Norepinephrine Leads to More Cardiopulmonary Toxicities than Epinephrine by Catecholamine Overdose in Rats

 , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. E and/or NE Injection
2.3. Ventricular Catheter Preparation
2.4. Detecting Rat Heart Rate and Ventricular Pressure
2.5. Catecholamine Levels
2.6. Histology and Quantitative Analysis of Immunohistochemistry
2.7. Echocardiography
2.8. Statistical Analysis
3. Results
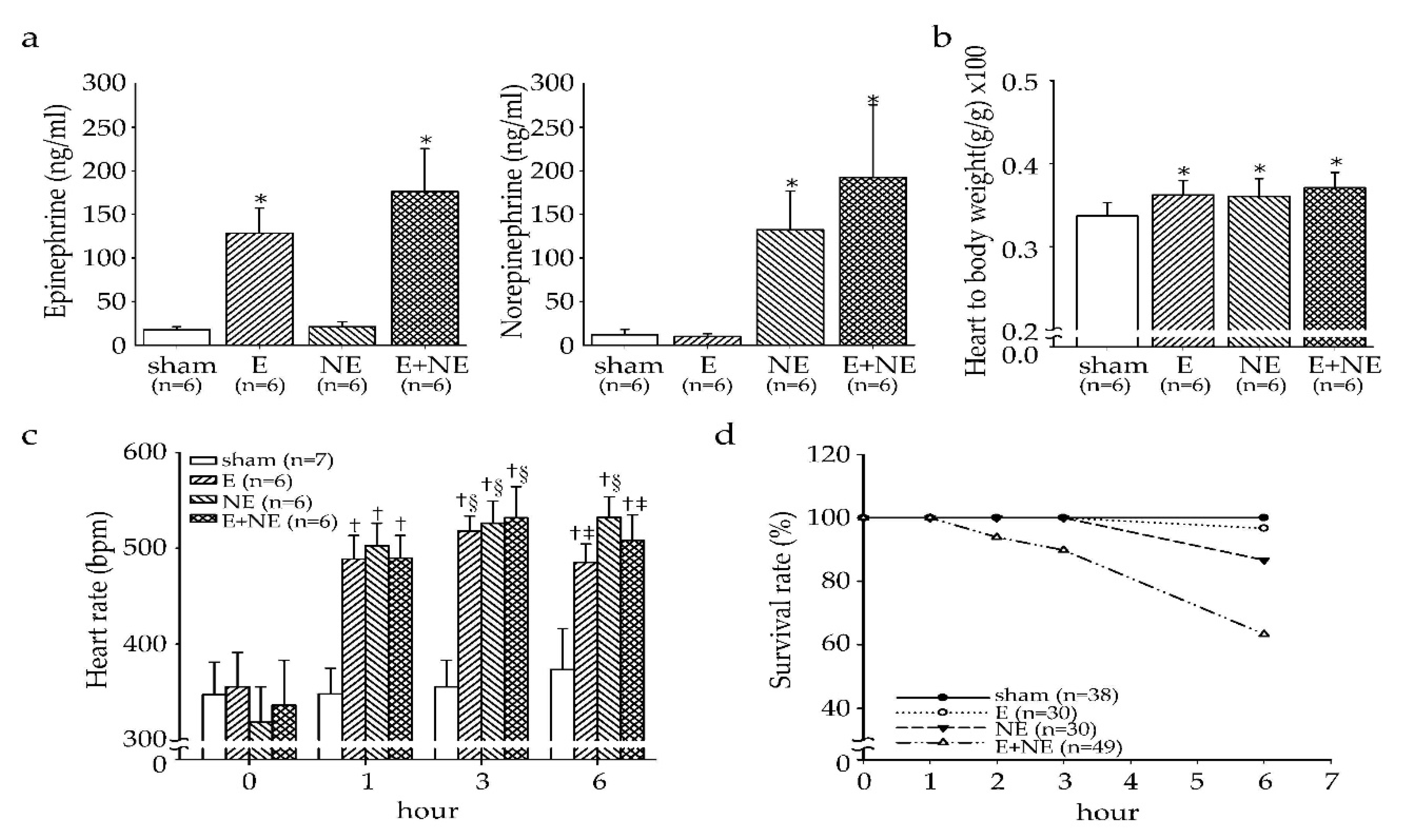
3.1. Overdose of NE and/or E Affected Heart Rate and Survival
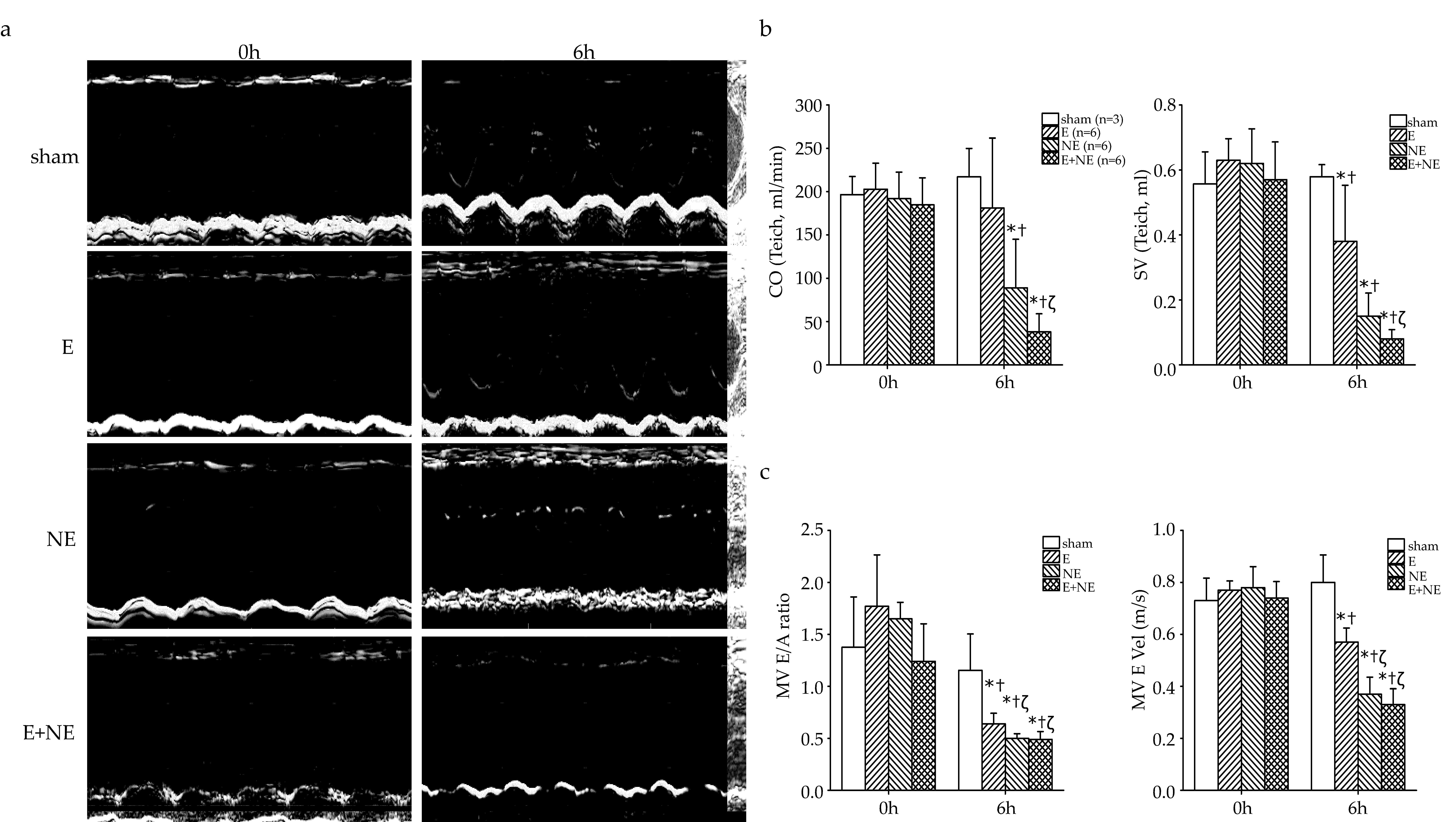
3.2. Overdose of NE Impaired Systolic and Diastolic Function More than E
3.3. Overdose of NE and/or E Led to Presence of Cardiac Damage Markers
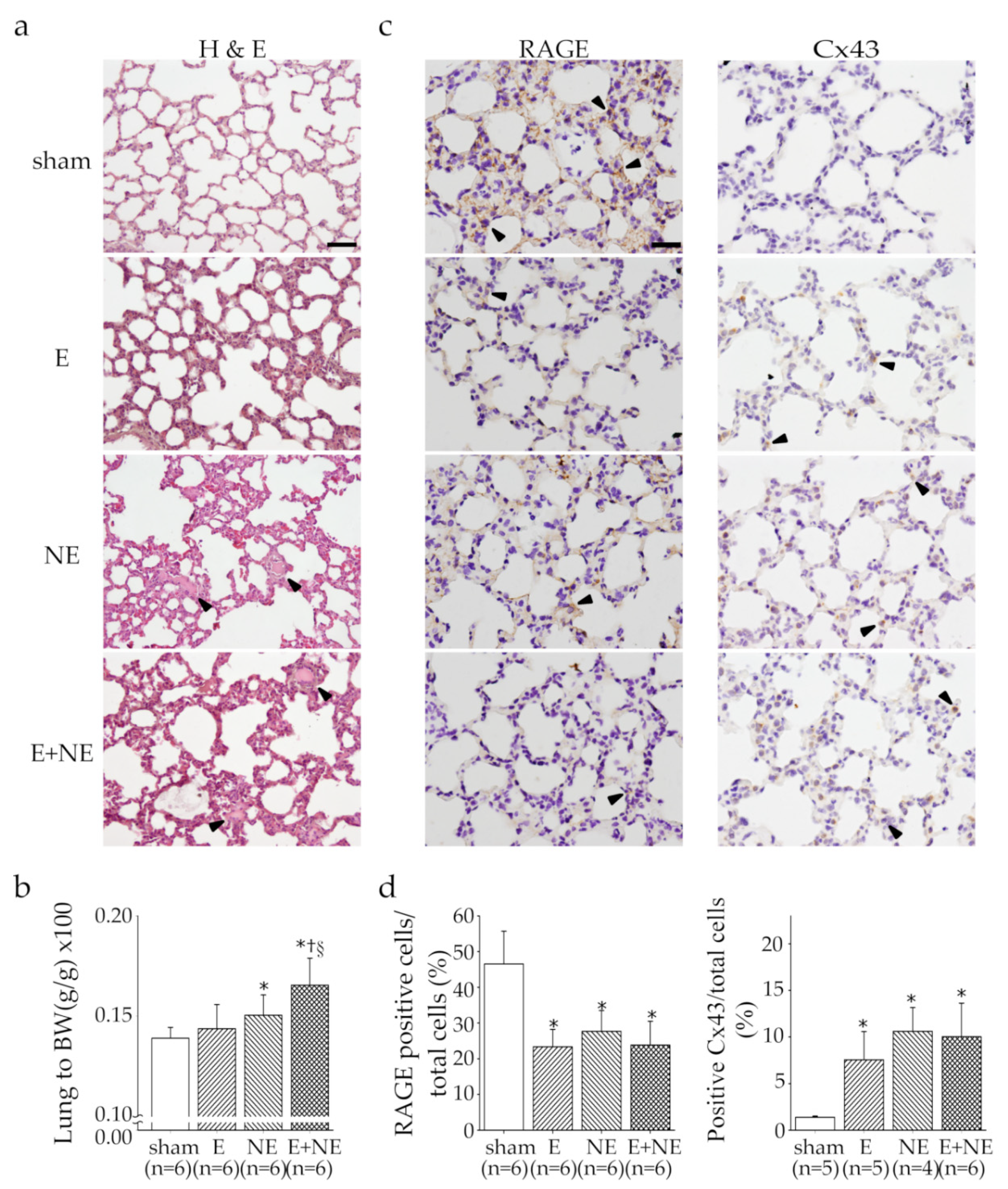
3.4. Overdose of NE rather than E Led to Pulmonary Edema and Damage
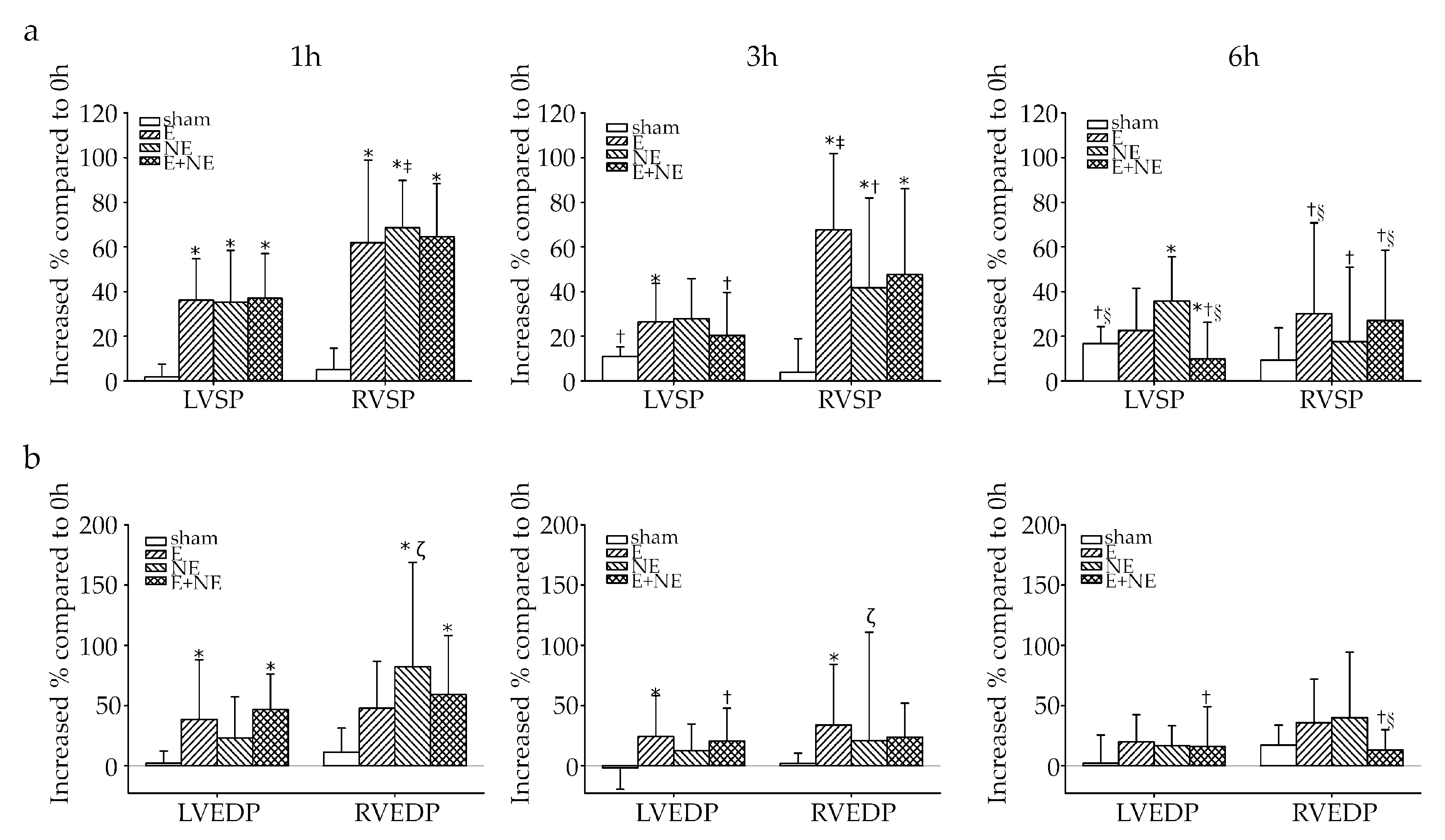
3.5. RV Was More Sensitive and More Vulnerable to Toxicity of Catecholamine Overdose
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Neukirchen, M.; Kienbaum, P. Sympathetic nervous system: Evaluation and importance for clinical general anesthesia. Anesthesiology 2008, 109, 1113–1131. [Google Scholar] [CrossRef] [PubMed]
- Panchal, A.R.; Berg, K.M.; Hirsch, K.G.; Kudenchuk, P.J.; Del Rios, M.; Cabanas, J.G.; Link, M.S.; Kurz, M.C.; Chan, P.S.; Morley, P.T.; et al. 2019 american heart association focused update on advanced cardiovascular life support: Use of advanced airways, vasopressors, and extracorporeal cardiopulmonary resuscitation during cardiac arrest: An update to the american heart association guidelines for cardiopulmonary resuscitation and emergency cardiovascular care. Circulation 2019, 140, e881–e894. [Google Scholar] [PubMed]
- Paradis, N.A.; Koscove, E.M. Epinephrine in cardiac arrest: A critical review. Ann. Emerg. Med. 1990, 19, 1288–1301. [Google Scholar] [CrossRef]
- Severs, W.B.; Summy-Long, J.Y. Norepinephrine-induced reflex bradycardia after central administration of angiotensin ii. Pharmacology 1977, 15, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, A.; Evans, L.E.; Alhazzani, W.; Levy, M.M.; Antonelli, M.; Ferrer, R.; Kumar, A.; Sevransky, J.E.; Sprung, C.L.; Nunnally, M.E.; et al. Surviving sepsis campaign: International guidelines for management of sepsis and septic shock: 2016. Intensive Care Med. 2017, 43, 304–377. [Google Scholar] [CrossRef]
- Xie, Q.; Li, S.; Gao, Y.; Jin, L.; Dai, C.; Song, J. Ergosterol attenuates isoproterenol-induced myocardial cardiotoxicity. Cardiovasc. Toxicol. 2020. [Google Scholar] [CrossRef]
- Boarescu, P.M.; Boarescu, I.; Bocsan, I.C.; Pop, R.M.; Gheban, D.; Bulboaca, A.E.; Nicula, C.; Rajnoveanu, R.M.; Bolboaca, S.D. Curcumin nanoparticles protect against isoproterenol induced myocardial infarction by alleviating myocardial tissue oxidative stress, electrocardiogram, and biological changes. Molecules 2019, 24, 2802. [Google Scholar] [CrossRef]
- Galetta, F.; Franzoni, F.; Bernini, G.; Poupak, F.; Carpi, A.; Cini, G.; Tocchini, L.; Antonelli, A.; Santoro, G. Cardiovascular complications in patients with pheochromocytoma: A mini-review. Biomed. Pharmacother. 2010, 64, 505–509. [Google Scholar] [CrossRef]
- Lu, W.H.; Hsieh, K.S.; Lu, P.J.; Wu, Y.S.; Ho, W.Y.; Lai, C.C.; Wang, J.S.; Ger, L.P.; Hsiao, M.; Tseng, C.J. Hexamethonium reverses the lethal cardiopulmonary damages in a rat model of brainstem lesions mimicking fatal enterovirus 71 encephalitis. Crit. Care Med. 2013, 41, 1276–1285. [Google Scholar] [CrossRef]
- Lu, W.H.; Hsieh, K.S.; Lu, P.J.; Wu, Y.S.; Ho, W.Y.; Cheng, P.W.; Lai, C.C.; Hsiao, M.; Tseng, C.J. Different impacts of alpha- and beta-blockers in neurogenic hypertension produced by brainstem lesions in rat. Anesthesiology 2014, 120, 1192–1204. [Google Scholar] [CrossRef]
- Krishnamoorthy, V.; Rowhani-Rahbar, A.; Gibbons, E.F.; Rivara, F.P.; Temkin, N.R.; Pontius, C.; Luk, K.; Graves, M.; Lozier, D.; Chaikittisilpa, N.; et al. Early systolic dysfunction following traumatic brain injury: A cohort study. Crit. Care Med. 2017, 45, 1028–1036. [Google Scholar] [CrossRef] [PubMed]
- Chi, C.Y.; Khanh, T.H.; Thoa le, P.K.; Tseng, F.C.; Wang, S.M.; Thinh le, Q.; Lin, C.C.; Wu, H.C.; Wang, J.R.; Hung, N.T.; et al. Milrinone therapy for enterovirus 71-induced pulmonary edema and/or neurogenic shock in children: A randomized controlled trial. Crit. Care Med. 2013, 41, 1754–1760. [Google Scholar] [CrossRef]
- Callaham, M.; Madsen, C.D.; Barton, C.W.; Saunders, C.E.; Pointer, J. A randomized clinical trial of high-dose epinephrine and norepinephrine vs standard-dose epinephrine in prehospital cardiac arrest. JAMA 1992, 268, 2667–2672. [Google Scholar] [CrossRef] [PubMed]
- Stiell, I.G.; Hebert, P.C.; Weitzman, B.N.; Wells, G.A.; Raman, S.; Stark, R.M.; Higginson, L.A.; Ahuja, J.; Dickinson, G.E. High-dose epinephrine in adult cardiac arrest. N. Engl. J. Med. 1992, 327, 1045–1050. [Google Scholar] [CrossRef]
- Zimmer, H.G.; Zierhut, W.; Seesko, R.C.; Varekamp, A.E. Right heart catheterization in rats with pulmonary hypertension and right ventricular hypertrophy. Basic Res. Cardiol. 1988, 83, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, H.G. Measurement of left ventricular hemodynamic parameters in closed-chest rats under control and various pathophysiologic conditions. Basic Res. Cardiol. 1983, 78, 77–84. [Google Scholar] [CrossRef]
- Feng, J.; Fitz, Y.; Li, Y.; Fernandez, M.; Cortes Puch, I.; Wang, D.; Pazniokas, S.; Bucher, B.; Cui, X.; Solomon, S.B. Catheterization of the carotid artery and jugular vein to perform hemodynamic measures, infusions and blood sampling in a conscious rat model. J. Vis. Exp. 2015. [Google Scholar] [CrossRef]
- Deten, A.; Millar, H.; Zimmer, H.G. Catheterization of pulmonary artery in rats with an ultraminiature catheter pressure transducer. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H2212–H2217. [Google Scholar] [CrossRef][Green Version]
- Prodromidis, G.; Nikitakis, N.G.; Sklavounou, A. Immunohistochemical analysis of the activation status of the akt/mtor/ps6 signaling pathway in oral lichen planus. Int. J. Dent. 2013, 2013, 743456. [Google Scholar] [CrossRef]
- Uzzaman, M.; Honjo, H.; Takagishi, Y.; Emdad, L.; Magee, A.I.; Severs, N.J.; Kodama, I. Remodeling of gap junctional coupling in hypertrophied right ventricles of rats with monocrotaline-induced pulmonary hypertension. Circ. Res. 2000, 86, 871–878. [Google Scholar] [CrossRef]
- Lee, C.C.; Lai, Y.T.; Chang, H.T.; Liao, J.W.; Shyu, W.C.; Li, C.Y.; Wang, C.N. Inhibition of high-mobility group box 1 in lung reduced airway inflammation and remodeling in a mouse model of chronic asthma. Biochem. Pharmacol. 2013, 86, 940–949. [Google Scholar] [CrossRef] [PubMed]
- Michela, P.; Velia, V.; Aldo, P.; Ada, P. Role of connexin 43 in cardiovascular diseases. Eur. J. Pharmacol. 2015, 768, 71–76. [Google Scholar] [CrossRef]
- Palatinus, J.A.; Rhett, J.M.; Gourdie, R.G. The connexin43 carboxyl terminus and cardiac gap junction organization. Biochim. Biophys. Acta 2012, 1818, 1831–1843. [Google Scholar] [CrossRef] [PubMed]
- Archan, S.; Fleisher, L.A. From creatine kinase-mb to troponin: The adoption of a new standard. Anesthesiology 2010, 112, 1005–1012. [Google Scholar] [CrossRef] [PubMed]
- Oczypok, E.A.; Perkins, T.N.; Oury, T.D. All the “rage” in lung disease: The receptor for advanced glycation endproducts (rage) is a major mediator of pulmonary inflammatory responses. Paediatr. Respir. Rev. 2017, 23, 40–49. [Google Scholar] [CrossRef]
- Kasper, M.; Traub, O.; Reimann, T.; Bjermer, L.; Grossmann, H.; Muller, M.; Wenzel, K.W. Upregulation of gap junction protein connexin43 in alveolar epithelial cells of rats with radiation-induced pulmonary fibrosis. Histochem. Cell Biol. 1996, 106, 419–424. [Google Scholar] [CrossRef]
- Campbell, R.L. Cardiovascular effects of epinephrine overdose: Case report. Anesth. Prog. 1977, 24, 190–193. [Google Scholar]
- Karch, S.B. Coronary artery spasm induced by intravenous epinephrine overdose. Am. J. Emerg. Med. 1989, 7, 485–488. [Google Scholar] [CrossRef]
- Liaudet, L.; Calderari, B.; Pacher, P. Pathophysiological mechanisms of catecholamine and cocaine-mediated cardiotoxicity. Heart Fail Rev. 2014, 19, 815–824. [Google Scholar] [CrossRef]
- Paur, H.; Wright, P.T.; Sikkel, M.B.; Tranter, M.H.; Mansfield, C.; O’Gara, P.; Stuckey, D.J.; Nikolaev, V.O.; Diakonov, I.; Pannell, L.; et al. High levels of circulating epinephrine trigger apical cardiodepression in a beta2-adrenergic receptor/gi-dependent manner: A new model of takotsubo cardiomyopathy. Circulation 2012, 126, 697–706. [Google Scholar] [CrossRef]
- Heubach, J.F.; Ravens, U.; Kaumann, A.J. Epinephrine activates both gs and gi pathways, but norepinephrine activates only the gs pathway through human beta2-adrenoceptors overexpressed in mouse heart. Mol. Pharmacol. 2004, 65, 1313–1322. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Goyal, P.; Idrees, S.; Aggarwal, S.; Bajaj, D.; Mattana, J. Association of endocrine conditions with takotsubo cardiomyopathy: A comprehensive review. J. Am. Heart Assoc. 2018, 7, e009003. [Google Scholar] [CrossRef] [PubMed]
- Lyon, A.R.; Rees, P.S.; Prasad, S.; Poole-Wilson, P.A.; Harding, S.E. Stress (takotsubo) cardiomyopathy—A novel pathophysiological hypothesis to explain catecholamine-induced acute myocardial stunning. Nat. Clin. Pract. Cardiovasc. Med. 2008, 5, 22–29. [Google Scholar] [CrossRef]
- Zaugg, M.; Xu, W.; Lucchinetti, E.; Shafiq, S.A.; Jamali, N.Z.; Siddiqui, M.A. Beta-adrenergic receptor subtypes differentially affect apoptosis in adult rat ventricular myocytes. Circulation 2000, 102, 344–350. [Google Scholar] [CrossRef] [PubMed]
- Nef, H.M.; Mollmann, H.; Akashi, Y.J.; Hamm, C.W. Mechanisms of stress (takotsubo) cardiomyopathy. Nat. Rev. Cardiol. 2010, 7, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Robinson, A.D.; Ramanathan, K.B.; McGee, J.E.; Newman, K.P.; Weber, K.T. Oxidative stress and cardiomyocyte necrosis with elevated serum troponins: Pathophysiologic mechanisms. Am. J. Med. Sci. 2011, 342, 129–134. [Google Scholar] [CrossRef]
- Smyth, J.W.; Hong, T.T.; Gao, D.; Vogan, J.M.; Jensen, B.C.; Fong, T.S.; Simpson, P.C.; Stainier, D.Y.; Chi, N.C.; Shaw, R.M. Limited forward trafficking of connexin 43 reduces cell-cell coupling in stressed human and mouse myocardium. J. Clin. Investig. 2010, 120, 266–279. [Google Scholar] [CrossRef]
- Macmillan, C.S.; Grant, I.S.; Andrews, P.J. Pulmonary and cardiac sequelae of subarachnoid haemorrhage: Time for active management? Intensive Care Med. 2002, 28, 1012–1023. [Google Scholar] [CrossRef]
- Inamasu, J.; Sugimoto, K.; Yamada, Y.; Ganaha, T.; Ito, K.; Watabe, T.; Hayashi, T.; Kato, Y.; Ozaki, Y.; Hirose, Y. The role of catecholamines in the pathogenesis of neurogenic pulmonary edema associated with subarachnoid hemorrhage. Acta Neurochir. 2012, 154, 2179–2184; discussion 2184–2185. [Google Scholar] [CrossRef]
- Rassler, B.; Reissig, C.; Briest, W.; Tannapfel, A.; Zimmer, H.G. Catecholamine-induced pulmonary edema and pleural effusion in rats—Alpha- and beta-adrenergic effects. Respir. Physiol. Neurobiol. 2003, 135, 25–37. [Google Scholar] [CrossRef]
- Barker, B.L.; Brightling, C.E. Phenotyping the heterogeneity of chronic obstructive pulmonary disease. Clin. Sci. 2013, 124, 371–387. [Google Scholar] [CrossRef] [PubMed]
- Guillory, A.N.; Clayton, R.P.; Prasai, A.; El Ayadi, A.; Herndon, D.N.; Finnerty, C.C. Biventricular differences in beta-adrenergic receptor signaling following burn injury. PLoS ONE 2017, 12, e0189527. [Google Scholar] [CrossRef] [PubMed]
- Quagliariello, V.; Passariello, M.; Coppola, C.; Rea, D.; Barbieri, A.; Scherillo, M.; Monti, M.G.; Iaffaioli, R.V.; De Laurentiis, M.; Ascierto, P.A.; et al. Cardiotoxicity and pro-inflammatory effects of the immune checkpoint inhibitor Pembrolizumab associated to Trastuzumab. Int. J. Cardiol. 2019, 292, 171–179. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Ventricular Pressure (mmHg) | ||||
|---|---|---|---|---|---|
| Hour | Sham (n = 7) | E (n = 7) | NE (n = 7) | E+NE (n = 8) | |
| LVSP | 0 h | 86.2 ± 11.1 | 90.3 ± 9.7 | 89.0 ± 11.1 | 87.0 ± 8.7 |
| 1 h | 87.8 ± 13.0 | 122.4 ± 18.7 *,† | 118.4 ± 8.2 *,† | 118.4 ± 16.4 *,† | |
| 3 h | 95.2 ± 10.0 †,‡ | 114.0 ± 20.3 *,† | 112.4 ± 11.5 *,† | 104.4 ± 16.9 †,‡ | |
| 6 h | 100.5 ± 14.21†,‡,§ | 111.1 ± 24.4 † | 119.3 ± 17.7 † | 95.1 ± 12.9 ‡,§,¥ | |
| LVEDP | 0 h | 11.9 ± 5.1 | 9.0 ± 2.8 | 11.8 ± 6.0 | 11.0 ± 3.9 |
| 1 h | 12.3 ± 5.5 | 16.7 ± 12.5 † | 19.9 ± 15.1 | 20.8 ± 8.3 *,† | |
| 3 h | 11.2 ± 5.6 | 14.4 ± 9.5 | 16.7 ± 11.8 | 15.2 ± 7.2 ‡ | |
| 6 h | 12.3 ± 7.1 | 13.1 ± 7.2 | 17.2 ± 11.3 | 13.3 ± 5.2 ‡ | |
| RVSP | 0 h | 17.2 ± 3.0 | 17.3 ± 3.6 | 16.8 ± 2.5 | 18.4 ± 3.9 |
| 1 h | 18.2 ± 4.2 | 27.3 ± 5.1 *,† | 27.8 ± 2.1 *,† | 29.8 ± 5.6 *,† | |
| 3 h | 17.9 ± 4.1 | 28.3 ± 5.1 *,† | 22.9 ± 2.9 *,†,‡,# | 26.4 ± 6.0 *,† | |
| 6 h | 18.8 ± 4.0 | 21.6 ± 5.3 ‡,§ | 19.3 ± 4.9 ‡ | 23.0 ± 6.2 †,‡,§ | |
| RVEDP | 0 h | 7.3 ± 3.1 | 8.0 ± 1.6 | 9.0 ± 4.1 | 8.1 ± 3.0 |
| 1 h | 8.2 ± 3.4 | 15.4 ± 5.2 *,† | 18.9 ± 4.0 *,† | 15.5 ± 4.5 *,† | |
| 3 h | 8.2 ± 3.5 | 17.0 ± 5.8 *,† | 15.1 ± 3.1 *,† | 13.2 ± 5.6 *,† | |
| 6 h | 9.2 ± 3.9 † | 13.2 ± 4.5 †,§ | 12.4 ± 4.1 ‡ | 10.2 ± 4.2 ‡,§ | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, W.-H.; Chen, H.-H.; Chen, B.-H.; Lee, J.-C.; Lai, C.-C.; Li, C.-H.; Tseng, C.-J. Norepinephrine Leads to More Cardiopulmonary Toxicities than Epinephrine by Catecholamine Overdose in Rats. Toxics 2020, 8, 69. https://doi.org/10.3390/toxics8030069
Lu W-H, Chen H-H, Chen B-H, Lee J-C, Lai C-C, Li C-H, Tseng C-J. Norepinephrine Leads to More Cardiopulmonary Toxicities than Epinephrine by Catecholamine Overdose in Rats. Toxics. 2020; 8(3):69. https://doi.org/10.3390/toxics8030069
Chicago/Turabian StyleLu, Wen-Hsien, Hsin-Hung Chen, Bo-Hau Chen, Jui-Chen Lee, Chi-Cheng Lai, Che-Hsing Li, and Ching-Jiunn Tseng. 2020. "Norepinephrine Leads to More Cardiopulmonary Toxicities than Epinephrine by Catecholamine Overdose in Rats" Toxics 8, no. 3: 69. https://doi.org/10.3390/toxics8030069
APA StyleLu, W.-H., Chen, H.-H., Chen, B.-H., Lee, J.-C., Lai, C.-C., Li, C.-H., & Tseng, C.-J. (2020). Norepinephrine Leads to More Cardiopulmonary Toxicities than Epinephrine by Catecholamine Overdose in Rats. Toxics, 8(3), 69. https://doi.org/10.3390/toxics8030069