
Exposure to Environmental Arsenic and Emerging Risk of Alzheimer’s Disease: Perspective Mechanisms, Management Strategy, and Future Directions

 ,
,  ,
,  ,
,  ,
, 

Abstract
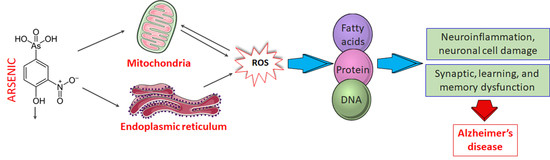
:
1. Introduction
2. Environmental Sources of Arsenic Exposure
3. Prevalence of Arsenic Exposure and Potential Risk of AD Development
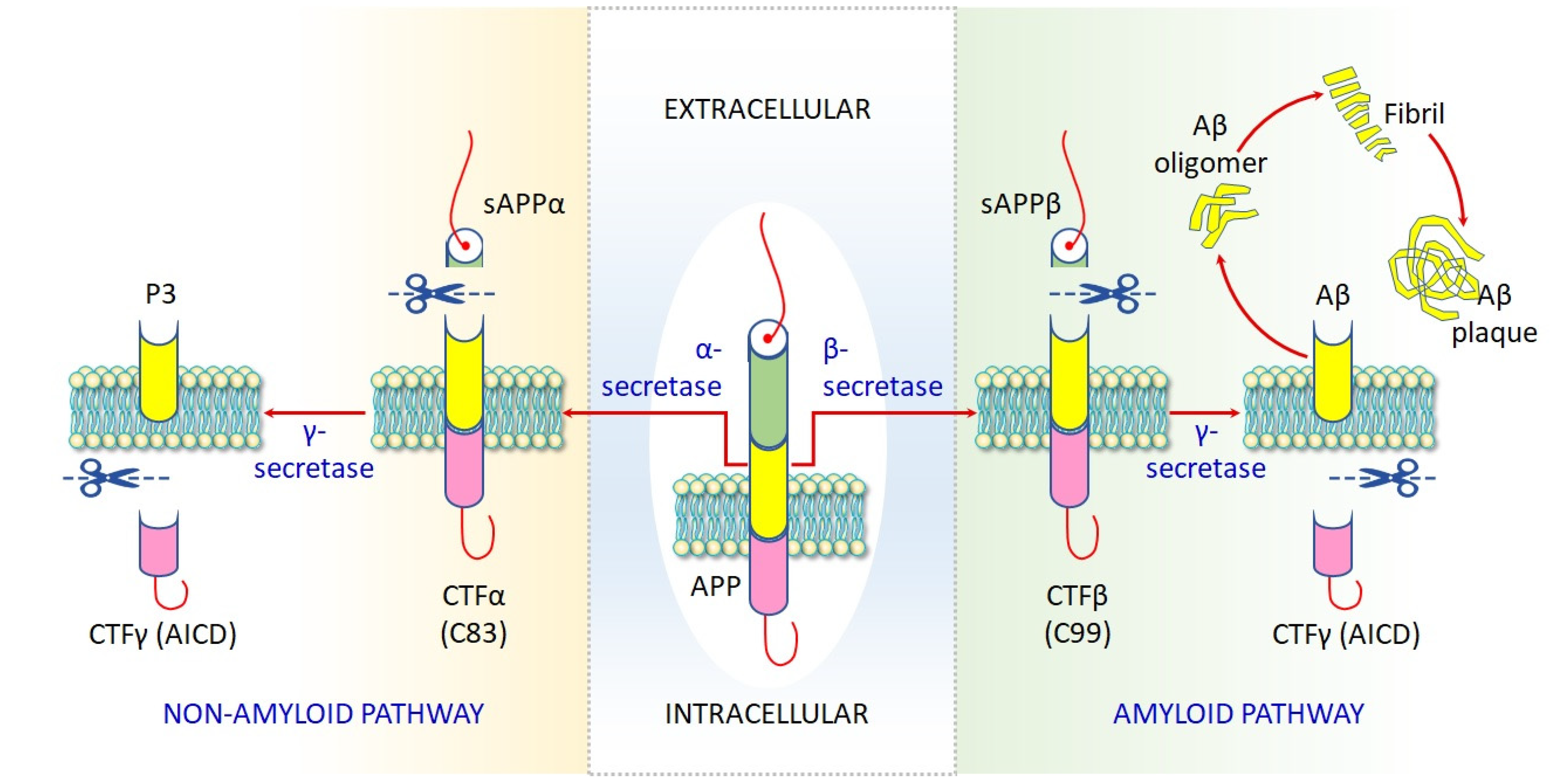
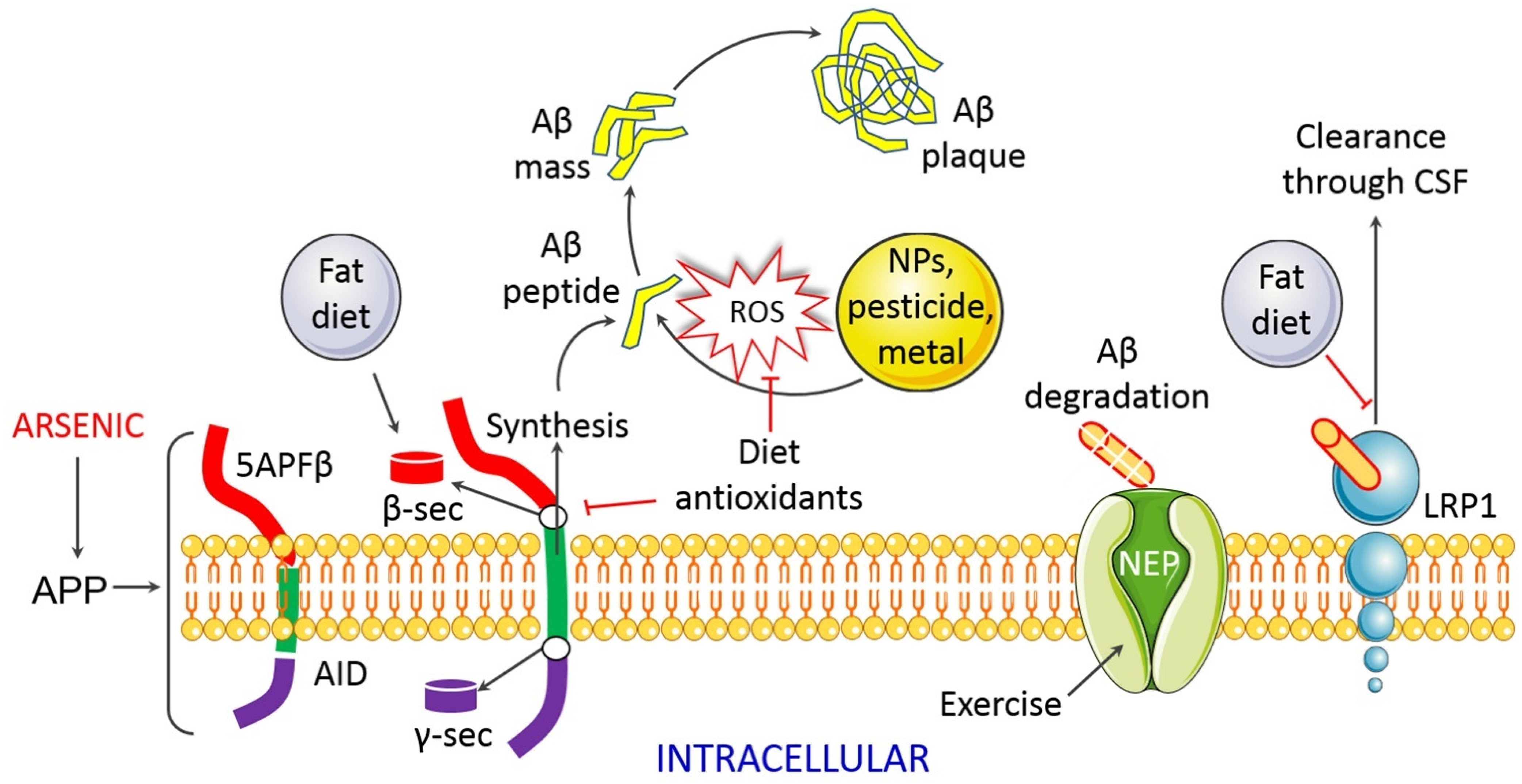
4. Molecular Basis of Arsenic Toxicity and Its Implication in AD Pathobiology
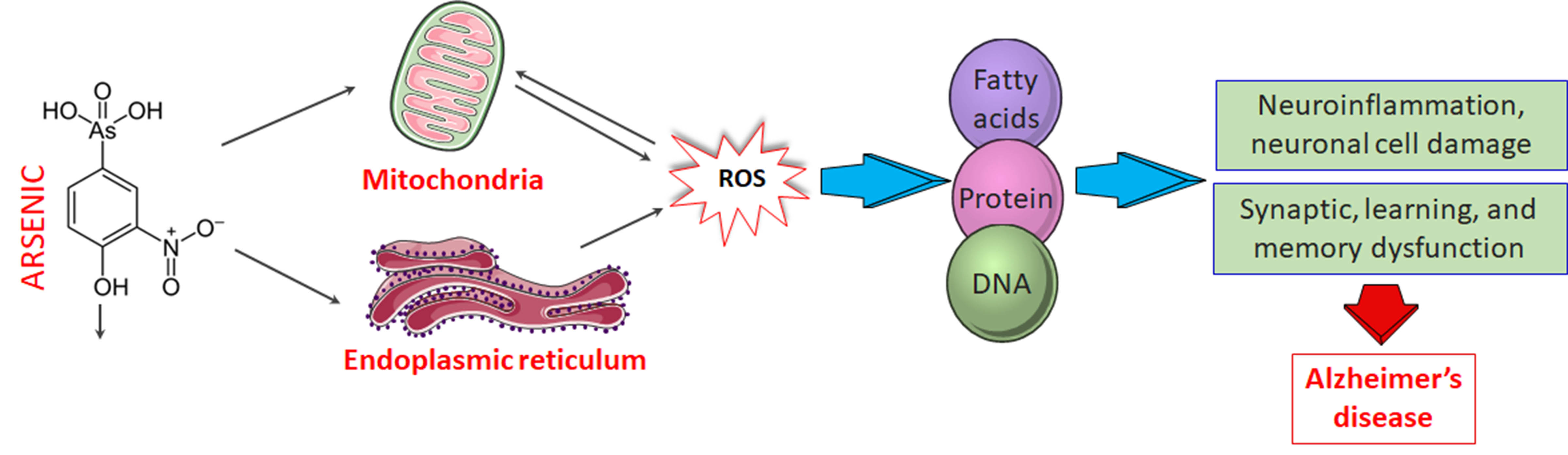
4.1. Mechanism of Arsenic-Induced Neurotoxicity
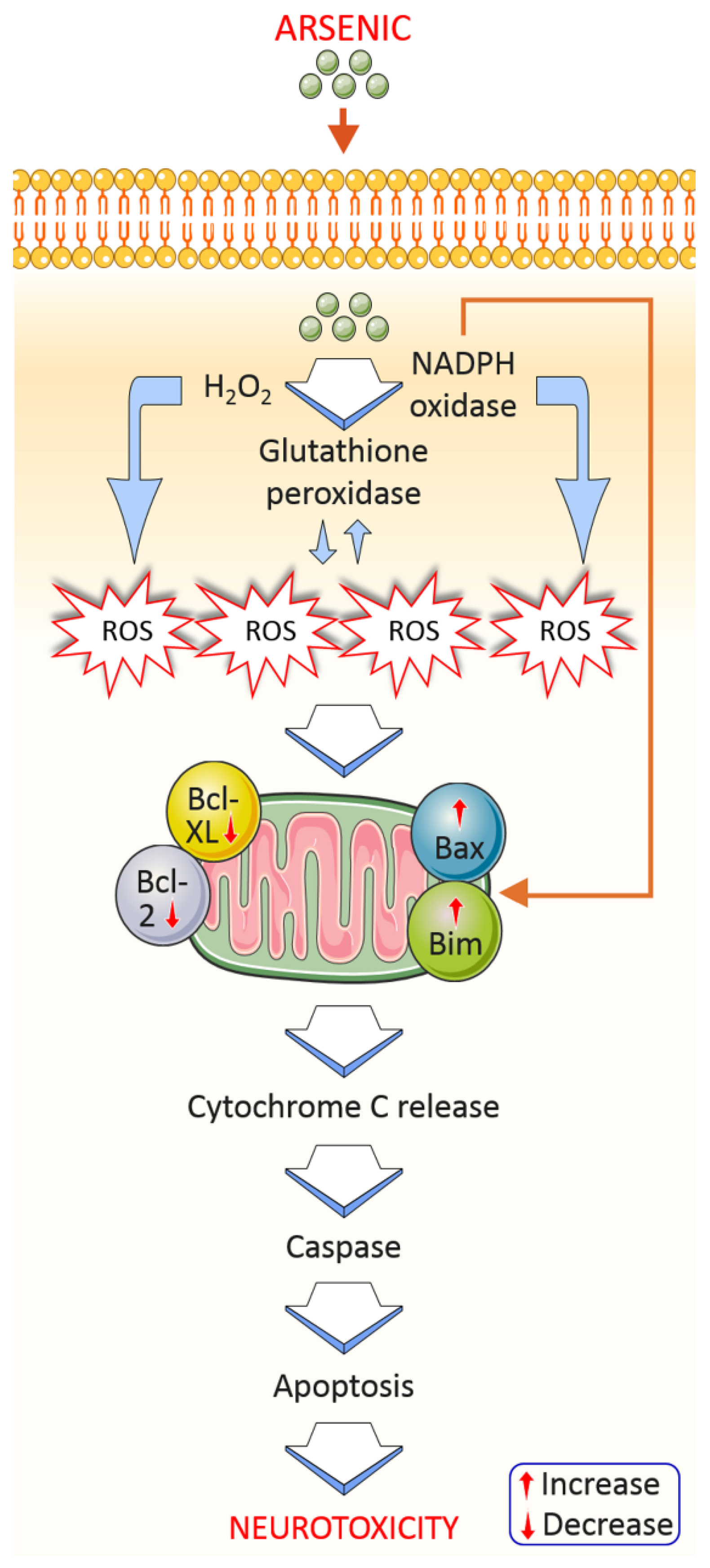
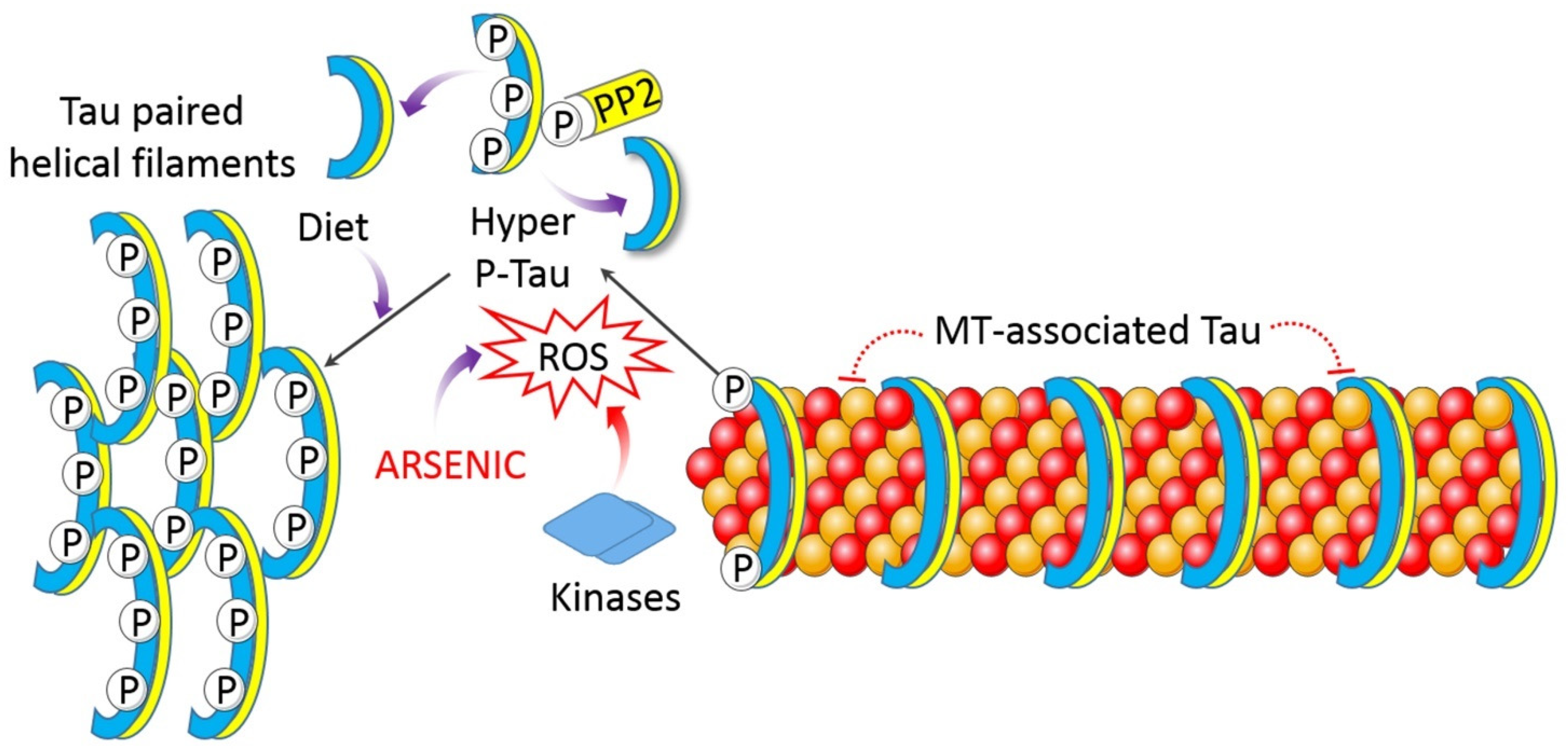
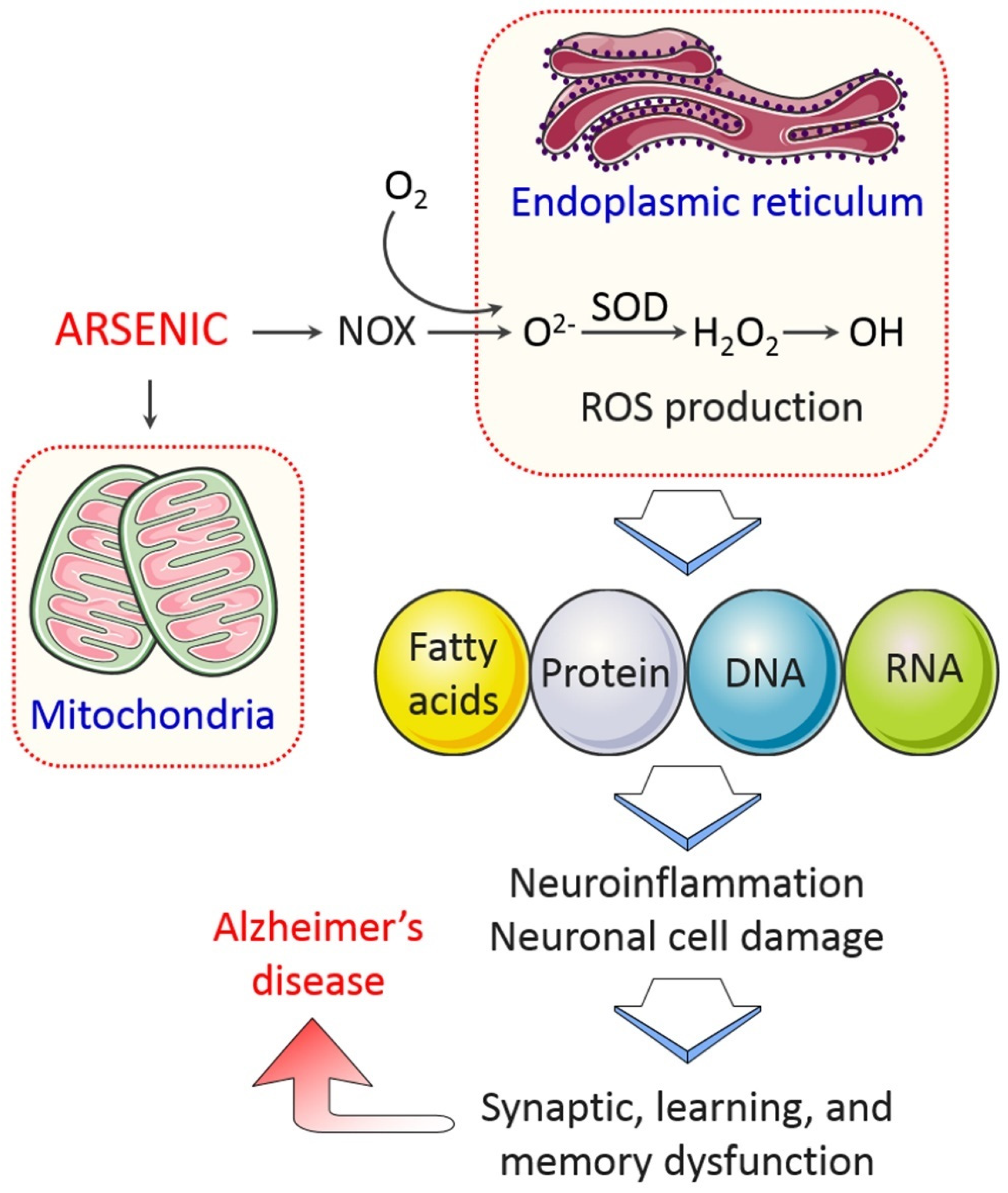
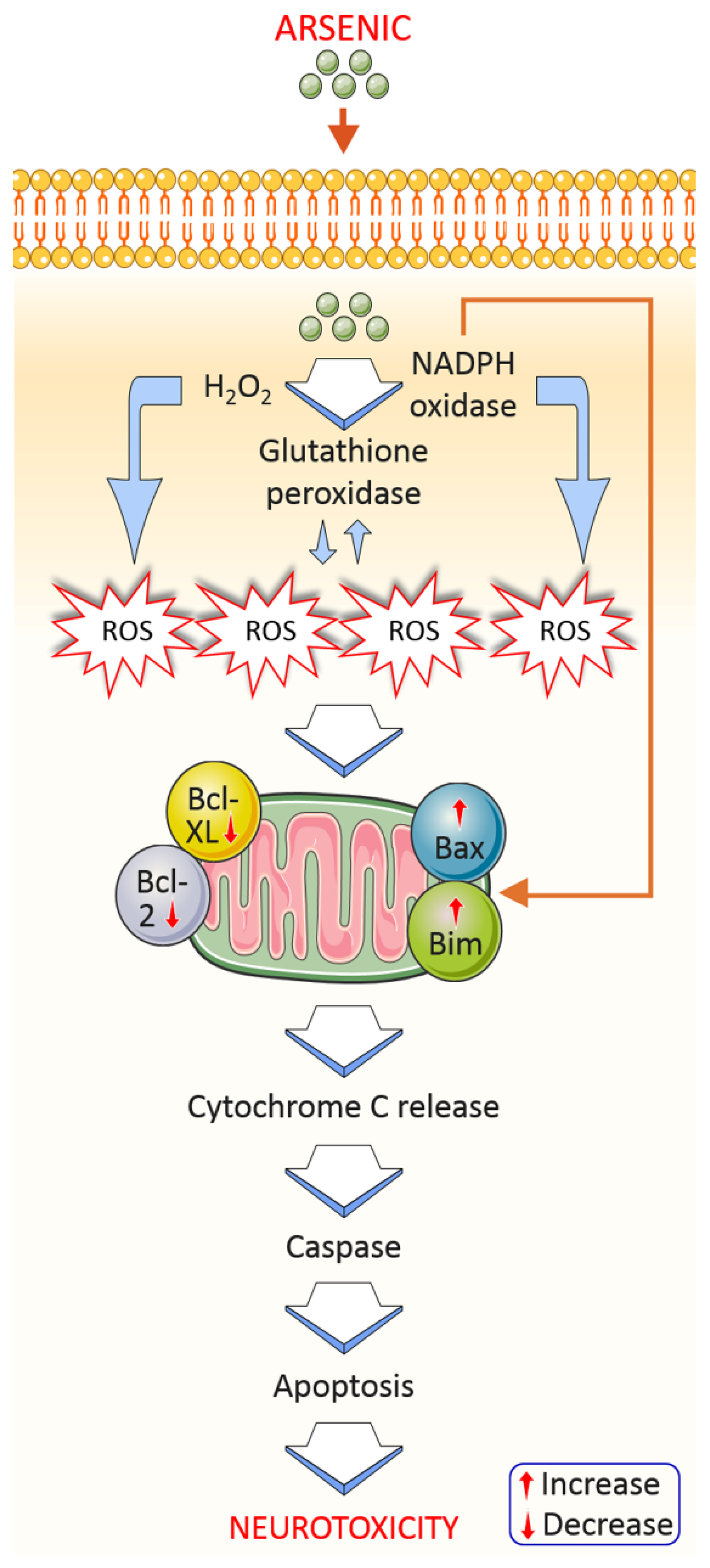
4.1.1. Oxidative Stress
4.1.2. Neuroinflammation
4.1.3. Mitochondrial Dysfunction
4.1.4. Endoplasmic Reticulum (ER) Stress
4.1.5. Apoptosis
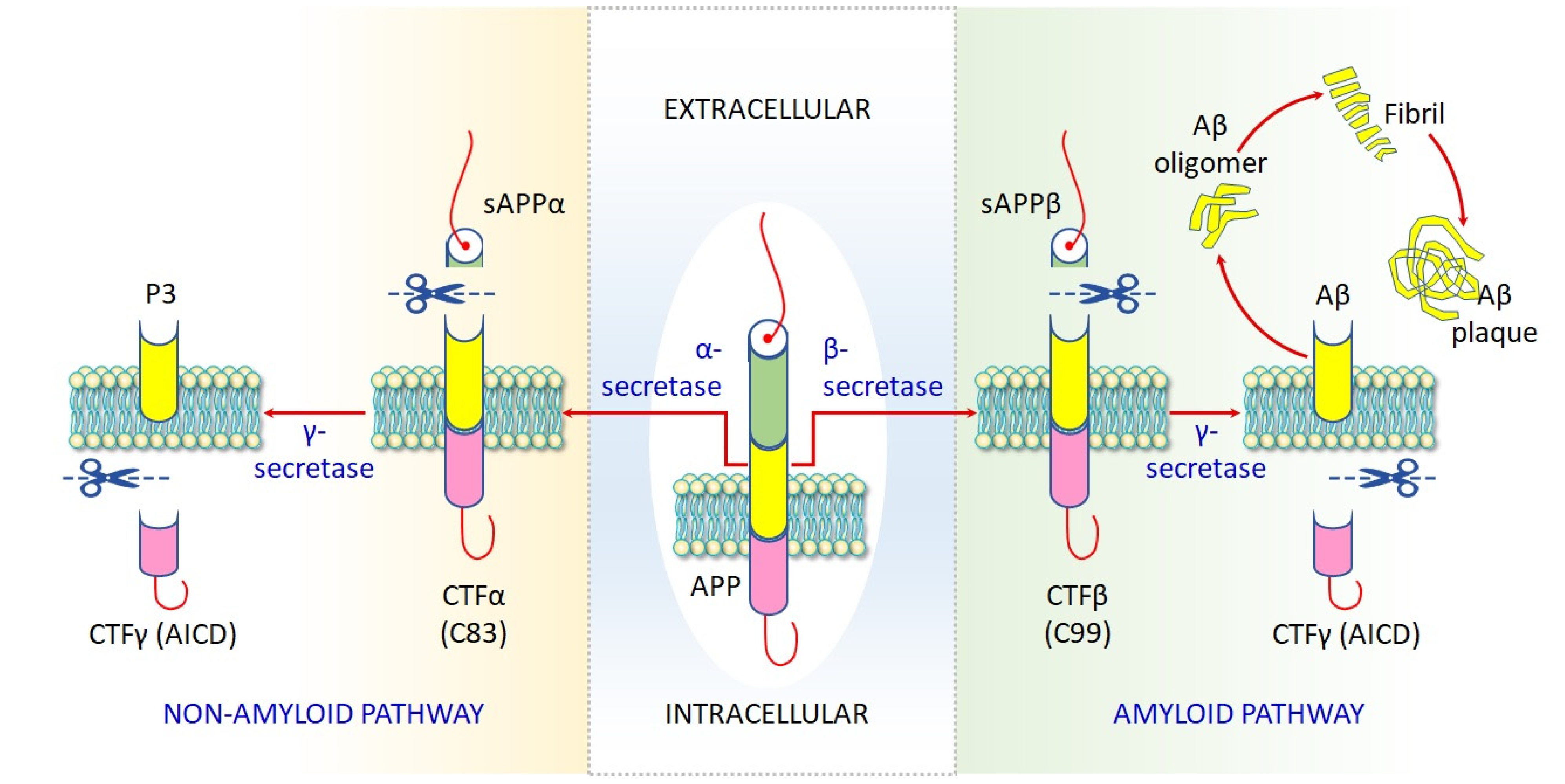
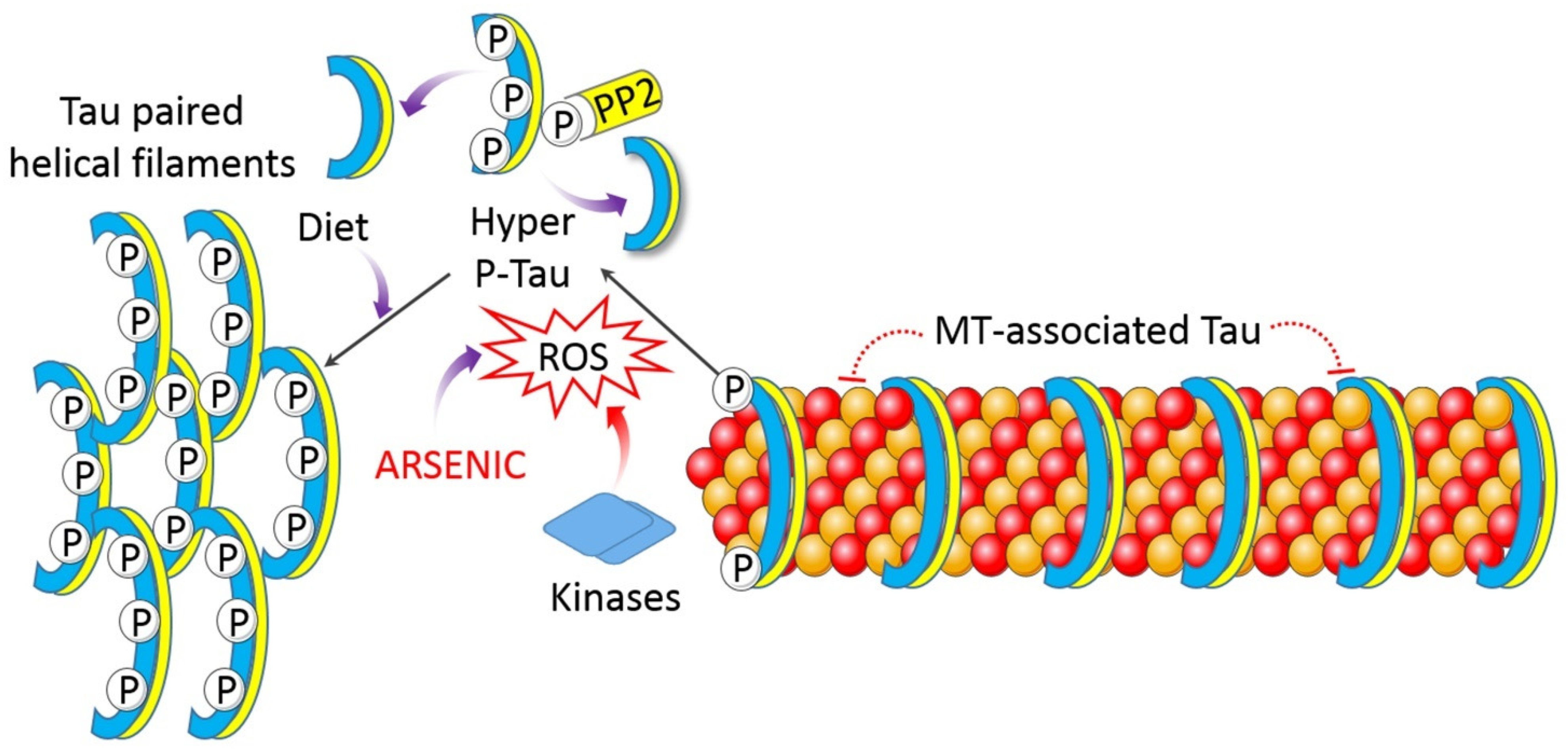
4.1.6. Impaired Proteostasis
4.1.7. Impaired Calcium Signaling
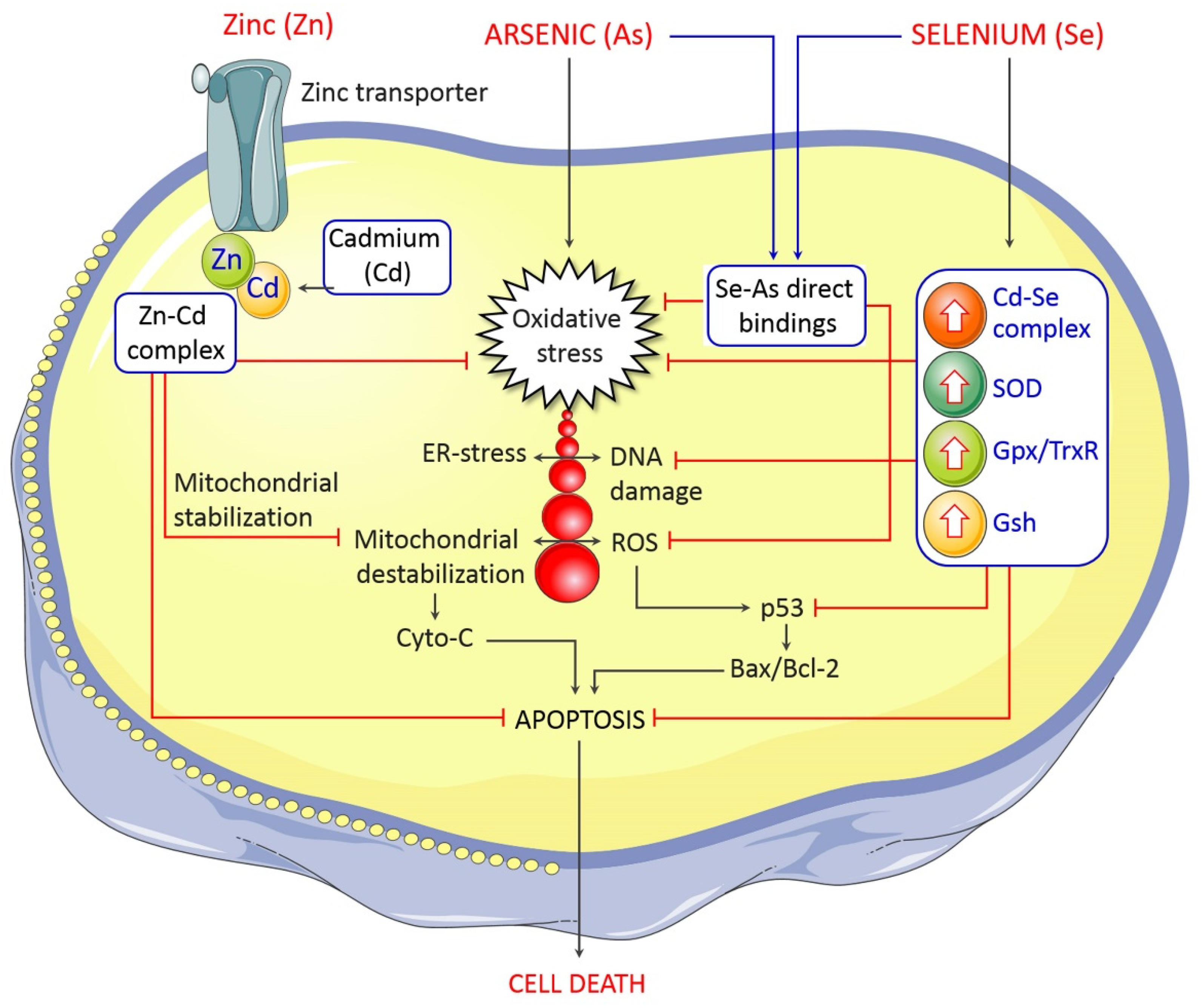
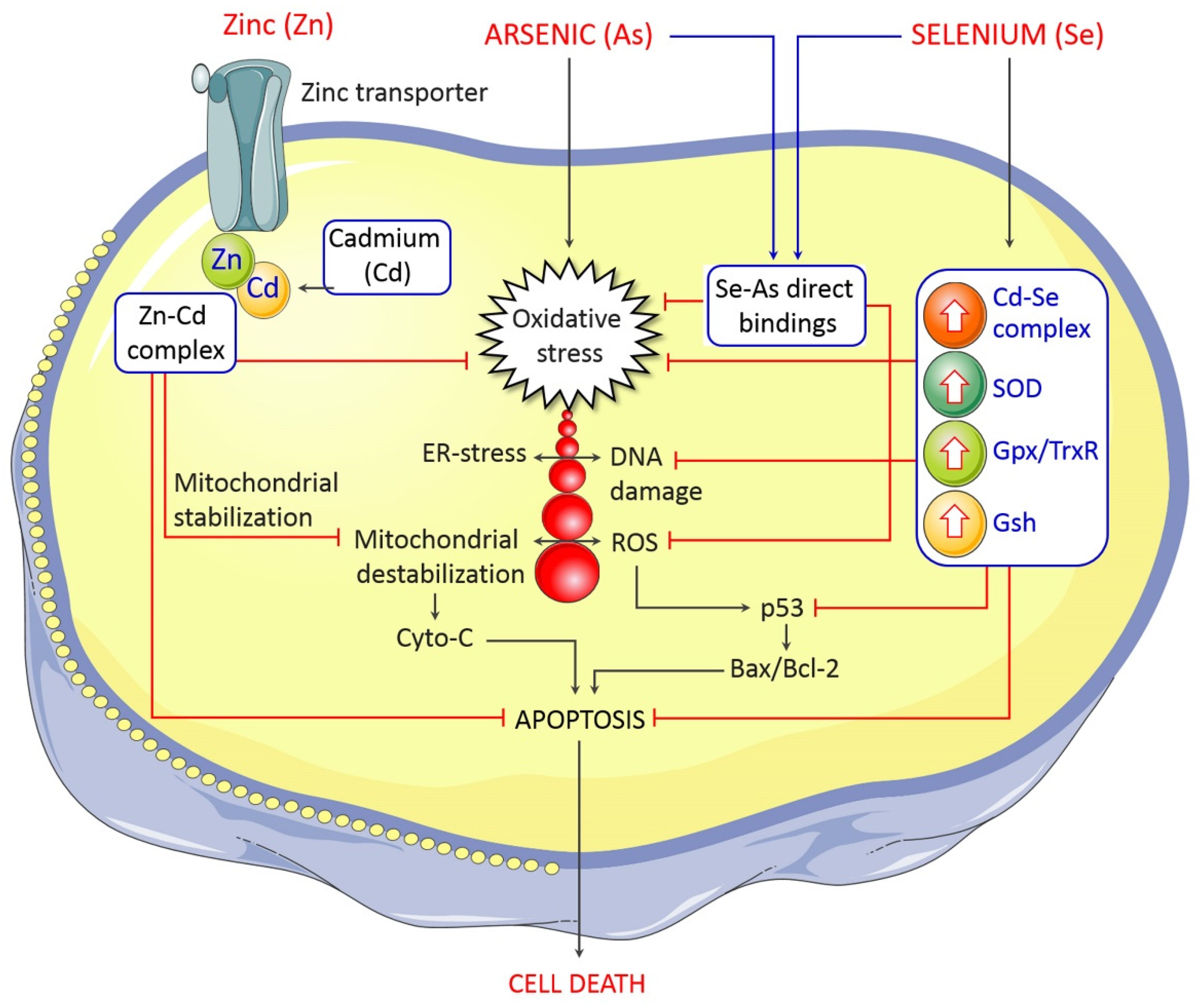
5. Management and Control of Arsenic-Induced Neurological Deficits
6. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rahman, M.A.; Rhim, H. Therapeutic implication of autophagy in neurodegenerative diseases. BMB Rep. 2017, 50, 345–354. [Google Scholar] [CrossRef] [Green Version]
- Moya-Alvarado, G.; Gershoni-Emek, N.; Perlson, E.; Bronfman, F.C. Neurodegeneration and Alzheimer’s disease (AD). What Can Proteomics Tell Us About the Alzheimer’s Brain? Mol. Cell. Proteom. 2016, 15, 409–425. [Google Scholar] [CrossRef] [Green Version]
- Rahman, M.A.; Rahman, M.S.; Uddin, M.J.; Mamum-Or-Rashid, A.N.M.; Pang, M.; Rhim, H. Emerging risk of environmental factors: Insight mechanisms of Alzheimer’s diseases. Environ. Sci. Pollut. Res. 2020, 27, 44659–44672. [Google Scholar] [CrossRef]
- Rahman, M.A.; Rahman, M.S.; Rahman, M.H.; Rasheduzzaman, M.; Mamun-Or-Rashid, A.N.M.; Uddin, M.J.; Rahman, M.R.; Hwang, H.; Pang, M.; Rhim, H. Modulatory Effects of Autophagy on APP Processing as a Potential Treatment Target for Alzheimer’s Disease. Biomedicines 2021, 9, 5. [Google Scholar] [CrossRef]
- Ghai, R.; Nagarajan, K.; Arora, M.; Grover, P.; Ali, N.; Kapoor, G. Current Strategies and Novel Drug Approaches for Alzheimer Disease. CNS Neurol. Disord. Drug Targets 2020, 19, 676–690. [Google Scholar] [CrossRef]
- Clayton, K.A.; Van Enoo, A.A.; Ikezu, T. Alzheimer’s Disease: The Role of Microglia in Brain Homeostasis and Proteopathy. Front. Neurosci. 2017, 11. [Google Scholar] [CrossRef]
- Tyler, C.R.; Allan, A.M. The Effects of Arsenic Exposure on Neurological and Cognitive Dysfunction in Human and Rodent Studies: A Review. Curr. Environ. Health Rep. 2014, 1, 132–147. [Google Scholar] [CrossRef] [Green Version]
- Ramos-Chavez, L.A.; Rendón-López, C.R.; Zepeda, A.; Silva-Adaya, D.; Del Razo, L.M.; Gonsebatt, M.E. Neurological effects of inorganic arsenic exposure: Altered cysteine/glutamate transport, NMDA expression and spatial memory impairment. Front. Cell. Neurosci. 2015, 9, 21. [Google Scholar] [CrossRef] [Green Version]
- Zarazua, S.; Bürger, S.; Delgado, J.M.; Jiménez-Capdeville, M.E.; Schliebs, R. Arsenic affects expression and processing of amyloid precursor protein (APP) in primary neuronal cells overexpressing the Swedish mutation of human APP. Int. J. Dev. Neurosci. 2011, 29, 389–396. [Google Scholar] [CrossRef]
- Nino, S.A.; Morales-Martínez, A.; Chi-Ahumada, E.; Carrizales, L.; Salgado-Delgado, R.; Pérez-Severiano, F.; Díaz-Cintra, S.; Jiménez-Capdeville, M.E.; Zarazúa, S. Arsenic Exposure Contributes to the Bioenergetic Damage in an Alzheimer’s Disease Model. ACS Chem. Neurosci. 2019, 10, 323–336. [Google Scholar] [CrossRef]
- Wisessaowapak, C.; Visitnonthachai, D.; Watcharasit, P.; Satayavivad, J. Prolonged arsenic exposure increases tau phosphorylation in differentiated SH-SY5Y cells: The contribution of GSK3 and ERK1/2. Environ. Toxicol. Pharmacol. 2021, 84, 103626. [Google Scholar] [CrossRef]
- Chung, J.Y.; Yu, S.D.; Hong, Y.S. Environmental source of arsenic exposure. J. Prev. Med. Public Health 2014, 47, 253–257. [Google Scholar] [CrossRef] [Green Version]
- IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Cobalt in hard metals and cobalt sulfate, gallium arsenide, indium phosphide and vanadium pentoxide. IARC Monogr. Eval. Carcinog. Risks Hum. 2006, 86, 1–294. [Google Scholar]
- Emadi, A.; Gore, S.D. Arsenic trioxide—An old drug rediscovered. Blood Rev. 2010, 24, 191–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, L.; Wang, B.; Jiang, J.W.; Fitzgerald, M.; Huang, Q.; Yu, Z.; Li, H.; Zhang, J.Q.; Wei, J.H.; Yang, C.Y.; et al. Heavy Metal Contaminations in Herbal Medicines: Determination, Comprehensive Risk Assessments, and Solutions. Front. Pharmacol. 2021, 11, 595335. [Google Scholar] [CrossRef] [PubMed]
- Borowska, S.; Brzoska, M.M. Metals in cosmetics: Implications for human health. J. Appl. Toxicol. 2015, 35, 551–572. [Google Scholar] [CrossRef]
- Briffa, J.; Sinagra, E.; Blundell, R. Heavy metal pollution in the environment and their toxicological effects on humans. Heliyon 2020, 6, e04691. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.Y.; Cobbina, S.J.; Mao, G.H.; Xu, H.; Zhang, Z.; Yang, L.Q. A review of toxicity and mechanisms of individual and mixtures of heavy metals in the environment. Environ. Sci. Pollut. Res. 2016, 23, 8244–8259. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Schoenau, J.J. Development of Resin Membranes as a Sensitive Indicator of Heavy-Metal Toxicity in the Soil Environment. Int. J. Environ. Anal. Chem. 1995, 59, 265–275. [Google Scholar] [CrossRef]
- Liu, Q.Q.; Leslie, E.M.; Le, X.C. Accumulation and Transport of Roxarsone, Arsenobetaine, and Inorganic Arsenic Using the Human Immortalized Caco-2 Cell Line. J. Agric. Food Chem. 2016, 64, 8902–8908. [Google Scholar] [CrossRef] [Green Version]
- Middleton, D.R.S.; Watts, M.J.; Hamilton, E.M.; Ander, E.L.; Close, R.M.; Exley, K.S.; Crabbe, H.; Leonardi, G.S.; Fletcher, T.; Polya, D.A. Urinary arsenic profiles reveal exposures to inorganic arsenic from private drinking water supplies in Cornwall, UK. Sci. Rep. 2016, 6, 25656. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, S.A.; Khan, M.H.; Haque, M. Arsenic contamination in groundwater in Bangladesh: Implications and challenges for healthcare policy. Risk Manag. Healthc. Policy 2018, 11, 251–261. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, A.; Sengupta, M.K.; Hossain, M.A.; Ahamed, S.; Das, B.; Nayak, B.; Lodh, D.; Rahman, M.M.; Chakraborti, D. Arsenic contamination in groundwater: A global perspective with emphasis on the Asian scenario. J. Health Popul. Nutr. 2006, 24, 142–163. [Google Scholar]
- Dani, S.U. Arsenic for the fool: An exponential connection. Sci. Total Environ. 2010, 408, 1842–1846. [Google Scholar] [CrossRef] [PubMed]
- O’Bryant, S.E.; Edwards, M.; Menon, C.V.; Gong, G.; Barber, R. Long-Term Low-Level Arsenic Exposure Is Associated with Poorer Neuropsychological Functioning: A Project FRONTIER Study. Int. J. Environ. Res. Public Health 2011, 8, 861–874. [Google Scholar] [CrossRef]
- Baum, L.; Chan, I.H.S.; Cheung, S.K.K.; Goggins, W.B.; Mok, V.; Lam, L.; Leung, V.; Hui, E.; Ng, C.; Woo, J.; et al. Serum zinc is decreased in Alzheimer’s disease and serum arsenic correlates positively with cognitive ability. Biometals 2010, 23, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Namgung, U.; Xia, Z. Arsenic induces apoptosis in rat cerebellar neurons via activation of JNK3 and p38 MAP kinases. Toxicol. Appl. Pharmacol. 2001, 174, 130–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, G.; O’Bryant, S.E. The Arsenic Exposure Hypothesis for Alzheimer Disease. Alzheimer Dis. Assoc. Disord. 2010, 24, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Bakulski, K.M.; Seo, Y.A.; Hickman, R.C.; Brandt, D.; Vadari, H.S.; Hu, H.; Park, S.K. Heavy Metals Exposure and Alzheimer’s Disease and Related Dementias. J. Alzheimers Dis. 2020, 76, 1215–1242. [Google Scholar] [CrossRef]
- Sharma, A.; Kumar, S. Arsenic exposure with reference to neurological impairment: An overview. Rev. Environ. Health 2019, 34, 403–414. [Google Scholar] [CrossRef]
- Alboghobeish, S.; Pashmforosh, M.; Zeidooni, L.; Samimi, A.; Rezaei, M. High fat diet deteriorates the memory impairment induced by arsenic in mice: A sub chronic in vivo study. Metab. Brain Dis. 2019, 34, 1595–1606. [Google Scholar] [CrossRef]
- Wai, K.M.; Umezaki, M.; Mar, O.; Umemura, M.; Watanabe, C. Arsenic exposure through drinking Water and oxidative stress Status: A cross-sectional study in the Ayeyarwady region, Myanmar. J. Trace. Elem. Med. Biol. 2019, 54, 103–109. [Google Scholar]
- Butterfield, D.A.; Boyd-Kimball, D. Oxidative Stress, Amyloid-beta Peptide, and Altered Key Molecular Pathways in the Pathogenesis and Progression of Alzheimer’s Disease. J. Alzheimers Dis. 2018, 62, 1345–1367. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.P.; Goel, R.K.; Kaur, T. Mechanisms pertaining to arsenic toxicity. Toxicol. Int. 2011, 18, 87–93. [Google Scholar] [PubMed] [Green Version]
- Ma, L.; Zhang, C.; Liu, W.J. Effects of arsenic on the offspring development in mice. Zhonghua Yu Fang Yi Xue Za Zhi 1994, 28, 20–23. [Google Scholar] [PubMed]
- Chin-Chan, M.; Navarro-Yepes, J.; Quintanilla-Vega, B. Environmental pollutants as risk factors for neurodegenerative disorders: Alzheimer and Parkinson diseases. Front. Cell. Neurosci. 2015, 9, 124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasaguri, H.; Nilsson, P.; Hashimoto, S.; Nagata, K.; Saito, T.; De Strooper, B.; Hardy, J.; Vassar, R.; Winblad, B.; Saido, T.C. APP mouse models for Alzheimer’s disease preclinical studies. EMBO J. 2017, 36, 2473–2487. [Google Scholar] [CrossRef] [PubMed]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef]
- Chandravanshi, L.P.; Gupta, R.; Shukla, R.K. Developmental Neurotoxicity of Arsenic: Involvement of Oxidative Stress and Mitochondrial Functions. Biol. Trace Elem. Res. 2018, 186, 185–198. [Google Scholar] [CrossRef]
- Hannan, M.A.; Dash, R.; Sohag, A.A.M.; Haque, M.N.; Moon, I.S. Neuroprotection against Oxidative Stress: Phytochemicals Targeting TrkB Signaling and the Nrf2-ARE Antioxidant System. Front. Mol. Neurosci. 2020, 13, 116. [Google Scholar] [CrossRef]
- Dwivedi, N.; Flora, S.J. Concomitant exposure to arsenic and organophosphates on tissue oxidative stress in rats. Food Chem. Toxicol. 2011, 49, 1152–1159. [Google Scholar] [CrossRef]
- Roy, N.K.; Murphy, A.; Costa, M. Arsenic Methyltransferase and Methylation of Inorganic Arsenic. Biomolecules 2020, 10, 1351. [Google Scholar] [CrossRef]
- Dash, R.; Mitra, S.; Ali, M.C.; Oktaviani, D.F.; Hannan, M.A.; Choi, S.M.; Moon, I.S. Phytosterols: Targeting neuroinflammation in neurodegeneration. Curr. Pharm. Des. 2021, 27, 383–401. [Google Scholar] [CrossRef]
- Medda, N.; Patra, R.; Ghosh, T.K.; Maiti, S. Neurotoxic Mechanism of Arsenic: Synergistic Effect of Mitochondrial Instability, Oxidative Stress, and Hormonal-Neurotransmitter Impairment. Biol. Trace Elem. Res. 2020, 198, 8–15. [Google Scholar] [CrossRef]
- Firdaus, F.; Zafeer, M.F.; Anis, E.; Ahmad, M.; Afzal, M. Ellagic acid attenuates arsenic induced neuro-inflammation and mitochondrial dysfunction associated apoptosis. Toxicol. Rep. 2018, 5, 411–417. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Chen, B.; Chen, S.; Zhang, M.; Duan, L.; Feng, X.; Chen, J.; Zhou, L.; Chen, L.; Duan, Y. MBP-activated autoimmunity plays a role in arsenic-induced peripheral neuropathy and the potential protective effect of mecobalamin. Environ. Toxicol. 2021, 36, 1243–1253. [Google Scholar] [CrossRef]
- Rahman, M.A.; Rahman, M.H.; Biswas, P.; Hossain, M.S.; Islam, R.; Hannan, M.A.; Uddin, M.J.; Rhim, H. Potential therapeutic role of phytochemicals to mitigate mitochondrial dysfunctions in Alzheimer’s disease. Antioxidants 2021, 10, 23. [Google Scholar] [CrossRef]
- Prakash, C.; Soni, M.; Kumar, V. Mitochondrial oxidative stress and dysfunction in arsenic neurotoxicity: A review. J. Appl. Toxicol. 2016, 36, 179–188. [Google Scholar] [CrossRef]
- Prakash, C.; Soni, M.; Kumar, V. Biochemical and Molecular Alterations Following Arsenic-Induced Oxidative Stress and Mitochondrial Dysfunction in Rat Brain. Biol. Trace Elem. Res. 2015, 167, 121–129. [Google Scholar] [CrossRef] [PubMed]
- King, A.P.; Wilson, J.J. Endoplasmic reticulum stress: An arising target for metal-based anticancer agents. Chem. Soc. Rev. 2020, 49, 8113–8136. [Google Scholar] [CrossRef]
- Liu, X.; Gao, Y.; Liu, Y.; Zhang, W.; Yang, Y.; Fu, X.; Sun, D.; Wang, J. Neuroglobin alleviates arsenic-induced neuronal damage. Environ. Toxicol. Pharmacol. 2021, 84, 103604. [Google Scholar] [CrossRef] [PubMed]
- Delaney, P.; Nair, A.R.; Palmer, C.; Khan, N.; Sadler, K.C. Arsenic induced redox imbalance triggers the unfolded protein response in the liver of zebrafish. Toxicol. Appl. Pharmacol. 2020, 409, 115307. [Google Scholar] [CrossRef]
- Rana, S.V.S. Endoplasmic Reticulum Stress Induced by Toxic Elements-a Review of Recent Developments. Biol. Trace Elem. Res. 2020, 196, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Qiu, T.; Yao, X.; Wang, N.; Jiang, L.; Jia, X.; Tao, Y.; Wang, Z.; Pei, P.; Zhang, J.; et al. Arsenic induces pancreatic dysfunction and ferroptosis via mitochondrial ROS-autophagy-lysosomal pathway. J. Hazard. Mater. 2020, 384, 121390. [Google Scholar] [CrossRef]
- Zhang, W.; Cui, X.; Gao, Y.; Sun, L.; Wang, J.; Yang, Y.; Liu, X.; Li, Y.; Guo, X.; Sun, D. Role of pigment epithelium-derived factor (PEDF) on arsenic-induced neuronal apoptosis. Chemosphere 2019, 215, 925–931. [Google Scholar] [CrossRef] [PubMed]
- Sinha, K.; Das, J.; Pal, P.B.; Sil, P.C. Oxidative stress: The mitochondria-dependent and mitochondria-independent pathways of apoptosis. Arch. Toxicol. 2013, 87, 1157–1180. [Google Scholar] [CrossRef]
- Fu, S.C.; Lin, J.W.; Liu, J.M.; Liu, S.H.; Fang, K.M.; Su, C.C.; Hsu, R.J.; Wu, C.C.; Huang, C.F.; Lee, K.I.; et al. Arsenic induces autophagy-dependent apoptosis via Akt inactivation and AMPK activation signaling pathways leading to neuronal cell death. Neurotoxicology 2021, 85, 133–144. [Google Scholar] [CrossRef]
- Dash, R.; Jahan, I.; Ali, M.C.; Mitra, S.; Munni, Y.A.; Timalsina, B.; Hannan, M.A.; Moon, I.S. Potential roles of natural products in the targeting of proteinopathic neurodegenerative diseases. Neurochem. Int. 2021, 145, 105011. [Google Scholar] [CrossRef]
- Dash, R.; Ali, M.C.; Jahan, I.; Munni, Y.A.; Mitra, S.; Hannan, M.A.; Timalsina, B.; Oktaviani, D.F.; Choi, H.J.; Moon, I.S. Emerging potential of cannabidiol in reversing proteinopathies. Ageing Res. Rev. 2021, 65, 101209. [Google Scholar] [CrossRef]
- Tam, L.M.; Wang, Y. Arsenic Exposure and Compromised Protein Quality Control. Chem. Res. Toxicol. 2020, 33, 1594–1604. [Google Scholar] [CrossRef]
- Andersson, S.; Romero, A.; Rodrigues, J.I.; Hua, S.S.; Hao, X.X.; Jacobson, T.; Karl, V.; Becker, N.; Ashouri, A.; Rauch, S.; et al. Genome-wide imaging screen uncovers molecular determinants of arsenite-induced protein aggregation and toxicity. J. Cell. Sci. 2021, 134, jcs258338. [Google Scholar] [CrossRef]
- Priya Wadgaonkar, F.C. Connections between endoplasmic reticulum stress-associated unfolded protein response, mitochondria, and autophagy in arsenic-induced carcinogenesis. Semin. Cancer Biol. 2021, in press. [Google Scholar] [CrossRef]
- Dodson, M.; Liu, P.F.; Jiang, T.; Ambrose, A.J.; Luo, G.; de la Vega, M.R.; Cholanians, A.B.; Wong, P.K.; Chapman, E.; Zhang, D.D. Increased O-GlcNAcylation of SNAP29 Drives Arsenic-Induced Autophagic Dysfunction. Mol. Cell. Biol. 2018, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodson, M.; de la Vega, M.R.; Harder, B.; Castro-Portuguez, R.; Rodrigues, S.D.; Wong, P.K.; Chapman, E.; Zhang, D.D. Low-level arsenic causes proteotoxic stress and not oxidative stress. Toxicol. Appl. Pharmacol. 2018, 143, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Bolt, A.M.; Zhao, F.; Pacheco, S.; Klimecki, W.T. Arsenite-induced autophagy is associated with proteotoxicity in human lymphoblastoid cells. Toxicol. Appl. Pharmacol. 2012, 264, 255–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weidling, I.; Swerdlow, R.H. Mitochondrial Dysfunction and Stress Responses in Alzheimer’s Disease. Biology 2019, 8, 39. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zhao, H.J.; Liu, Y.C.; Guo, M.H.; Tian, Y.; Huang, P.Y.; Xing, M.W. Arsenite induce neurotoxicity of common carp: Involvement of blood brain barrier, apoptosis and autophagy, and subsequently relieved by zinc (Ⅱ) supplementation. Aquat. Toxicol. 2021, 232, 105765. [Google Scholar] [CrossRef]
- Zhao, H.; Wang, Y.; Liu, J.; Guo, M.; Fei, D.; Yu, H.; Xing, M. The cardiotoxicity of the common carp (Cyprinus carpio) exposed to environmentally relevant concentrations of arsenic and subsequently relieved by zinc supplementation. Environ. Pollut. 2019, 253, 741–748. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, H.; Liu, Y.; Nie, X.; Xing, M. Zinc exerts its renal protection effect on arsenic-exposed common carp: A signaling network comprising Nrf2, NF-κB and MAPK pathways. Fish Shellfish. Immunol. 2020, 104, 383–390. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, H.; Nie, X.; Guo, M.; Jiang, G.; Xing, M. Zinc application alleviates the adverse renal effects of arsenic stress in a protein quality control way in common carp. Environ. Res. 2020, 191, 110063. [Google Scholar] [CrossRef]
- Zhao, H.; Wang, Y.; Guo, M.; Fei, D.; Mu, M.; Yu, H.; Xing, M. Hepatoprotective effects of zinc (II) via cytochrome P-450/reactive oxygen species and canonical apoptosis pathways after arsenite waterborne exposure in common carp. Chemosphere 2019, 236, 124869. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, H.; Mu, M.; Guo, M.; Xing, M. Zinc offers splenic protection through suppressing PERK/IRE1-driven apoptosis pathway in common carp (Cyprinus carpio) under arsenic stress. Ecotoxicol. Environ. Saf. 2021, 208, 111473. [Google Scholar] [CrossRef] [PubMed]
- Cao, A.L.; Beaver, L.M.; Wong, C.P.; Hudson, L.G.; Ho, E. Zinc deficiency alters the susceptibility of pancreatic beta cells (INS-1) to arsenic exposure. Biometals 2019, 32, 845–859. [Google Scholar] [CrossRef]
- Garla, R.; Sharma, N.; Shamli; Kaushal, N.; Garg, M.L. Effect of Zinc on Hepatic and Renal Tissues of Chronically Arsenic Exposed Rats: A Biochemical and Histopathological Study. Biol. Trace Elem. Res. 2021. [Google Scholar] [CrossRef] [PubMed]
- Milton, A.G.; Zalewski, P.D.; Ratnaike, R.N. Zinc protects against arsenic-induced apoptosis in a neuronal cell line, measured by DEVD-caspase activity. Biometals 2004, 17, 707–713. [Google Scholar] [CrossRef]
- Wei, Y.Y.; Huang, H.; Xia, Y.K.; Wei, L.M.; Chen, X.; Zhang, R.Y.; Duan, W.W.; Su, L.; Rahman, M.L.; Rahman, M.; et al. Antagonistic effect of early stage zinc on arsenic toxicity induced preterm birth during pregnancy: Evidence from a rural Bangladesh birth cohort. Chin. Med. J. 2021, 134, 619–621. [Google Scholar] [CrossRef]
- Adedara, I.A.; Fabunmi, A.T.; Ayenitaju, F.C.; Atanda, O.E.; Adebowale, A.A.; Ajayi, B.O.; Owoeye, O.; Rocha, J.B.T.; Farombi, E.O. Neuroprotective mechanisms of selenium against arsenic-induced behavioral impairments in rats. Neurotoxicology 2020, 76, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Samad, N.; Rao, T.; Rehman, M.H.u.; Bhatti, S.A.; Imran, I. Inhibitory Effects of Selenium on Arsenic-Induced Anxiety-/Depression-Like Behavior and Memory Impairment. Biol. Trace Elem. Res. 2021. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.; Deng, H.; Wu, Q.; Jia, G.; Wen, N.; Deng, Y.; Zhu, L.; Zuo, Z.; Deng, J. Effect of Selenium on Brain Injury in Chickens with Subacute Arsenic Poisoning. Biol. Trace Elem. Res. 2021. [Google Scholar] [CrossRef]
- Ren, Z.; Deng, H.; Deng, Y.; Tang, W.; Wu, Q.; Zuo, Z.; Cui, H.; Hu, Y.; Yu, S.; Xu, S.Y.; et al. Effects of Selenium on Arsenic-Induced Liver Lesions in Broilers. Biol. Trace Elem. Res. 2021, 199, 1080–1089. [Google Scholar] [CrossRef] [PubMed]
- Adedara, I.A.; Adebowale, A.A.; Atanda, O.E.; Fabunmi, A.T.; Ayenitaju, A.C.; Rocha, J.B.T.; Farombi, E.O. Selenium abates reproductive dysfunction via attenuation of biometal accumulation, oxido-inflammatory stress and caspase-3 activation in male rats exposed to arsenic. Environ. Pollut. 2019, 254 Pt B, 113079. [Google Scholar] [CrossRef]
- Zwolak, I. The Role of Selenium in Arsenic and Cadmium Toxicity: An Updated Review of Scientific Literature. Biol. Trace Elem. Res. 2020, 193, 44–63. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, N.; Das, B.; Bishayee, A.; Sinha, D. Arsenal of Phytochemicals to Combat Against Arsenic-Induced Mitochondrial Stress and Cancer. Antioxid. Redox Signal. 2020, 33, 1230–1256. [Google Scholar] [CrossRef]
- Zhang, Q.Y.; Wang, F.X.; Jia, K.K.; Kong, L.D. Natural Product Interventions for Chemotherapy and Radiotherapy-Induced Side Effects. Front. Pharmacol. 2018, 9, 1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salehi, B.; Machin, L.; Monzote, L.; Sharifi-Rad, J.; Ezzat, S.M.; Salem, M.A.; Merghany, R.M.; El Mahdy, N.M.; Kılıç, C.S.; Sytar, O.; et al. Therapeutic Potential of Quercetin: New Insights and Perspectives for Human Health. ACS Omega 2020, 5, 11849–11872. [Google Scholar] [CrossRef]
- Sarkozi, K.; Papp, A.; Horváth, E.; Máté, Z.; Ferencz, A.; Hermesz, E.; Krisch, J.; Paulik, E.; Szabó, A. Green tea and vitamin C ameliorate some neuro-functional and biochemical signs of arsenic toxicity in rats. Nutr. Neurosci. 2016, 19, 102–109. [Google Scholar] [CrossRef]
- Vazhappilly, C.G.; Devanga, R.N.K.; Palamadai, K.S.; Rangasamy, A.K. In Vitro Protective Potentials of Annona muricata Leaf Extracts Against Sodium Arsenite-induced Toxicity. Curr. Drug Discov. Technol. 2015, 12, 59–63. [Google Scholar]
- Jomova, K.; Jenisova, Z.; Feszterova, M.; Baros, S.; Liska, J.; Hudecova, D.; Rhodes, C.J.; Valko, M. Arsenic: Toxicity, oxidative stress and human disease. J. Appl. Toxicol. 2011, 31, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Das, B.; Mandal, S.; Chaudhuri, K. Role of arginine, a component of aqueous garlic extract, in remediation of sodium arsenite induced toxicity in A375 cells. Toxicol. Res. 2014, 3, 191–196. [Google Scholar] [CrossRef]
- Das, T.; Roychoudhury, A.; Sharma, A.; Talukder, G. Modification of clastogenicity of three known clastogens by garlic extract in mice in vivo. Environ. Mol. Mutagen. 1993, 21, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Aslani, M.R.; Najarnezhad, V.; Mohri, M. Individual and combined effect of meso-2,3-dimercaptosuccinic acid and allicin on blood and tissue lead content in mice. Planta Med. 2010, 76, 241–244. [Google Scholar] [CrossRef] [PubMed]
- Amagase, H.; Petesch, B.L.; Matsuura, H.; Kasuga, S.; Itakura, Y. Intake of garlic and its bioactive components. J. Nutr. 2001, 131, 955S–962S. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Flora, S.J. Therapeutic value of Hippophae rhamnoides L. against subchronic arsenic toxicity in mice. J. Med. Food 2005, 8, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Flora, S.J. Effect of Centella asiatica on arsenic induced oxidative stress and metal distribution in rats. J. Appl. Toxicol. 2006, 26, 213–222. [Google Scholar] [CrossRef]
- Tiwari, H.; Rao, M.V. Curcumin supplementation protects from genotoxic effects of arsenic and fluoride. Food Chem. Toxicol. 2010, 48, 1234–1238. [Google Scholar] [CrossRef] [PubMed]
- Mishra, D.; Flora, S.J. Quercetin administration during chelation therapy protects arsenic-induced oxidative stress in mice. Biol. Trace Elem. Res. 2008, 122, 137–147. [Google Scholar] [CrossRef]
- Ghosh, A.; Mandal, A.K.; Sarkar, S.; Panda, S.; Das, N. Nanoencapsulation of quercetin enhances its dietary efficacy in combating arsenic-induced oxidative damage in liver and brain of rats. Life Sci. 2009, 84, 75–80. [Google Scholar] [CrossRef]
- Bjorklund, G.; Oliinyk, P.; Lysiuk, R.; Rahaman, M.S.; Antonyak, H.; Lozynska, I.; Lenchyk, L.; Peana, M. Arsenic intoxication: General aspects and chelating agents. Arch. Toxicol. 2020, 94, 1879–1897. [Google Scholar] [CrossRef]
- Aziz, S.N.; Boyle, K.J.; Rahman, M. Knowledge of arsenic in drinking-water: Risks and avoidance in Matlab, Bangladesh. J. Health Popul. Nutr. 2006, 24, 327–335. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dose and Level of Arsenic | Study Model | Effects/Molecular Mechanism | References |
|---|---|---|---|
| 13–15 mg/kg. | Mortality data by WHO, epidemiological and geological data. | Induce AD and other dementias as a composite morbi-mortality index. | [24] |
| Sodium arsenite (10 µM). | Cerebellar granule neurons of rats. | Activation of p38 and JNK3 MAP kinases cause cerebellar granule neurotoxicity and apoptosis. | [27] |
| Groundwater long exposure of 240.15 ± 182.96 µg/L. | Longitudinal epidemiological human study. | Low and long As exposure linked to global cognition function. | [25] |
| Drinking water (10 µg/L). | Rat and human brain. | Tau hyperphosphorylation and APP over transcription. | [28] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rahman, M.A.; Hannan, M.A.; Uddin, M.J.; Rahman, M.S.; Rashid, M.M.; Kim, B. Exposure to Environmental Arsenic and Emerging Risk of Alzheimer’s Disease: Perspective Mechanisms, Management Strategy, and Future Directions. Toxics 2021, 9, 188. https://doi.org/10.3390/toxics9080188
Rahman MA, Hannan MA, Uddin MJ, Rahman MS, Rashid MM, Kim B. Exposure to Environmental Arsenic and Emerging Risk of Alzheimer’s Disease: Perspective Mechanisms, Management Strategy, and Future Directions. Toxics. 2021; 9(8):188. https://doi.org/10.3390/toxics9080188
Chicago/Turabian StyleRahman, Md. Ataur, Md. Abdul Hannan, Md Jamal Uddin, Md Saidur Rahman, Md Mamunur Rashid, and Bonglee Kim. 2021. "Exposure to Environmental Arsenic and Emerging Risk of Alzheimer’s Disease: Perspective Mechanisms, Management Strategy, and Future Directions" Toxics 9, no. 8: 188. https://doi.org/10.3390/toxics9080188
APA StyleRahman, M. A., Hannan, M. A., Uddin, M. J., Rahman, M. S., Rashid, M. M., & Kim, B. (2021). Exposure to Environmental Arsenic and Emerging Risk of Alzheimer’s Disease: Perspective Mechanisms, Management Strategy, and Future Directions. Toxics, 9(8), 188. https://doi.org/10.3390/toxics9080188