Impact of Maternal Air Pollution Exposure on Children’s Lung Health: An Indian Perspective



Abstract
1. Introduction
2. Evidence of Maternal Air Pollution-Induced Health Effects in Offspring
3. Maternal Air Pollution Exposure and Airway Disease
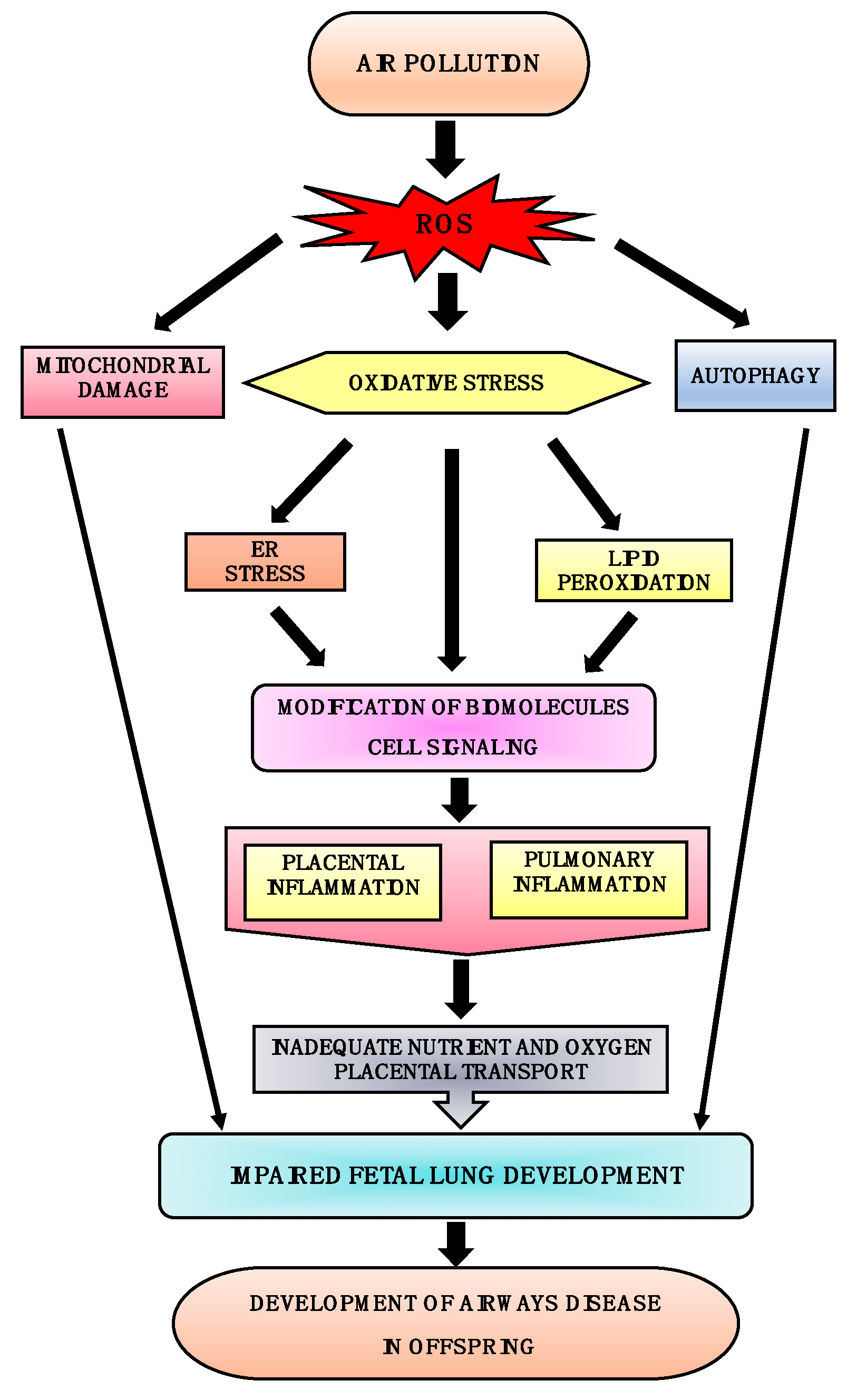
4. In Utero Cellular Mechanisms Involved in the Development of Airway Disease
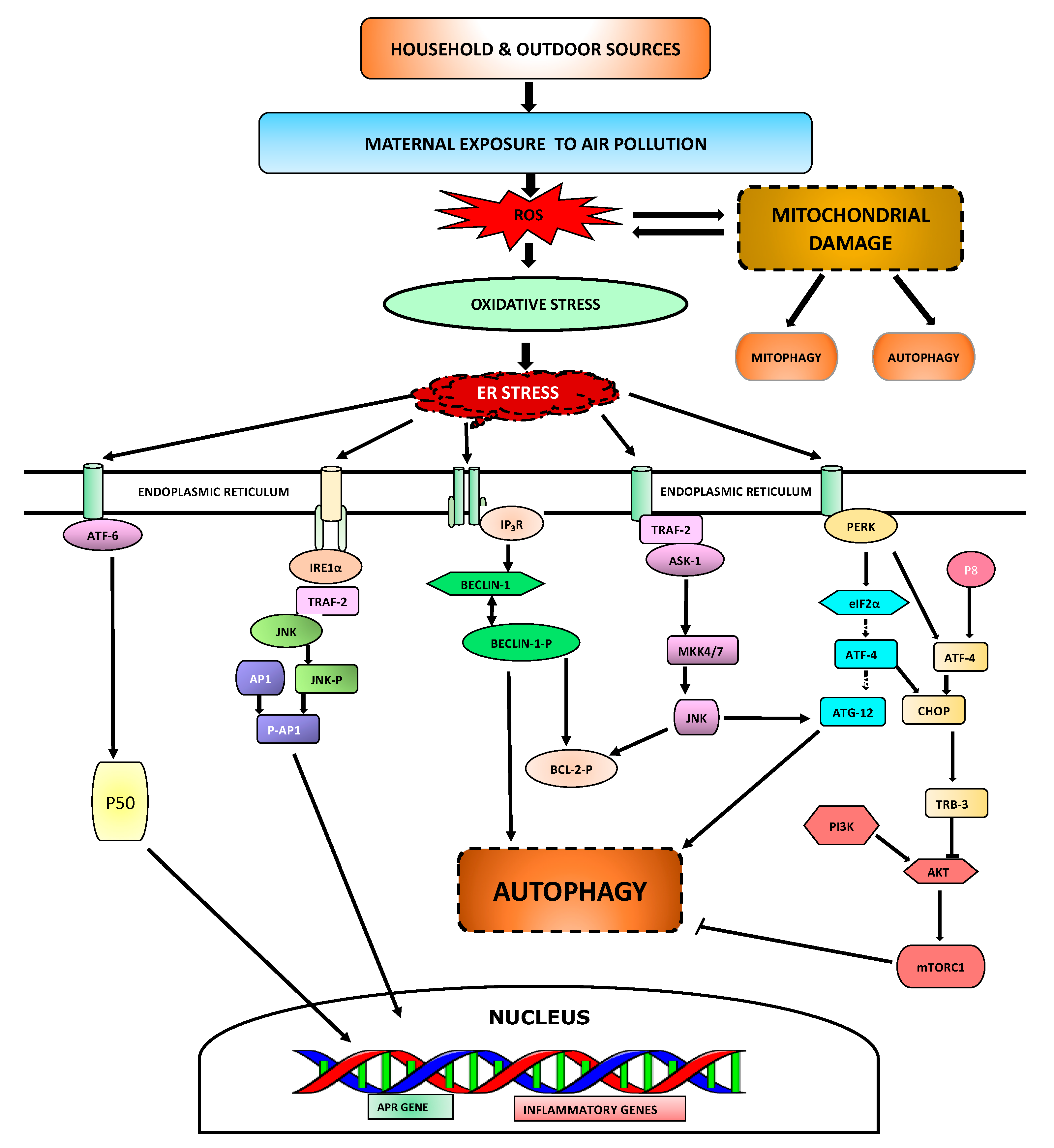
4.1. ER Stress
4.2. Autophagy
4.3. Mitochondrial Damage
5. Conclusion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kelly, F.J.; Fussell, J.C. Air pollution and public health: Emerging hazards and improved understanding of risk. Environ. Geochem. Health 2015, 37, 631–649. [Google Scholar] [CrossRef] [PubMed]
- Forouzanfar, M.H.; Afshin, A.; Alexander, L.T.; Anderson, H.R.; Bhutta, Z.A.; Biryukov, S.; Brauer, M.; Burnett, R.; Cercy, K.; Charlson, F.J.; et al. Global, regional, and national comparative risk assessment of 79 behavioural, environmental and occupational, and metabolic risks or clusters of risks, 1990–2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet 2016, 388, 1659–1724. [Google Scholar] [CrossRef]
- Jiang, X.-Q.; Mei, X.-D.; Feng, D. Air pollution and chronic airway diseases: What should people know and do? J. Thorac. Dis. 2016, 8, E31–E40. [Google Scholar] [PubMed]
- Kim, K.-H.; Kabir, E.; Kabir, S. A review on the human health impact of airborne particulate matter. Environ. Int. 2015, 74, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.D.; Woodruff, T.J.; Basu, R.; Schoendorf, K.C. Air pollution and birth weight among term infants in California. Pediatrics 2005, 115, 121–128. [Google Scholar] [CrossRef] [PubMed]
- WHO. First WHO Conference on Air Poolution and Health. In Improving Air Quality, Combatting Climate Change–Saving Lives; WHO: Geneva, Switzerland, 2018. [Google Scholar]
- WHO. Ambient Air Pollution: Health Impacts. 23 October 2018. Available online: https://www.who.int/airpollution/ambient/health-impacts/en/ (accessed on 4 November 2018).
- Landrigan, P.J.; Fuller, R.; Acosta, N.J.; Adeyi, O.; Arnold, R.; Baldé, A.B.; Bertollini, R.; Bose-O’Reilly, S.; Boufford, J.I.; Breysse, P.N.; et al. The Lancet Commission on pollution and health. Lancet 2017, 391, 462–512. [Google Scholar] [CrossRef]
- Dadvand, P.; Parker, J.; Bell, M.L.; Bonzini, M.; Brauer, M.; Darrow, L.A.; Gehring, U.; Glinianaia, S.V.; Gouveia, N.; Ha, E.-H.; et al. Maternal exposure to particulate air pollution and term birth weight: A multi-country evaluation of effect and heterogeneity. Environ. Health Perspect. 2013, 121, 267–373. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.S.; Balkhair, T.; Knowledge Synthesis Group on Determinants of Preterm/LBW births. Air pollution and birth outcomes: A systematic review. Environ. Int. 2011, 37, 498–516. [Google Scholar] [CrossRef] [PubMed]
- Stieb, D.M.; Chen, L.; Eshoul, M.; Judek, S. Ambient air pollution, birth weight and preterm birth: A systematic review and meta-analysis. Environ. Res. 2012, 117, 100–111. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Liang, S.; Zhao, J.; Qian, Z.; Bassig, B.A.; Yang, R.; Zhang, Y.; Hu, K.; Xu, S.; Zheng, T. Maternal exposure to air pollutant PM 2.5 and PM 10 during pregnancy and risk of congenital heart defects. J. Expo. Sci. Environ. Epidemiol. 2016, 26, 422–427. [Google Scholar] [CrossRef] [PubMed]
- Rich, D.Q.; Demissie, K.; Lu, S.-E.; Kamat, L.; Wartenberg, D.; Rhoads, G.G. Ambient air pollutant concentrations during pregnancy and the risk of fetal growth restriction. J. Epidemiol. Community Health 2009, 63, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Rauh, V.; Garfinkel, R.; Tu, Y.; Perera, F.P. Prenatal exposure to airborne polycyclic aromatic hydrocarbons and risk of intrauterine growth restriction. Environ. Health Perspect. 2008, 116, 658–665. [Google Scholar] [CrossRef] [PubMed]
- Hyland, A.; Piazza, K.M.; Hovey, K.M.; Ockene, J.K.; Andrews, C.A.; Rivard, C.; Wactawski-Wende, J. Associations of lifetime active and passive smoking with spontaneous abortion, stillbirth and tubal ectopic pregnancy: A cross-sectional analysis of historical data from the Women’s Health Initiative. Tob. Control 2015, 24, 328–335. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Hu, Y.; Chen, C.; Yang, F.; Li, Z.; Fang, Z.; Wang, L.; Chen, D. Passive smoking, metabolic gene polymorphisms, and infant birth weight in a prospective cohort study of Chinese women. Am. J. Epidemiol. 2007, 166, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Siddique, S.; Banerjee, M.; Ray, M.R.; Lahiri, T. Attention-deficit hyperactivity disorder in children chronically exposed to high level of vehicular pollution. Eur. J. Pediatr. 2011, 170, 923–929. [Google Scholar] [CrossRef] [PubMed]
- Newman, N.C.; Ryan, P.; LeMasters, G.; Levin, L.; Bernstein, D.; Hershey, G.K.K.; Lockey, J.E.; Villareal, M.; Reponen, T.; Grinshpun, S. Traffic-related air pollution exposure in the first year of life and behavioral scores at 7 years of age. Environ. Health Perspect. 2013, 121, 731–736. [Google Scholar] [CrossRef] [PubMed]
- Gong, T.; Almqvist, C.; Bölte, S.; Lichtenstein, P.; Anckarsäter, H.; Lind, T.; Lundholm, C.; Pershagen, G. Exposure to air pollution from traffic and neurodevelopmental disorders in Swedish twins. Twin Res. Hum. Genet. 2014, 17, 553–562. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Ha, S.U.; Basnet, R. A review of epidemiological research on adverse neurological effects of exposure to ambient air pollution. Front. Public Health 2016, 4, 157. [Google Scholar] [CrossRef] [PubMed]
- Jedrychowski, W.; Perera, F.; Maugeri, U.; Mrozek-Budzyn, D.; Miller, R.L.; Flak, E.; Mroz, E.; Jacek, R.; Spengler, J.D. Effects of prenatal and perinatal exposure to fine air pollutants and maternal fish consumption on the occurrence of infantile eczema. Int. Arch. Allergy Immunol. 2011, 155, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Lavigne, É.; Bélair, M.-A.; Duque, D.R.; Do, M.T.; Stieb, D.M.; Hystad, P.; van Donkelaar, A.; Martin, R.V.; Crouse, D.L.; Crighton, E. Effect modification of perinatal exposure to air pollution and childhood asthma incidence. Eur. Respir. J. 2018, 1701884. [Google Scholar] [CrossRef] [PubMed]
- Juda-Rezler, K.; Reizer, M.; Oudinet, J.-P. Determination and analysis of PM10 source apportionment during episodes of air pollution in Central Eastern European urban areas: The case of wintertime 2006. Atmos. Environ. 2011, 45, 6557–6566. [Google Scholar] [CrossRef]
- Apte, K.; Salvi, S. Household air pollution and its effects on health. F1000Research 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, R.W.; Fuller, G.W.; Anderson, H.R.; Harrison, R.M.; Armstrong, B. Urban ambient particle metrics and health: A time-series analysis. Epidemiology 2010, 21, 501–511. [Google Scholar] [CrossRef] [PubMed]
- Srimuruganandam, B.; Nagendra, S.S. Source characterization of PM10 and PM2.5 mass using a chemical mass balance model at urban roadside. Sci. Total Environ. 2012, 433, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Misra, C.; Geller, M.D.; Shah, P.; Sioutas, C.; Solomon, P.A. Development and Evaluation of a Continuous Coarse (PM10–PM25) Particle Monitor. J. Air Waste Manag. Assoc. 2001, 51, 1309–1317. [Google Scholar] [CrossRef] [PubMed]
- Air Pollution: 10 Countries with the World’s Dirtiest Air. Available online: http://www.abc.net.au/news/2014-05-08/10-countries-with-the-worlds-dirtiest-air/5438872 (accessed on 8 October 2018).
- Brauer, M.; Freedman, G.; Frostad, J.; Van Donkelaar, A.; Martin, R.V.; Dentener, F.; Dingenen, R.V.; Estep, K.; Amini, H.; Apte, J.S.; et al. Ambient air pollution exposure estimation for the global burden of disease 2013. Environ. Sci. Technol. 2015, 50, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Pant, P.; Guttikunda, S.K.; Peltier, R.E. Exposure to particulate matter in India: A synthesis of findings and future directions. Environ. Res. 2016, 147, 480–496. [Google Scholar] [CrossRef] [PubMed]
- Dey, S.; Di Girolamo, L.; van Donkelaar, A.; Tripathi, S.; Gupta, T.; Mohan, M. Variability of outdoor fine particulate (PM2.5) concentration in the Indian Subcontinent: A remote sensing approach. Remote Sens. Environ. 2012, 127, 153–161. [Google Scholar] [CrossRef]
- Lim, S.S.; Vos, T.; Flaxman, A.D.; Danaei, G.; Shibuya, K.; Adair-Rohani, H.; AlMazroa, M.A.; Amann, M.; Anderson, H.R.; Andrews, K.G. A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990–2010: A systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012, 380, 2224–2260. [Google Scholar] [CrossRef]
- Balakrishnan, K.; Ramaswamy, P.; Sambandam, S.; Thangavel, G.; Ghosh, S.; Johnson, P.; Mukhopadhyay, K.; Venugopal, V.; Thanasekaraan, V. Air pollution from household solid fuel combustion in India: An overview of exposure and health related information to inform health research priorities. Glob. Health Action 2011, 4, 5638. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, S.; Hopke, P.K.; Thimmaiah, D.; Dumka, U.C.; Srivastava, A.K.; Bisht, D.S.; Rao, P.S.; Chate, D.M.; Srivastava, M.K.; Tripathi, S.N. Nature and sources of ionic species in precipitation across the Indo-Gangetic Plains, India. Aerosol Air Qual. Res. 2016, 16, 943–957. [Google Scholar] [CrossRef]
- Attri, S. Atlas of Hourly Mixing Height and Assimilative Capacity of Atmosphere in India; India Meteorological Department: New Delhi, India, 2008. [Google Scholar]
- Barbier, E.B. Natural Resources and Economic Development; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar]
- Guaita, R.; Pichiule, M.; Maté, T.; Linares, C.; Díaz, J. Short-term impact of particulate matter (PM2.5) on respiratory mortality in Madrid. Int. J. Environ. Health Res. 2011, 21, 260–274. [Google Scholar] [CrossRef] [PubMed]
- Joseph, A.; Sawant, A.; Srivastava, A. PM10 and its impacts on health-a case study in Mumbai. Int. J. Environ. Health Res. 2003, 13, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Balakrishnan, K.; Sambandam, S.; Ramaswamy, P.; Ghosh, S.; Venkatesan, V.; Thangavel, G.; Mukhopadhyay, K.; Johnson, P.; Paul, S.; Puttaswamy, N.; et al. Establishing integrated rural–urban cohorts to assess air pollution-related health effects in pregnant women, children and adults in Southern India: An overview of objectives, design and methods in the Tamil Nadu Air Pollution and Health Effects (TAPHE) study. BMJ Open 2015, 5, e008090. [Google Scholar] [CrossRef] [PubMed]
- Tobollik, M.; Razum, O.; Wintermeyer, D.; Plass, D. Burden of Outdoor Air Pollution in Kerala, India—A First Health Risk Assessment at State Level. Int. J. Environ. Res. Public Health 2015, 12, 10602–10619. [Google Scholar] [CrossRef] [PubMed]
- Salvi, S.; Kumar, G.A.; Dhaliwal, R.S.; Paulson, K.; Agrawal, A.; Koul, P.A.; Mahesh, P.A.; Nair, S.; Singh, V.; Aggarwal, A.N.; et al. The burden of chronic respiratory diseases and their heterogeneity across the states of India: the Global Burden of Disease Study 1990–2016. Lancet Glob. Health 2018, 6, e1363–e1374. [Google Scholar] [CrossRef]
- Awasthi, A.; Singh, N.; Mittal, S.; Gupta, P.K.; Agarwal, R. Effects of agriculture crop residue burning on children and young on PFTs in North West India. Sci. Total Environ. 2010, 408, 4440–4445. [Google Scholar] [CrossRef] [PubMed]
- Balakrishnan, K.; Sankar, S.; Parikh, J.; Padmavathi, R.; Srividya, K.; Venugopal, V.; Prasad, S.; Pandey, V.L. Daily average exposures to respirable particulate matter from combustion of biomass fuels in rural households of southern India. Environ. Health Perspect. 2002, 110, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Padhy, P.K.; Padhi, B.K. Effects of biomass combustion smoke on hematological and antioxidant profile among children (8–13 years) in India. Inhal. Toxicol. 2009, 21, 705–711. [Google Scholar] [CrossRef] [PubMed]
- 14 of World’s 15 Most Polluted Cities in India. Available online: https://timesofindia.indiatimes.com/city/delhi/14-of-worlds-15-most-polluted-cities-in-india/articleshow/63993356.cms (accessed on 8 October 2018).
- Kumar, R.; Nagar, J.K.; Goel, N.; Kumar, P.; Kushwah, A.S.; Gaur, S.N. Indoor air pollution and asthma in children at Delhi, India. Adv. Respir. Med. 2015, 83, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Sharma, B.B.; Sharma, S.; Sabir, M.; Singh, V.; Investigators, I.C. Prevalence and severity of asthma among Indian school children aged between 6 and 14 years: Associations with parental smoking and traffic pollution. J. Asthma 2016, 53, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Murlidhar, V. Case Report: An 11-year-old boy with silico-tuberculosis attributable to secondary exposure to sandstone mining in central India. BMJ Case Rep. 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Rumchev, K.; Zhao, Y.; Spickett, J. Health Risk Assessment of Indoor Air Quality, Socioeconomic and House Characteristics on Respiratory Health among Women and Children of Tirupur, South India. Int. J. Environ. Res. Public Health 2017, 14, 429. [Google Scholar] [CrossRef] [PubMed]
- Perez, L.; Tobías, A.; Querol, X.; Pey, J.; Alastuey, A.; Díaz, J.; Sunyer, J. Saharan dust, particulate matter and cause-specific mortality: A case–crossover study in Barcelona (Spain). Environ. Int. 2012, 48, 150–155. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Luo, X.; Zhao, C.; Zhang, B.; Tao, J.; Yang, Z.; Ma, W.; Liu, T. The associations between birth weight and exposure to fine particulate matter (PM2.5) and its chemical constituents during pregnancy: A meta-analysis. Environ. Pollut. 2016, 211, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Perera, F.P.; Whyatt, R.M.; Jedrychowski, W.; Rauh, V.; Manchester, D.; Santella, R.M.; Ottman, R. Recent developments in molecular epidemiology: A study of the effects of environmental polycyclic aromatic hydrocarbons on birth outcomes in Poland. Am. J. Epidemiol. 1998, 147, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Dejmek, J.; Solanský, I.; Benes, I.; Lenícek, J.; Srám, R.J. The impact of polycyclic aromatic hydrocarbons and fine particles on pregnancy outcome. Environ. Health Perspect. 2000, 108, 1159–1164. [Google Scholar] [CrossRef] [PubMed]
- Risom, L.; Møller, P.; Loft, S. Oxidative stress-induced DNA damage by particulate air pollution. Mutat. Res. 2005, 592, 119–137. [Google Scholar] [CrossRef] [PubMed]
- Bobak, M. Outdoor air pollution, low birth weight, and prematurity. Environ. Health Perspect. 2000, 108, 173–176. [Google Scholar] [CrossRef] [PubMed]
- Kannan, S.; Misra, D.P.; Dvonch, J.T.; Krishnakumar, A. Exposures to airborne particulate matter and adverse perinatal outcomes: A biologically plausible mechanistic framework for exploring potential. Cienc. Saude Coletiva 2007, 12, 1591–1602. [Google Scholar] [CrossRef]
- Valkonen, V.-P.; Päivä, H.; Salonen, J.T.; Lakka, T.A.; Lehtimäki, T.; Laakso, J.; Laaksonen, R. Risk of acute coronary events and serum concentration of asymmetrical dimethylarginine. Lancet 2001, 358, 2127–2128. [Google Scholar] [CrossRef]
- Leone, A.; Moncada, S.; Vallance, P.; Calver, A.; Collier, J. Accumulation of an endogenous inhibitor of nitric oxide synthesis in chronic renal failure. Lancet 1992, 339, 572–575. [Google Scholar] [CrossRef]
- Ibald-Mulli, A.; Stieber, J.; Wichmann, H.-E.; Koenig, W.; Peters, A. Effects of air pollution on blood pressure: A population-based approach. Am. J. Public Health 2001, 91, 571–577. [Google Scholar] [PubMed]
- Jedrychowski, W.A.; Perera, F.P.; Maugeri, U.; Spengler, J.; Mroz, E.; Flak, E.; Stigter, L.; Majewska, R.; Kaim, I.; Sowa, A. Prohypertensive effect of gestational personal exposure to fine particulate matter. Prospective cohort study in non-smoking and non-obese pregnant women. Cardiovasc. Toxicol. 2012, 12, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Van den Eeden, L.; Lambrechts, N.; Verheyen, V.; Berth, M.; Schoeters, G.; Jacquemyn, Y. Impact of particulate matter on mothers and babies in Antwerp (IPANEMA): A prospective cohort study on the impact of pollutants and particulate matter in pregnancy. BMJ Open 2018, 8, e020028. [Google Scholar] [CrossRef] [PubMed]
- Mölter, A.; Agius, R.M.; de Vocht, F.; Lindley, S.; Gerrard, W.; Lowe, L.; Belgrave, D.; Custovic, A.; Simpson, A. Long-term exposure to PM10 and NO2 in association with lung volume and airway resistance in the MAAS birth cohort. Environ. Health Perspect. 2013, 121, 1232–1238. [Google Scholar] [CrossRef] [PubMed]
- Paulin, L.; Hansel, N. Particulate air pollution and impaired lung function. F1000Research 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Moshammer, H.; Hutter, H.; Hauck, H.; Neuberger, M. Low levels of air pollution induce changes of lung function in a panel of school children. Eur. Respir. J. 2006, 27, 1138–1143. [Google Scholar] [CrossRef] [PubMed]
- Misra, D.P. The effect of the pregnancy-induced hypertension on fetal growth: A review of the literature. Paediatr. Périnat. Epidemiol. 1996, 10, 244–263. [Google Scholar] [CrossRef] [PubMed]
- Duvekot, J.J.; Cheriex, E.C.; Pieters, F.A.; Peeters, L.L. Severely impaired fetal growth is preceded by maternal hemodynamic maladaptation in very early pregnancy. Acta Obstet. Gynecol. Scand. 1995, 74, 693–697. [Google Scholar] [CrossRef] [PubMed]
- Keelan, J.; Blumenstein, M.; Helliwell, R.; Sato, T.; Marvin, K.; Mitchell, M. Cytokines, prostaglandins and parturition—A review. Placenta 2003, 24, S33–S46. [Google Scholar] [CrossRef] [PubMed]
- Hansel, N.N.; McCormack, M.C.; Kim, V. The effects of air pollution and temperature on COPD. COPD J. Chronic Obstr. Pulm. Dis. 2016, 13, 372–379. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, G.; Seemungal, T.; Bhowmik, A.; Wedzicha, J. Relationship between exacerbation frequency and lung function decline in chronic obstructive pulmonary disease. Thorax 2002, 57, 847–852. [Google Scholar] [CrossRef] [PubMed]
- Seemungal, T.A.; Donaldson, G.C.; Paul, E.A.; Bestall, J.C.; Jeffries, D.J.; Wedzicha, J.A. Effect of exacerbation on quality of life in patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 1998, 157, 1418–1422. [Google Scholar] [CrossRef] [PubMed]
- Eapen, M.S.; Myers, S.; Walters, E.H.; Sohal, S.S. Airway inflammation in chronic obstructive pulmonary disease (COPD): A true paradox. Expert Rev. Respir. Med. 2017, 11, 827–839. [Google Scholar] [CrossRef] [PubMed]
- Sohal, S.; Ward, C.; Danial, W.; Wood-Baker, R.; Walters, E. Recent advances in understanding inflammation and remodeling in the airways in chronic obstructive pulmonary disease. Expert Rev. Respir. Med. 2013, 7, 275–288. [Google Scholar] [CrossRef] [PubMed]
- Connors, A.F., Jr.; Dawson, N.V.; Thomas, C.; Harrell, F.E., Jr.; Desbiens, N.; Fulkerson, W.J.; Kussin, P.; Bellamy, P.; Goldman, L.; Knaus, W.A. Outcomes following acute exacerbation of severe chronic obstructive lung disease. The SUPPORT investigators (Study to Understand Prognoses and Preferences for Outcomes and Risks of Treatments). Am. J. Respir. Crit. Care Med. 1996, 154, 959–967. [Google Scholar] [CrossRef] [PubMed]
- Sahiner, U.M.; Birben, E.; Erzurum, S.; Sackesen, C.; Kalayci, O. Oxidative stress in asthma. World Allergy Organ. J. 2011, 4, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Hogg, J.C.; Pare, P.D.; Hackett, T.L. The Contribution of Small Airway Obstruction to the Pathogenesis of Chronic Obstructive Pulmonary Disease. Physiol. Rev. 2017, 97, 529–552. [Google Scholar] [CrossRef] [PubMed]
- Gasana, J.; Dillikar, D.; Mendy, A.; Forno, E.; Vieira, E.R. Motor vehicle air pollution and asthma in children: A meta-analysis. Environ. Res. 2012, 117, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Leon Hsu, H.-H.; Mathilda Chiu, Y.-H.; Coull, B.A.; Kloog, I.; Schwartz, J.; Lee, A.; Wright, R.O.; Wright, R.J. Prenatal particulate air pollution and asthma onset in urban children. Identifying sensitive windows and sex differences. Am. J. Respir. Crit. Care Med. 2015, 192, 1052–1059. [Google Scholar] [CrossRef] [PubMed]
- Clark, N.A.; Demers, P.A.; Karr, C.J.; Koehoorn, M.; Lencar, C.; Tamburic, L.; Brauer, M. Effect of early life exposure to air pollution on development of childhood asthma. Environ. Health Perspect. 2010, 118, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Sbihi, H.; Tamburic, L.; Koehoorn, M.; Brauer, M. Perinatal air pollution exposure and development of asthma from birth to age 10 years. Eur. Respir. J. 2016, 47, 1062–1071. [Google Scholar] [CrossRef] [PubMed]
- Deng, Q.; Lu, C.; Li, Y.; Sundell, J.; Norbäck, D. Exposure to outdoor air pollution during trimesters of pregnancy and childhood asthma, allergic rhinitis, and eczema. Environ. Res. 2016, 150, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Barrett, E.G. Maternal influence in the transmission of asthma susceptibility. Pulm. Pharmacol. Ther. 2008, 21, 474–484. [Google Scholar] [CrossRef] [PubMed]
- Kirkham, P.A.; Barnes, P.J. Oxidative stress in COPD. Chest 2013, 144, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Xia, T.; Kovochich, M.; Nel, A.E. Impairment of mitochondrial function by particulate matter (PM) and their toxic components: Implications for PM-induced cardiovascular and lung disease. Front. Biosci. 2007, 12, 1238–1246. [Google Scholar] [CrossRef] [PubMed]
- van der Toorn, M.; Rezayat, D.; Kauffman, H.F.; Bakker, S.J.; Gans, R.O.; Koëter, G.H.; Choi, A.M.; van Oosterhout, A.J.; Slebos, D.-J. Lipid-soluble components in cigarette smoke induce mitochondrial production of reactive oxygen species in lung epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 297, L109–L114. [Google Scholar] [CrossRef] [PubMed]
- Aguilera-Aguirre, L.; Bacsi, A.; Saavedra-Molina, A.; Kurosky, A.; Sur, S.; Boldogh, I. Mitochondrial dysfunction increases allergic airway inflammation. J. Immunol. 2009, 183, 5379–5387. [Google Scholar] [CrossRef] [PubMed]
- Pinamonti, S.; Leis, M.; Barbieri, A.; Leoni, D.; Muzzoli, M.; Sostero, S.; Chicca, M.C.; Carrieri, A.; Ravenna, F.; Fabbri, L.M.; et al. Detection of xanthine oxidase activity products by EPR and HPLC in bronchoalveolar lavage fluid from patients with chronic obstructive pulmonary disease. Free Radic. Boil. Med. 1998, 25, 771–779. [Google Scholar] [CrossRef]
- Aaron, S.D.; Angel, J.B.; Lunau, M.; Wright, K.; Fex, C.; Le Saux, N.; Dales, R.E. Granulocyte inflammatory markers and airway infection during acute exacerbation of chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2001, 163, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Wood, L.; Gibson, P.; Garg, M. Biomarkers of lipid peroxidation, airway inflammation and asthma. Eur. Respir. J. 2003, 21, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J. The endoplasmic reticulum: A multifunctional signaling organelle. Cell Calcium 2002, 32, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Görlach, A.; Klappa, P.; Kietzmann, D.T. The endoplasmic reticulum: Folding, calcium homeostasis, signaling, and redox control. Antioxid. Redox Signal. 2006, 8, 1391–1418. [Google Scholar] [CrossRef] [PubMed]
- Sevier, C.S.; Kaiser, C.A. Ero1 and redox homeostasis in the endoplasmic reticulum. Biochim. Biophys. Acta Mol. Cell Res. 2008, 1783, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Szegezdi, E.; Logue, S.E.; Gorman, A.M.; Samali, A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006, 7, 880–885. [Google Scholar] [CrossRef] [PubMed]
- Kouroku, Y.; Fujita, E.; Tanida, I.; Ueno, T.; Isoai, A.; Kumagai, H.; Ogawa, S.; Kaufman, R.; Kominami, E.; Momoi, T. ER stress (PERK/eIF2α phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ. 2007, 14, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.A.; Turner, M.J.; DeLay, M.L.; Klenk, E.I.; Sowders, D.P.; Colbert, R.A. Endoplasmic reticulum stress and the unfolded protein response are linked to synergistic IFN-β induction via X-box binding protein 1. Eur. J. Immunol. 2008, 38, 1194–1203. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.R.; Im Kim, D.; Kang, M.R.; Lee, K.S.; Park, S.Y.; Jeong, J.S.; Lee, Y.C. Endoplasmic reticulum stress influences bronchial asthma pathogenesis by modulating nuclear factor κB activation. J. Allergy Clin. Immunol. 2013, 132, 1397–1408. [Google Scholar] [CrossRef] [PubMed]
- Flodby, P.; Li, C.; Liu, Y.; Wang, H.; Marconett, C.N.; Laird-Offringa, I.A.; Minoo, P.; Lee, A.S.; Zhou, B. The 78-kD glucose-regulated protein regulates endoplasmic reticulum homeostasis and distal epithelial cell survival during lung development. Am. J. Respir. Cell Mol. Boil. 2016, 55, 135–149. [Google Scholar] [CrossRef] [PubMed]
- Morrisey, E.E.; Hogan, B.L. Preparing for the first breath: Genetic and cellular mechanisms in lung development. Dev. Cell 2010, 18, 8–23. [Google Scholar] [CrossRef] [PubMed]
- Adamson, I.; Bowden, D. Derivation of type 1 epithelium from type 2 cells in the developing rat lung. Lab. Investig. 1975, 32, 736–745. [Google Scholar] [PubMed]
- Barkauskas, C.E.; Cronce, M.J.; Rackley, C.R.; Bowie, E.J.; Keene, D.R.; Stripp, B.R.; Randell, S.H.; Noble, P.W.; Hogan, B.L. Type 2 alveolar cells are stem cells in adult lung. J. Clin. Investig. 2013, 123, 3025–3036. [Google Scholar] [CrossRef] [PubMed]
- Mimura, N.; Hamada, H.; Kashio, M.; Jin, H.; Toyama, Y.; Kimura, K.; Iida, M.; Goto, S.; Saisho, H.; Toshimori, K.; et al. Aberrant quality control in the endoplasmic reticulum impairs the biosynthesis of pulmonary surfactant in mice expressing mutant BiP. Cell Death Differ. 2007, 14, 1475–1485. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I.; Ron, D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 2011, 13, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Sureshbabu, A.; Syed, M.A.; Boddupalli, C.S.; Dhodapkar, M.V.; Homer, R.J.; Minoo, P.; Bhandari, V. Conditional overexpression of TGFβ1 promotes pulmonary inflammation, apoptosis and mortality via TGFβR2 in the developing mouse lung. Respir. Res. 2015, 16, 4. [Google Scholar] [CrossRef] [PubMed]
- Kota, A.; Deshpande, D.; Haghi, M.; Oliver, B.; Sharma, P. Autophagy and airway fibrosis: Is there a link? F1000Research 2017, 6, 4. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Klionsky, D.J. Development by self-digestion: Molecular mechanisms and biological functions of autophagy. Dev. Cell 2004, 6, 463–477. [Google Scholar] [CrossRef]
- Zeki, A.; Yeganeh, B.; Kenyon, N.; Post, M.; Ghavami, S. Autophagy in airway diseases: A new frontier in human asthma? Allergy 2016, 71, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Poon, A.H.; Chouiali, F.; Tse, S.M.; Litonjua, A.A.; Hussain, S.N.; Baglole, C.J.; Eidelman, D.H.; Olivenstein, R.; Martin, J.G.; Weiss, S.T. Genetic and histologic evidence for autophagy in asthma pathogenesis. J. Allergy Clin. Immunol. 2012, 129, 569–571. [Google Scholar] [CrossRef] [PubMed]
- Poon, A.; Eidelman, D.; Laprise, C.; Hamid, Q. ATG5, autophagy and lung function in asthma. Autophagy 2012, 8, 694–695. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.-H.; Kim, H.P.; Sciurba, F.C.; Lee, S.-J.; Feghali-Bostwick, C.; Stolz, D.B.; Dhir, R.; Landreneau, R.J.; Schuchert, M.J.; Yousem, S.A.; et al. Egr-1 regulates autophagy in cigarette smoke-induced chronic obstructive pulmonary disease. PLoS ONE 2008, 3, e3316. [Google Scholar] [CrossRef] [PubMed]
- Mizumura, K.; Cloonan, S.M.; Nakahira, K.; Bhashyam, A.R.; Cervo, M.; Kitada, T.; Glass, K.; Owen, C.A.; Mahmood, A.; Washko, G.R. Mitophagy-dependent necroptosis contributes to the pathogenesis of COPD. J. Clin. Investig. 2014, 124, 3987–4003. [Google Scholar] [CrossRef] [PubMed]
- Lam, H.C.; Cloonan, S.M.; Bhashyam, A.R.; Haspel, J.A.; Singh, A.; Sathirapongsasuti, J.F.; Cervo, M.; Yao, H.; Chung, A.L.; Mizumura, K. Histone deacetylase 6–mediated selective autophagy regulates COPD-associated cilia dysfunction. J. Clin. Investig. 2013, 123, 5212–5230. [Google Scholar] [CrossRef] [PubMed]
- Webber, J.L.; Young, A.R.; Tooze, S.A. Atg9 trafficking in mammalian cells. Autophagy 2007, 3, 54–56. [Google Scholar] [CrossRef] [PubMed]
- Ropolo, A.; Grasso, D.; Pardo, R.; Sacchetti, M.L.; Archange, C.; Re, A.L.; Seux, M.; Nowak, J.; Gonzalez, C.D.; Iovanna, J.L.; et al. The pancreatitis-induced vacuole membrane protein 1 triggers autophagy in mammalian cells. J. Boil. Chem. 2007, 282, 37124–37133. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.-P.; Hu, L.-F.; Zheng, H.-F.; Mao, C.-J.; Hu, W.-D.; Xiong, K.-P.; Wang, F.; Liu, C.-F. Application and interpretation of current autophagy inhibitors and activators. Acta Pharmacol. Sin. 2013, 34, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Ban, G.Y.; Pham, D.; Trinh, T.; Lee, S.I.; Suh, D.H.; Yang, E.M.; Ye, Y.M.; Shin, Y.; Chwae, Y.J.; Park, H.S. Autophagy mechanisms in sputum and peripheral blood cells of patients with severe asthma: A new therapeutic target. Clin. Exp. Allergy 2016, 46, 48–59. [Google Scholar] [CrossRef] [PubMed]
- Fimia, G.M.; Stoykova, A.; Romagnoli, A.; Giunta, L.; Di Bartolomeo, S.; Nardacci, R.; Corazzari, M.; Fuoco, C.; Ucar, A.; Schwartz, P.; et al. Ambra1 regulates autophagy and development of the nervous system. Nature 2007, 447, 1121–1125. [Google Scholar] [PubMed]
- Nakashima, A.; Aoki, A.; Kusabiraki, T.; Shima, T.; Yoshino, O.; Cheng, S.B.; Sharma, S.; Saito, S. Role of autophagy in oocytogenesis, embryogenesis, implantation, and pathophysiology of pre-eclampsia. J. Obstet. Gynaecol. Res. 2017, 43, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Raha, S.; Robinson, B.H. Mitochondria, oxygen free radicals, disease and ageing. Trends Biochem. Sci. 2000, 25, 502–508. [Google Scholar] [CrossRef]
- Andreyev, A.Y.; Kushnareva, Y.E.; Starkov, A. Mitochondrial metabolism of reactive oxygen species. Biochemical 2005, 70, 200–214. [Google Scholar] [CrossRef]
- Papa, S.; Skulachev, V. Reactive oxygen species, mitochondria, apoptosis and aging. In Detection of Mitochondrial Diseases; Springer: Berlin, Germany, 1997; pp. 305–319. [Google Scholar]
- Walter, L.; Nogueira, V.; Leverve, X.; Heitz, M.-P.; Bernardi, P.; Fontaine, E. Three classes of ubiquinone analogs regulate the mitochondrial permeability transition pore through a common site. J. Boil. Chem. 2000, 275, 29521–29527. [Google Scholar] [CrossRef] [PubMed]
- Fontaine, E.; Ichas, F.; Bernardi, P. A ubiquinone-binding site regulates the mitochondrial permeability transition pore. J. Boil. Chem. 1998, 273, 25734–25740. [Google Scholar] [CrossRef]
- Xia, T.; Korge, P.; Weiss, J.N.; Li, N.; Venkatesen, M.I.; Sioutas, C.; Nel, A. Quinones and aromatic chemical compounds in particulate matter induce mitochondrial dysfunction: Implications for ultrafine particle toxicity. Environ. Health Perspect. 2004, 112, 1347–1358. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, P.; Petronilli, V.; Di Lisa, F.; Forte, M. A mitochondrial perspective on cell death. Trends Biochem. Sci. 2001, 26, 112–117. [Google Scholar] [CrossRef]
- Zamzami, N.; Kroemer, G. The mitochondrion in apoptosis: How Pandora’s box opens. Nat. Rev. Mol. Cell Biol. 2001, 2, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Wang, X. Cytochrome C-mediated apoptosis. Annu. Rev. Biochem. 2004, 73, 87–106. [Google Scholar] [CrossRef] [PubMed]
- Ichas, F.; Mazat, J.-P. From calcium signaling to cell death: Two conformations for the mitochondrial permeability transition pore. Switching from low-to high-conductance state. Biochim. Biophys. Acta 1998, 1366, 33–50. [Google Scholar] [CrossRef]
- Redondo, P.C.; Salido, G.M.; Rosado, J.A.; Pariente, J.A. Effect of hydrogen peroxide on Ca2+ mobilisation in human platelets through sulphydryl oxidation dependent and independent mechanisms. Biochem. Pharmacol. 2004, 67, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Brookes, P.S.; Yoon, Y.; Robotham, J.L.; Anders, M.; Sheu, S.-S. Calcium, ATP, and ROS: A mitochondrial love-hate triangle. Am. J. Physiol. Cell Physiol. 2004, 287, C817–C833. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C.; Mariño, G.; Kroemer, G. Autophagy and aging. Cell 2011, 146, 682–695. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| RANK | CITY | PM2.5 LEVEL (Annual Mean, µg/m3) |
|---|---|---|
| 1 | Kanpur | 173 |
| 2 | Faridabad | 172 |
| 3 | Varanasi | 151 |
| 4 | Gaya | 149 |
| 5 | Patna | 144 |
| 6 | Delhi | 143 |
| 7 | Lucknow | 138 |
| 8 | Agra | 131 |
| 9 | Muzaffarpur | 120 |
| 10 | Srinagar | 113 |
| 11 | Gurgaon | 113 |
| 12 | Jaipur | 105 |
| 13 | Patiala | 101 |
| 14 | Jodhpur | 98 |
| S. No. | Author | Study Design | Sample Size | Exposure | Parameter Studied | Comments and Association |
|---|---|---|---|---|---|---|
| 1. | Padhy et al., 2009 [44] | Case-control | Control (105) Biomass user (115) | Biomass smoke | Respiratory symptoms Oxidative stress Hematological changes | Exposure to biomass smoke significantly associated with respiratory diseases, oxidative stress, and hematological changes |
| 2. | Awasthi et al., 2010 [42] | Cohort | 23 children (10–13 years of age) | Agriculture crop residue burning (ACRB) | Pulmonary function | Decrease in pulmonary function with an increase in air pollutant levels due to ACRB |
| 3. | Kumar et al., 2015 [46] | Cohort | 3104 children | Indoor suspended particulate matter (SPM) | Asthma | Indoor SPM level was significantly higher in asthmatic children’s houses |
| 4. | Singh et al., 2015 [47] | Cross-sectional, multicenter | 44,928 (6–7 year age group); 48,088 (13–14 year age group) | Traffic pollution, maternal and paternal smoking | Asthma | Traffic pollution and maternal and paternal smoking is associated with increased prevalence of asthma |
| 5. | Murlidhar et al., 2015 [48] | Case-report | 11-year-old boy, malnourished | Secondary exposure to sandstone mining | Silico-tuberculosis | Mother started working in the mines soon after her marriage and the family lives close to the mines |
| 6. | Rumchev et al., 2017 [49] | Cohort | 170 children between 1 and 15 years | Indoor exposure to PM2.5 | Respiratory symptoms | No significant association between PM-exposure and respiratory symptoms even though odds are high |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saha, P.; Johny, E.; Dangi, A.; Shinde, S.; Brake, S.; Eapen, M.S.; Sohal, S.S.; Naidu, V.; Sharma, P. Impact of Maternal Air Pollution Exposure on Children’s Lung Health: An Indian Perspective. Toxics 2018, 6, 68. https://doi.org/10.3390/toxics6040068
Saha P, Johny E, Dangi A, Shinde S, Brake S, Eapen MS, Sohal SS, Naidu V, Sharma P. Impact of Maternal Air Pollution Exposure on Children’s Lung Health: An Indian Perspective. Toxics. 2018; 6(4):68. https://doi.org/10.3390/toxics6040068
Chicago/Turabian StyleSaha, Pritam, Ebin Johny, Ashish Dangi, Sopan Shinde, Samuel Brake, Mathew Suji Eapen, Sukhwinder Singh Sohal, VGM Naidu, and Pawan Sharma. 2018. "Impact of Maternal Air Pollution Exposure on Children’s Lung Health: An Indian Perspective" Toxics 6, no. 4: 68. https://doi.org/10.3390/toxics6040068
APA StyleSaha, P., Johny, E., Dangi, A., Shinde, S., Brake, S., Eapen, M. S., Sohal, S. S., Naidu, V., & Sharma, P. (2018). Impact of Maternal Air Pollution Exposure on Children’s Lung Health: An Indian Perspective. Toxics, 6(4), 68. https://doi.org/10.3390/toxics6040068