. Lett. 2012, 211, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Chávez, L.A.; Rendón-López, C.R.; Zepeda, A.; Adaya, I.D.S.; Del Razo, L.M.; Gonsebatt, M.E.; Rendón-López, C.R.R. Neurological effects of inorganic arsenic exposure: Altered cysteine/glutamate transport, NMDA expression and spatial memory impairment. Front. Cell. Neurosci. 2015, 9, 21. [Google Scholar] [CrossRef] [PubMed]
- Silva-Adaya, D.; Ramos-Chávez, L.A.; Petrosyan, P.; González-Alfonso, W.L.; Pérez-Acosta, A.; Gonsebatt, M.E. Early Neurotoxic Effects of Inorganic Arsenic Modulate Cortical GSH Levels Associated with the Activation of the Nrf2 and NFκB Pathways, Expression of Amino Acid Transporters and NMDA Receptors and the Production of Hydrogen Sulfide. Front. Cell. Neurosci. 2020, 14. [Google Scholar] [CrossRef] [PubMed]
- Nelson-Mora, J.; Escobar, M.L.; Rodríguez-Durán, L.; Massieu, L.; Montiel, T.; Rodriguez, V.; Hernández-Mercado, K.; Gonsebatt, M.E. Gestational exposure to inorganic arsenic (iAs3+) alters glutamate disposition in the mouse hippocampus and ionotropic glutamate receptor expression leading to memory impairment. Arch. Toxicol. 2017, 92, 1037–1048. [Google Scholar] [CrossRef] [PubMed]
- Maekawa, F.; Tsuboi, T.; Oya, M.; Aung, K.H.; Tsukahara, S.; Pellerin, L.; Nohara, K. Effects of sodium arsenite on neurite outgrowth and glutamate AMPA receptor expression in mouse cortical neurons. NeuroToxicology 2013, 37, 197–206. [Google Scholar] [CrossRef]
- Mónaco, N.M.; Bartos, M.; Dominguez, S.; Gallegos, C.; Bras, C.; Esandi, M.D.C.; Bouzat, C.; Giannuzzi, L.; Minetti, A.; Gumilar, F. Low arsenic concentrations impair memory in rat offpring exposed during pregnancy and lactation: Role of α7 nicotinic receptor, glutamate and oxidative stress. NeuroToxicology 2018, 67, 37–45. [Google Scholar] [CrossRef]
- Chandravanshi, L.P.; Gupta, R.; Shukla, R.K. Arsenic-Induced Neurotoxicity by Dysfunctioning Cholinergic and Dopaminergic System in Brain of Developing Rats. Biol. Trace Element Res. 2019, 189, 118–133. [Google Scholar] [CrossRef]
- Ávila, C.L.M.; Limón-Pacheco, J.H.; Giordano, M.; Rodríguez, V.M. Chronic Exposure to Arsenic in Drinking Water Causes Alterations in Locomotor Activity and Decreases Striatal mRNA for the D2 Dopamine Receptor in CD1 Male Mice. J. Toxicol. 2016, 2016, 1–10. [Google Scholar] [CrossRef]
- Usai, C.; Barberis, A.; Moccagatta, L.; Marchetti, C.; Neurochem, J. Pathways of cadmium influx in mammalian neurons. J. Neurochem. 2008, 72, 2154–2161. [Google Scholar] [CrossRef] [PubMed]
- Tu, Y.-C.; Yang, Y.-C.; Kuo, C.-C. Modulation of NMDA channel gating by Ca2+ and Cd2+ binding to the external pore mouth. Sci. Rep. 2016, 6, 37029. [Google Scholar] [CrossRef]
- Watanabe, J.; Beck, C.; Kuner, T.; Premkumar, L.S.; Wollmuth, L.P. DRPEER: A Motif in the Extracellular Vestibule Conferring High Ca2+Flux Rates in NMDA Receptor Channels. J. Neurosci. 2002, 22, 10209–10216. [Google Scholar] [CrossRef]
- Wang, S.; Hu, P.; Wang, H.; Wang, M.; Chen, J.; Tang, J.; Ruan, D. Effects of Cd2+ on AMPA receptor-mediated synaptic transmission in rat hippocampal CA1 area. Toxicol. Lett. 2008, 176, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Del Pino, J.; Zeballos, G.; Anadon, M.J.; Díaz, M.J.; Moya, M.T.F.; Díaz, G.G.; García, J.; Lobo, M.; Frejo, M.T. Muscarinic M1 receptor partially modulates higher sensitivity to cadmium-induced cell death in primary basal forebrain cholinergic neurons: A cholinesterase variants dependent mechanism. Toxicol. 2016, 361-362, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Moyano, P.; de Frias, M.; Lobo, M.; Anadon, M.J.; Sola, E.; Pelayo, A.; Díaz, M.J.; Frejo, M.T.; Del Pino, J. Cadmium induced ROS alters M1 and M3 receptors, leading to SN56 cholinergic neuronal loss, through AChE variants disruption. Toxicol. 2018, 394, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Celentano, J.J.; Gyenes, M.; Gibbs, T.; Farb, D. Negative modulation of the gamma-aminobutyric acid response by extracellular zinc. Mol. Pharmacol. 1991, 40, 766–773. [Google Scholar] [PubMed]
- Zhao, Q.; Gao, L.; Liu, Q.; Cao, Y.; He, Y.; Hu, A.; Chen, W.; Cao, J.; Hu, C.; Li, L.; et al. Impairment of learning and memory of mice offspring at puberty, young adulthood, and adulthood by low-dose Cd exposure during pregnancy and lactation via GABAAR α5 and δ subunits. Ecotoxicol. Environ. Saf. 2018, 166, 336–344. [Google Scholar] [CrossRef]
- Gupta, R.; Shukla, R.K.; Pandey, A.; Sharma, T.; Dhuriya, Y.; Srivastava, P.; Singh, M.P.; Siddiqi, M.I.; Pant, A.B.; Khanna, V.K. Involvement of PKA/DARPP-32/PP1α and β- arrestin/Akt/GSK-3β Signaling in Cadmium-Induced DA-D2 Receptor-Mediated Motor Dysfunctions: Protective Role of Quercetin. Sci. Rep. 2018, 8, 1–18. [Google Scholar] [CrossRef]
- Hansen, K.B.; Yi, F.; Perszyk, R.; Furukawa, H.; Wollmuth, L.P.; Gibb, A.; Traynelis, S.F. Structure, function, and allosteric modulation of NMDA receptors. J. Gen. Physiol. 2018, 150, 1081–1105. [Google Scholar] [CrossRef]
- Büsselberg, D.; Michael, D.; Platt, B. Pb2+ reduces voltage- andN-methyl-d-aspartate (NMDA)-activated calcium channel currents. Cell. Mol. Neurobiol. 1994, 14, 711–722. [Google Scholar] [CrossRef]
- Büsselberg, D. Calcium channels as target sites of heavy metals. Toxicol. Lett. 1995, 82, 255–261. [Google Scholar] [CrossRef]
- Ordemann, J.M.; Austin, R.N. Lead neurotoxicity: Exploring the potential impact of lead substitution in zinc-finger proteins on mental health. Metallomics 2016, 8, 579–588. [Google Scholar] [CrossRef] [PubMed]
- Jalali-Yazdi, F.; Chowdhury, S.; Yoshioka, C.; Gouaux, E. Mechanisms for Zinc and Proton Inhibition of the GluN1/GluN2A NMDA Receptor. Cell 2018, 175, 1520–1532.e15. [Google Scholar] [CrossRef] [PubMed]
- Guilarte, T.R.; Miceli, R.C.; Jett, D.A. Biochemical evidence of an interaction of lead at the zinc allosteric sites of the NMDA re-ceptor complex: Effects of neuronal development. Neurotoxicology 1995, 16, 63–71. [Google Scholar] [PubMed]
- Gavazzo, P.; Zanardi, I.; Baranowska-Bosiacka, I.; Marchetti, C. Molecular determinants of Pb2+ interaction with NMDA receptor channels. Neurochem. Int. 2008, 52, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Neal, A.P.; Guilarte, T.R. Mechanisms of lead and manganese neurotoxicity. Toxicol. Res. 2013, 2, 99–114. [Google Scholar] [CrossRef]
- Neal, A.P.; Worley, P.F.; Guilarte, T.R. Lead exposure during synaptogenesis alters NMDA receptor targeting via NMDA receptor inhibition. NeuroToxicology 2011, 32, 281–289. [Google Scholar] [CrossRef]
- Guilarte, T.R.; McGlothan, J.L. Hippocampal NMDA receptor mRNA undergoes subunit specific changes during devel-opmental lead exposure. Brain Res. 1998, 790, 98–107. [Google Scholar] [CrossRef]
- Nihei, M.K.; Desmond, N.L.; McGlothan, J.L.; Kuhlmann, A.C.; Guilarte, T.R. N-methyl-d-aspartate receptor subunit changes are associated with lead-induced deficits of long-term potentiation and spatial learning. Neuroscience 2000, 99, 233–242. [Google Scholar] [CrossRef]
- Zhang, X.-Y.; Liu, A.-P.; Ruan, D.-Y.; Liu, J. Effect of developmental lead exposure on the expression of specific NMDA receptor subunit mRNAs in the hippocampus of neonatal rats by digoxigenin-labeled in situ hybridization histochemistry. Neurotoxicology Teratol. 2002, 24, 149–160. [Google Scholar] [CrossRef]
- Guilarte, T.R.; McGlothan, J.L.; Nihei, M.K. Hippocampal expression of N-methyl-d-aspartate receptor (NMDAR1) subunit splice variant mRNA is altered by developmental exposure to Pb2+. Mol. Brain Res. 2000, 76, 299–305. [Google Scholar] [CrossRef]
- Basha, R.; Wei, W.; Brydie, M.; Razmiafshari, M.; Zawia, N. Lead-induced developmental perturbations in hippocampal Sp1 DNA-binding are prevented by zinc supplementation: In vivo evidence for Pb and Zn competition. Int. J. Dev. Neurosci. 2003, 21, 1–12. [Google Scholar] [CrossRef]
- Wang, T.; Guan, R.-L.; Liu, M.-C.; Shen, X.-F.; Chen, J.Y.; Zhao, M.-G.; Luo, W.-J. Lead Exposure Impairs Hippocampus Related Learning and Memory by Altering Synaptic Plasticity and Morphology During Juvenile Period. Mol. Neurobiol. 2015, 53, 3740–3752. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, M.; Lasley, S. Developmental lead (Pb) exposure reduces the ability of the NMDA antagonist MK-801 to suppress long-term potentiation (LTP) in the rat dentate gyrus, in vivo. Neurotoxicology Teratol. 2007, 29, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Neal, A.P.; Guilarte, T.R. Molecular Neurobiology of Lead (Pb2+): Effects on Synaptic Function. Mol. Neurobiol. 2010, 42, 151–160. [Google Scholar] [CrossRef] [PubMed]
- White, L.; Cory-Slechta, D.; Gilbert, M.; Tiffany-Castiglioni, E.; Zawia, N.; Virgolini, M.; Rossi-George, A.; Lasley, S.; Qian, Y.; Basha, R. New and evolving concepts in the neurotoxicology of lead. Toxicol. Appl. Pharmacol. 2007, 225, 1–27. [Google Scholar] [CrossRef]
- Ishida, K.; Kotake, Y.; Miyara, M.; Aoki, K.; Sanoh, S.; Kanda, Y.; Ohta, S. Involvement of decreased glutamate receptor subunit GluR2 expression in lead-induced neuronal cell death. J. Toxicol. Sci. 2013, 38, 513–521. [Google Scholar] [CrossRef]
- Ishida, K.; Kotake, Y.; Sanoh, S.; Ohta, S. Lead-Induced ERK Activation Is Mediated by GluR2 Non-containing AMPA Receptor in Cortical Neurons. Biol. Pharm. Bull. 2017, 40, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.-J.; Zou, R.-X.; He, H.-M.; Tang, Y.-Q.; Wang, H.-L. Effect of Pb Exposure on Synaptic Scaling Through Regulation of AMPA Receptor Surface Trafficking. Toxicol. Sci. 2018, 165, 224–231. [Google Scholar] [CrossRef]
- Devi, C.; Reddy, G.; Prasanthi, R.; Chetty, C.; Reddy, G. Developmental lead exposure alters mitochondrial monoamine oxidase and synaptosomal catecholamine levels in rat brain. Int. J. Dev. Neurosci. 2005, 23, 375–381. [Google Scholar] [CrossRef]
- Zhang, S.; Zhou, Z.; Fu, J. Effect of manganese chloride exposure on liver and brain mitochondria function in rats. Environ. Res. 2003, 93, 149–157. [Google Scholar] [CrossRef]
- Carmona, A.; Malard, V.; Avazeri, E.; Roudeau, S.; Porcaro, F.; Paredes, E.; Vidaud, C.; Bresson, C.; Ortega, R. Uranium exposure of human dopaminergic cells results in low cytotoxicity, accumulation within sub-cytoplasmic regions, and down regulation of MAO-B. NeuroToxicology 2018, 68, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Lasley, S.M.; Gilbert, M.E. Rat Hippocampal Glutamate and GABA Release Exhibit Biphasic Effects as a Function of Chronic Lead Exposure Level. Toxicol. Sci. 2002, 66, 139–147. [Google Scholar] [CrossRef]
- Xiao, C.; Gu, Y.; Zhou, C.-Y.; Wang, L.; Zhang, M.-M.; Ruan, D.-Y. Pb2+ impairs GABAergic synaptic transmission in rat hippocampal slices: A possible involvement of presynaptic calcium channels. Brain Res. 2006, 1088, 93–100. [Google Scholar] [CrossRef]
- Finkelstein, Y.; Milatovic, D.; Aschner, M. Modulation of cholinergic systems by manganese. NeuroToxicology 2007, 28, 1003–1014. [Google Scholar] [CrossRef]
- Goncalves Soares, A.T.; Silva, A.D.C.; Tinkov, A.A.; Khan, H.; Santamaría, A.; Skalnaya, M.G.; Skalny, A.V.; Tsatsakis, A.; Bowman, A.B.; Aschner, M.; et al. The impact of manganese on neurotransmitter systems. J. Trace Elements Med. Biol. 2020, 61, 126554. [Google Scholar] [CrossRef]
- Tinkov, A.; Paoliello, M.; Mazilina, A.; Skalny, A.; Martins, A.; Voskresenskaya, O.; Aaseth, J.; Santamaria, A.; Notova, S.; Tsatsakis, A.; et al. Molecular Targets of Manganese-Induced Neurotoxicity: A Five-Year Update. Int. J. Mol. Sci. 2021, 22, 4646. [Google Scholar] [CrossRef]
- Mayer, M.L.; Westbrook, G.L. Permeation and block of N-methyl-D-aspartic acid receptor channels by divalent cations in mouse cultured central neurones. J. Physiol. 1987, 394, 501–527. [Google Scholar] [CrossRef]
- Guilarte, T.R.; Chen, M.-K. Manganese inhibits NMDA receptor channel function: Implications to psychiatric and cognitive effects. NeuroToxicology 2007, 28, 1147–1152. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Xu, Z.-F.; Deng, Y. Effect of manganese exposure on intracellular Ca2+ homeostasis and expression of NMDA receptor subunits in primary cultured neurons. NeuroToxicology 2009, 30, 941–949. [Google Scholar] [CrossRef] [PubMed]
- Song, Q.; Deng, Y.; Yang, X.; Bai, Y.; Xu, B.; Liu, W.; Zheng, W.; Wang, C.; Zhang, M.; Xu, Z. Manganese-Disrupted Interaction of Dopamine D1 and NMDAR in the Striatum to Injury Learning and Memory Ability of Mice. Mol. Neurobiol. 2015, 53, 6745–6758. [Google Scholar] [CrossRef]
- Wang, L.; Fu, H.; Liu, B.; Liu, X.; Chen, W.; Yu, X. The effect of postnatal manganese exposure on the NMDA receptor signaling pathway in rat hippocampus. J. Biochem. Mol. Toxicol. 2017, 31, e21969. [Google Scholar] [CrossRef]
- Calabresiab, P.; Ammassari-Teule, M.; Gubelliniad, P.; Sancesarioa, G.; Morello, M.; Centonze, D.; Marfia, G.A.; Saullea, E.; Passinoc, E.; Picconi, B.; et al. A Synaptic Mechanism Underlying the Behavioral Abnormalities Induced by Manganese Intoxication. Neurobiol. Dis. 2001, 8, 419–432. [Google Scholar] [CrossRef] [PubMed]
- Burton, N.C.; Schneider, J.S.; Syversen, T.; Guilarte, T.R. Effects of Chronic Manganese Exposure on Glutamatergic and GABAergic Neurotransmitter Markers in the Nonhuman Primate Brain. Toxicol. Sci. 2009, 111, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Sakata, M.; Watanabe, M.; Aikawa, Y.; Fujii, H. The entry of manganese ions into the brain is accelerated by the activation of N-methyl-d-aspartate receptors. Neurosci. 2008, 154, 732–740. [Google Scholar] [CrossRef]
- Ma, Z.; Liu, K.; Li, X.-R.; Wang, C.; Liu, C.; Yan, D.-Y.; Deng, Y.; Liu, W.; Xu, B. Alpha-synuclein is involved in manganese-induced spatial memory and synaptic plasticity impairments via TrkB/Akt/Fyn-mediated phosphorylation of NMDA receptors. Cell Death Dis. 2020, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Tan, J.; Xu, X.; Yang, H.; Wu, F.; Xu, B.; Liu, W.; Shi, P.; Xu, Z.; Deng, Y. Prepubertal overexposure to manganese induce precocious puberty through GABAA receptor/nitric oxide pathway in immature female rats. Ecotoxicol. Environ. Saf. 2020, 188, 109898. [Google Scholar] [CrossRef]
- Sun, Y.; He, Y.; Yang, L.; Liang, D.; Shi, W.; Zhu, X.; Jiang, Y.; Ou, C. Manganese induced nervous injury by α-synuclein accumulation via ATP-sensitive K(+) channels and GABA receptors. Toxicol. Lett. 2020, 332, 164–170. [Google Scholar] [CrossRef]
- Stredrick, D.L.; Stokes, A.H.; Worst, T.J.; Freeman, W.; Johnson, E.A.; Lash, L.; Aschner, M.; Vrana, K.E. Manganese-Induced Cytotoxicity in Dopamine-Producing Cells. NeuroToxicology 2004, 25, 543–553. [Google Scholar] [CrossRef]
- Nam, J.; Kim, K. Abnormal Motor Function and the Expression of Striatal Dopamine D2 Receptors in Manganese-Treated Mice. Biol. Pharm. Bull. 2008, 31, 1894–1897. [Google Scholar] [CrossRef][Green Version]
- Kern, C.H.; Stanwood, G.; Smith, D.R. Preweaning manganese exposure causes hyperactivity, disinhibition, and spatial learning and memory deficits associated with altered dopamine receptor and transporter levels. Synapse 2010, 64, 363–378. [Google Scholar] [CrossRef]
- McDougall, S.A.; Der-Ghazarian, T.; Britt, C.E.; Varela, F.A.; Crawford, C.A. Postnatal manganese exposure alters the expression of D2L and D2S receptor isoforms: Relationship to PKA activity and Akt levels. Synapse 2010, 65, 583–591. [Google Scholar] [CrossRef] [PubMed]
- Criswell, S.R.; Warden, M.N.; Nielsen, S.S.; Perlmutter, J.S.; Moerlein, S.M.; Sheppard, L.; Lenox-Krug, J.; Checkoway, H.; Racette, B.A. Selective D2 receptor PET in manganese-exposed workers. Neurology 2018, 91, e1022–e1030. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, K.-I.; Nakanishi, H.; Moriguchi, S.; Fukuyama, N.; Eto, K.; Wakamiya, J.; Murao, K.; Arimura, K.; Osame, M. Involvement of enhanced sensitivity of N-methyl-D-aspartate receptors in vulnerability of developing cortical neurons to methylmercury neurotoxicity. Brain Res. 2001, 901, 252–258. [Google Scholar] [CrossRef]
- Juárez, B.I.; Portillo-Salazar, H.; González-Amaro, R.; Mandeville, P.; Aguirre, J.R.; Jiménez, M.E. Participation of N-methyl-d-aspartate receptors on methylmercury-induced DNA damage in rat frontal cortex. Toxicology 2005, 207, 223–229. [Google Scholar] [CrossRef]
- Vidal, L.; Duran, R.; Faro, L.; Campos, F.; Cervantes, R.; Alfonso, M. Protection from inorganic mercury effects on the in vivo dopamine release by ionotropic glutamate receptor antagonists and nitric oxide synthase inhibitors. Toxicology 2007, 238, 140–146. [Google Scholar] [CrossRef]
- Wyrembek, P.; Szczuraszek, K.; Majewska, M.D.; Mozrzymas, J. Intermingled modulatory and neurotoxic effects of thimerosal and mercuric ions on electrophysiological responses to GABA and NMDA in hippocampal neurons. J. Physiol. Pharmacol. 2010, 61, 753–758. [Google Scholar]
- Yuan, Y.; Atchison, W.D. Methylmercury Differentially Affects GABA A Receptor-Mediated Spontaneous IPSCs in Purkinje and Granule Cells of Rat Cerebellar Slices. J. Physiol. 2003, 550 Pt 1, 191–204. [Google Scholar] [CrossRef]
- Olczak, M.; Duszczyk, M.; Mierzejewski, P.; Meyza, K.; Majewska, M.D. Persistent behavioral impairments and alterations of brain dopamine system after early postnatal administration of thimerosal in rats. Behav. Brain Res. 2011, 223, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Coccini, T.; Roda, E.; Castoldi, A.F.; Poli, D.; Goldoni, M.; Vettori, M.V.; Mutti, A.; Manzo, L. Developmental exposure to methylmercury and 2,2′,4,4′,5,5′-hexachlorobiphenyl (PCB153) affects cerebral dopamine D1-like and D2-like receptors of weanling and pubertal rats. Arch. Toxicol. 2011, 85, 1281–1294. [Google Scholar] [CrossRef]
- Scheuhammer, A.M.; Cherian, M. Effects of heavy metal cations, sulfhydryl reagents and other chemical agents on striatal D2 dopamine receptors. Biochem. Pharmacol. 1985, 34, 3405–3413. [Google Scholar] [CrossRef]
- Basu, N.; Kwan, M.; Chan, H.M. Mercury but not Organochlorines Inhibits Muscarinic Cholinergic Receptor Binding in the Cerebrum of Ringed Seals (Phoca hispida). J. Toxicol. Environ. Health Part A 2006, 69, 1133–1143. [Google Scholar] [CrossRef]
- Basu, N.; Scheuhammer, A.M.; Rouvinen-Watt, K.; Evans, R.D.; Grochowina, N.; Chan, L.H. The effects of mercury on muscarinic cholinergic receptor subtypes (M1 and M2) in captive mink. NeuroToxicology 2008, 29, 328–334. [Google Scholar] [CrossRef]
- Sarowar, T.; Grabrucker, A.M. Actin-Dependent Alterations of Dendritic Spine Morphology in Shankopathies. Neural Plast. 2016, 2016, 1–15. [Google Scholar] [CrossRef]
- Joensuu, M.; Lanoue, V.; Hotulainen, P. Dendritic spine actin cytoskeleton in autism spectrum disorder. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2018, 84 Pt B, 362–381. [Google Scholar] [CrossRef]
- Lasser, M.; Tiber, J.; Lowery, L.A. The Role of the Microtubule Cytoskeleton in Neurodevelopmental Disorders. Front. Cell. Neurosci. 2018, 12, 165. [Google Scholar] [CrossRef]
- Dent, E.W. Of microtubules and memory: Implications for microtubule dynamics in dendrites and spines. Mol. Biol. Cell 2017, 28, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Pelucchi, S.; Stringhi, R.; Marcello, E. Dendritic Spines in Alzheimer’s Disease: How the Actin Cytoskeleton Contributes to Synaptic Failure. Int. J. Mol. Sci. 2020, 21, 908. [Google Scholar] [CrossRef] [PubMed]
- Perrin, L.; Roudeau, S.; Carmona, A.; Domart, F.; Petersen, J.D.; Bohic, S.; Yang, Y.; Cloetens, P.; Ortega, R. Zinc and Copper Effects on Stability of Tubulin and Actin Networks in Dendrites and Spines of Hippocampal Neurons. ACS Chem. Neurosci. 2017, 8, 1490–1499. [Google Scholar] [CrossRef] [PubMed]
- Domart, F.; Cloetens, P.; Roudeau, S.; Carmona, A.; Verdier, E.; Choquet, D.; Ortega, R. Correlating STED and synchrotron XRF nano-imaging unveils cosegregation of metals and cytoskeleton proteins in dendrites. eLife 2020, 9. [Google Scholar] [CrossRef]
- Baron, M.K.; Boeckers, T.M.; Vaida, B.; Faham, S.; Gingery, M.; Sawaya, M.R.; Salyer, D.; Gundelfinger, E.D.; Bowie, J.U. An Architectural Framework That May Lie at the Core of the Postsynaptic Density. Science 2006, 311, 531–535. [Google Scholar] [CrossRef] [PubMed]
- Bucher, M.; Fanutza, T.; Mikhaylova, M. Cytoskeletal makeup of the synapse: Shaft versus spine. Cytoskeleton 2019, 77, 55–64. [Google Scholar] [CrossRef]
- Parato, J.; Bartolini, F. The microtubule cytoskeleton at the synapse. Neurosci. Lett. 2021, 753, 135850. [Google Scholar] [CrossRef] [PubMed]
- Durand, C.M.; Betancur, C.; Boeckers, T.M.; Bockmann, J.; Chaste, P.; Fauchereau, F.; Nygren, G.; Rastam, M.; Gillberg, I.C.; Anckarsäter, H.; et al. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat. Genet. 2006, 39, 25–27. [Google Scholar] [CrossRef] [PubMed]
- Grabrucker, A.; Knight, M.J.; Proepper, C.; Bockmann, J.; Joubert, M.; Rowan, M.; Nienhaus, G.U.; Garner, C.; Bowie, J.U.; Kreutz, M.R.; et al. Concerted action of zinc and ProSAP/Shank in synaptogenesis and synapse maturation. EMBO J. 2011, 30, 569–581. [Google Scholar] [CrossRef]
- Grabrucker, S.; Jannetti, L.; Eckert, M.; Gaub, S.; Chhabra, R.; Pfaender, S.; Mangus, K.; Reddy, P.P.; Rankovic, V.; Schmeisser, M.J.; et al. Zinc deficiency dysregulates the synaptic ProSAP/Shank scaffold and might contribute to autism spectrum disorders. Brain 2014, 137 Pt 1, 137–152. [Google Scholar] [CrossRef]
- Hagmeyer, S.; Sauer, A.K.; Grabrucker, A.M. Prospects of Zinc Supplementation in Autism Spectrum Disorders and Shankopathies Such as Phelan McDermid Syndrome. Front. Synaptic Neurosci. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Fourie, C.; Vyas, Y.; Lee, K.; Jung, Y.; Garner, C.; Montgomery, J.M. Dietary Zinc Supplementation Prevents Autism Related Behaviors and Striatal Synaptic Dysfunction in Shank3 Exon 13–16 Mutant Mice. Front. Cell. Neurosci. 2018, 12, 374. [Google Scholar] [CrossRef] [PubMed]
- Hagmeyer, S.; Mangus, K.; Boeckers, T.M.; Grabrucker, A.M. Effects of Trace Metal Profiles Characteristic for Autism on Synapses in Cultured Neurons. Neural Plast. 2015, 2015, 1–16. [Google Scholar] [CrossRef]
- Vahidnia, A.; Romijn, F.; van der Voet, G.; de Wolff, F. Arsenic-induced neurotoxicity in relation to toxicokinetics: Effects on sciatic nerve proteins. Chem. Interact. 2008, 176, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Aung, K.H.; Kurihara, R.; Nakashima, S.; Maekawa, F.; Nohara, K.; Kobayashi, T.; Tsukahara, S. Inhibition of neurite outgrowth and alteration of cytoskeletal gene expression by sodium arsenite. NeuroToxicology 2012, 34, 226–235. [Google Scholar] [CrossRef]
- Stern, M.; Gierse, A.; Tan, S.; Bicker, G. Human Ntera2 cells as a predictive in vitro test system for developmental neurotoxicity. Arch. Toxicol. 2013, 88, 127–136. [Google Scholar] [CrossRef]
- Zhao, Y.; Toselli, P.; Li, W. Microtubules as a Critical Target for Arsenic Toxicity in Lung Cells in Vitro and in Vivo. Int. J. Environ. Res. Public Health 2012, 9, 474–495. [Google Scholar] [CrossRef]
- Ge, Y.; Song, X.; Chen, L.; Hu, D.; Hua, L.; Cui, Y.; Liu, J.; An, Z.; Yin, Z.; Ning, H. Cadmium induces actin cytoskeleton alterations and dysfunction in Neuro-2a cells. Environ. Toxicol. 2019, 34, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Di Liegro, I.; Gerspacher, C.; Scheuber, U.; Schiera, G.; Proia, P.; Gygax, D. The effect of cadmium on brain cells in culture. Int. J. Mol. Med. 2009, 24, 311–318. [Google Scholar] [CrossRef][Green Version]
- Wang, T.; Wang, Q.; Song, R.; Zhang, Y.; Yang, J.; Wang, Y.; Yuan, Y.; Bian, J.; Liu, X.; Gu, J.; et al. Cadmium induced inhibition of autophagy is associated with microtubule disruption and mitochondrial dysfunction in primary rat cerebral cortical neurons. Neurotoxicology Teratol. 2016, 53, 11–18. [Google Scholar] [CrossRef]
- Forcella, M.; Lau, P.; Oldani, M.; Melchioretto, P.; Bogni, A.; Gribaldo, L.; Fusi, P.; Urani, C. Neuronal specific and non-specific re-sponses to cadmium possibly involved in neurodegeneration: A toxicogenomics study in a human neuronal cell model. Neu-rotoxicology 2020, 76, 162–173. [Google Scholar] [CrossRef]
- Go, Y.-M.; Orr, M.; Jones, D.P. Actin cytoskeleton redox proteome oxidation by cadmium. Am. J. Physiol. Cell. Mol. Physiol. 2013, 305, L831–L843. [Google Scholar] [CrossRef] [PubMed]
- Choi, W.-S.; Kim, S.-J.; Kim, J.S. Inorganic lead (Pb)- and mercury (Hg)-induced neuronal cell death involves cytoskeletal reorganization. Lab. Anim. Res. 2011, 27, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, H.-P.; Faulstich, H.; Hänsch, G.; Doenges, K.; Stournaras, C. The interaction of triethyl lead with tubulin and microtubules. Mutat. Res. Mol. Mech. Mutagen. 1988, 201, 293–302. [Google Scholar] [CrossRef]
- Stanwood, G.D.; Leitch, D.B.; Savchenko, V.; Wu, J.; Fitsanakis, V.A.; Anderson, D.J.; Stankowski, J.N.; Aschner, M.; McLaughlin, B. Manganese exposure is cytotoxic and alters dopaminergic and GABAergic neurons within the basal ganglia. J. Neurochem. 2009, 110, 378–389. [Google Scholar] [CrossRef]
- Parsons-White, A.B.; Spitzer, N. Environmentally relevant manganese overexposure alters neural cell morphology and differentiation in vitro. Toxicol. Vitr. 2018, 50, 22–28. [Google Scholar] [CrossRef]
- Stoiber, T.; Degen, G.H.; Bolt, H.M.; Unger, E. Interaction of mercury(II) with the microtubule cytoskeleton in IMR-32 neuroblastoma cells. Toxicol. Lett. 2004, 151, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; He, B.; Hu, L.; Huang, X.; Yun, Z.; Liu, R.; Zhou, Q.; Jiang, G. Characterization of mercury-binding proteins in human neuroblastoma SK-N-SH cells with immobilized metal affinity chromatography. Talanta 2018, 178, 811–817. [Google Scholar] [CrossRef] [PubMed]
- Nong, Q.; Dong, H.; Liu, Y.; Liu, L.; He, B.; Huang, Y.; Jiang, J.; Luan, T.; Chen, B.; Hu, L. Characterization of the mercury-binding proteins in tuna and salmon sashimi: Implications for health risk of mercury in food. Chemosphere 2021, 263, 128110. [Google Scholar] [CrossRef]
- Ajsuvakova, O.P.; Tinkov, A.A.; Aschner, M.; Rocha, J.B.; Michalke, B.; Skalnaya, M.G.; Skalny, A.V.; Butnariu, M.; Dadar, M.; Sarac, I.; et al. Sulfhydryl groups as targets of mercury toxicity. Coord. Chem. Rev. 2020, 417, 213343. [Google Scholar] [CrossRef]
- Castoldi, A.F.; Barni, S.; Turin, I.; Gandini, C.; Manzo, L. Early acute necrosis, delayed apoptosis and cytoskeletal breakdown in cultured cerebellar granule neurons exposed to methylmercury. J. Neurosci. Res. 2000, 59, 775–787. [Google Scholar] [CrossRef]
- Xu, F.; Farkas, S.; Kortbeek, S.; Zhang, F.-X.; Chen, L.; Zamponi, G.W.; Syed, N.I. Mercury-induced toxicity of rat cortical neurons is mediated through N-methyl-D-Aspartate receptors. Mol. Brain 2012, 5, 30. [Google Scholar] [CrossRef]
- Moran, J.; Sabanero, M.; Meza, I.; Pasantes-Morales, H. Changes of actin cytoskeleton during swelling and regulatory volume decrease in cultured astrocytes. Am. J. Physiol. Physiol. 1996, 271, C1901–C1907. [Google Scholar] [CrossRef] [PubMed]
- Rao, K.R.; Reddy, P.; Hazell, A.; Norenberg, M. Manganese induces cell swelling in cultured astrocytes. NeuroToxicology 2007, 28, 807–812. [Google Scholar] [CrossRef]
- Mori, H.; Sasaki, G.; Nishikawa, M.; Hara, M. Effects of subcytotoxic cadmium on morphology of glial fibrillary acidic protein network in astrocytes derived from murine neural stem/progenitor cells. Environ. Toxicol. Pharmacol. 2015, 40, 639–644. [Google Scholar] [CrossRef] [PubMed]
- Perea, G.; Navarrete, M.; Araque, A. Tripartite synapses: Astrocytes process and control synaptic information. Trends Neurosci. 2009, 32, 421–431. [Google Scholar] [CrossRef] [PubMed]
- Arendt, T. Synaptic degeneration in Alzheimer’s disease. Acta Neuropathol. 2009, 118, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Crouch, P.J.; Barnham, K.J. Therapeutic Redistribution of Metal Ions To Treat Alzheimer’s Disease. Accounts Chem. Res. 2012, 45, 1604–1611. [Google Scholar] [CrossRef] [PubMed]
- Squitti, R.; Pal, A.; Picozza, M.; Avan, A.; Ventriglia, M.; Rongioletti, M.C.; Hoogenraad, T. Zinc Therapy in Early Alzheimer’s Disease: Safety and Potential Therapeutic Efficacy. Biomology 2020, 10, 1164. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carmona, A.; Roudeau, S.; Ortega, R. Molecular Mechanisms of Environmental Metal Neurotoxicity: A Focus on the Interactions of Metals with Synapse Structure and Function. Toxics 2021, 9, 198. https://doi.org/10.3390/toxics9090198
Carmona A, Roudeau S, Ortega R. Molecular Mechanisms of Environmental Metal Neurotoxicity: A Focus on the Interactions of Metals with Synapse Structure and Function. Toxics. 2021; 9(9):198. https://doi.org/10.3390/toxics9090198
Chicago/Turabian StyleCarmona, Asuncion, Stéphane Roudeau, and Richard Ortega. 2021. "Molecular Mechanisms of Environmental Metal Neurotoxicity: A Focus on the Interactions of Metals with Synapse Structure and Function" Toxics 9, no. 9: 198. https://doi.org/10.3390/toxics9090198
APA StyleCarmona, A., Roudeau, S., & Ortega, R. (2021). Molecular Mechanisms of Environmental Metal Neurotoxicity: A Focus on the Interactions of Metals with Synapse Structure and Function. Toxics, 9(9), 198. https://doi.org/10.3390/toxics9090198