
Glutamate-Mediated Neural Alterations in Lead Exposure: Mechanisms, Pathways, and Phenotypes



Abstract

1. Introduction
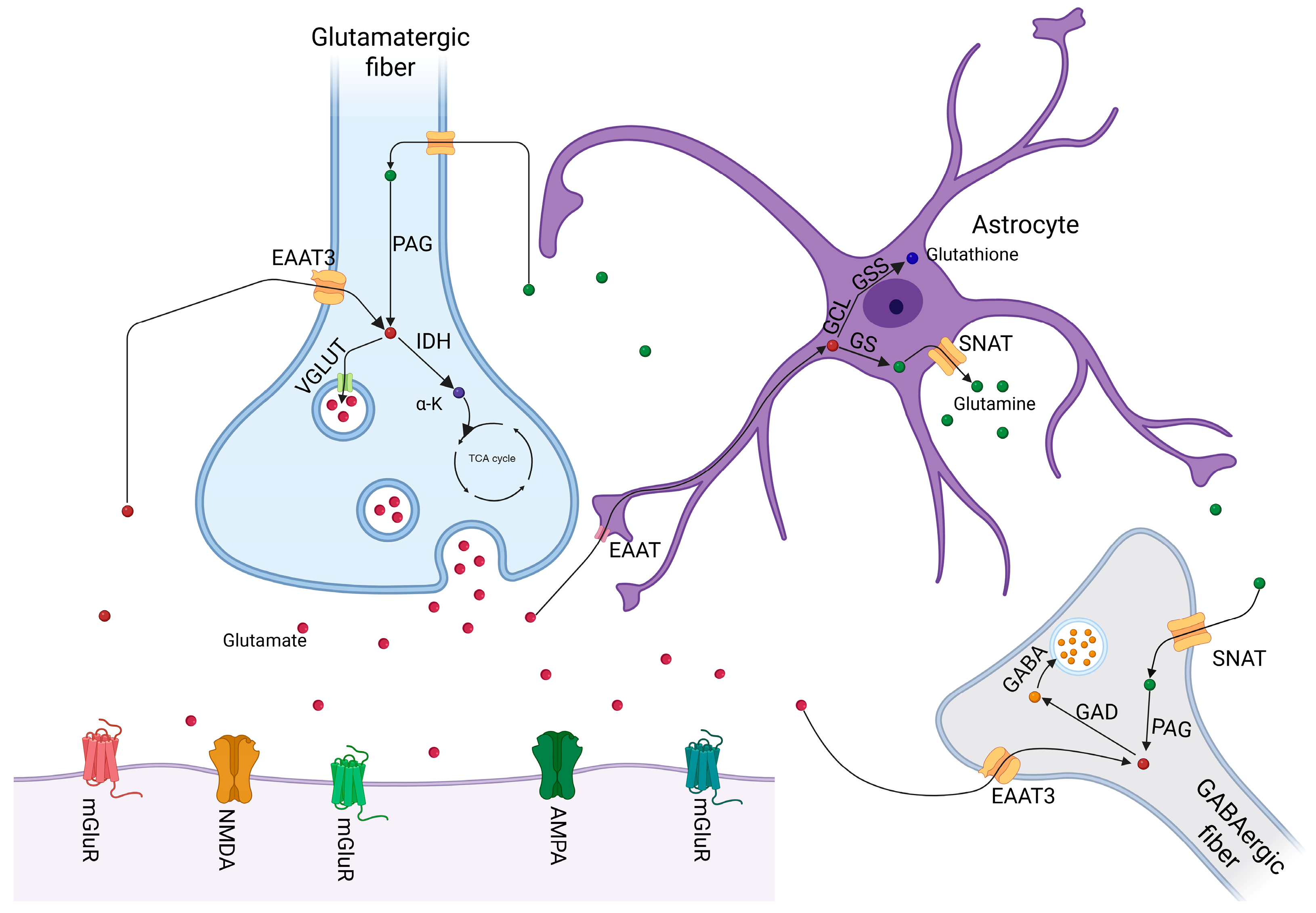
2. Overview of Glutamatergic Neurotransmission
Glutamate Dysregulation and Behavioral Impairments
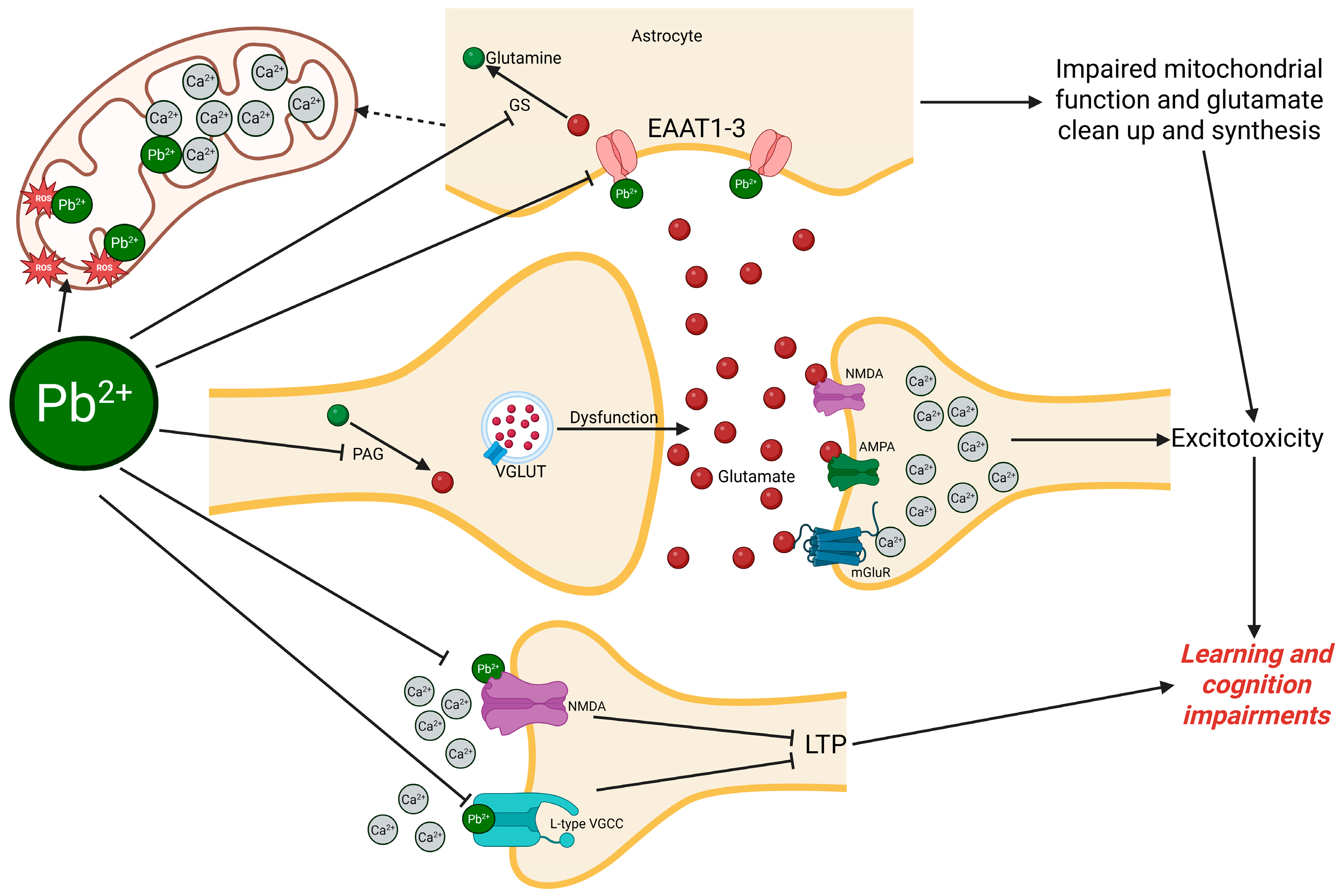
3. Pb as a Neurotoxicant Targeting Glutamatergic Signaling
3.1. Calcium Dysregulation and Glutamate Excitotoxicity Induced by Pb
3.2. Receptor-Mediated Glutamatergic Neurotoxicity by Pb
3.2.1. Ionotropic Glutamate Receptors (NMDA and AMPA)
3.2.2. Metabotropic Glutamate Receptors (mGluRs)
3.3. Transporter-Mediated Disruption of Glutamate Homeostasis
3.3.1. Excitatory Amino Acid Transporters (EAATs)
3.3.2. Sodium-Coupled Neutral Amino Acid Transporters (SNATs)
3.4. Glutamate Release Machinery Disruption by Pb
3.4.1. Vesicular Glutamate Transporters (VGLUTs)
3.4.2. Synaptic Vesicle Docking and Exocytosis Machinery
3.4.3. SNARE Complex
3.4.4. Pb Disruption of Key Regulators of Vesicle Priming, Calcium Sensing, and Fusion
3.5. Glutamate Metabolism and Cycling Disruption in Pb Exposure
3.5.1. Tricarboxylic Acid (TCA) Cycle and Glutamate Biosynthesis
3.5.2. Glutamate–Glutamine Shuttle
3.5.3. Glutamate–GABA Conversion
3.5.4. Glutamate–Glutathione Pathway and Oxidative Stress
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AMPA | α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid |
| BDNF | brain-derived neurotrophic factor |
| CaMKII | calcium/calmodulin-dependent protein kinase II |
| CREB | cAMP response element-binding protein |
| EAATs | excitatory amino acid transporter |
| GABA | gamma-aminobutyric acid |
| GPCRs | G-protein-coupled receptors |
| GSH | glutathione |
| LTD | long-term depression |
| LTP | long-term potentiation |
| mGluRs | metabotropic glutamate receptors |
| NMDA | N-methyl-D-aspartate |
| Pb | lead |
| ROS | reactive oxygen species |
| RNS | reactive nitrogen species |
| SNARE | soluble NSF attachment protein receptor |
| SNATs | sodium-coupled neutral amino acid transporters |
| TCA | tricarboxylic acid cycle |
| VGLUTs | vesicular glutamate transporters |
| VSCCs | voltage-sensitive calcium channels |
References
- Chen, Y.; Liu, M. Evaluation of Cytotoxicity of Pb2+ Ion-Adsorbed Amino-Functionalized Magnetic Mesoporous Silica Nanoparticles: An In Vitro Study. Front. Mater. 2022, 9, 914009. [Google Scholar] [CrossRef]
- Zhu, Z.; Wang, J.; Cheng, H.; Zhao, H.; Liu, C.; Zhou, X.; Yang, J. Combined Toxicity Assessment of Deoxynivalenol and Pb2+ on HK-2 Cells Involved in Excessive ROS-Induced Ferroptosis. J. Agric. Food Chem. 2025, 73, 2573–2584. [Google Scholar] [CrossRef] [PubMed]
- Caetano, E.L.A.; Novoa San Miguel, F.J.; Errázuriz León, R.; Grotto, D.; Hornos Carneiro, M.F. Exploring the Impact of Agaricus bisporus on Mitigating Lead Reproductive Toxicity Using the Caenorhabditis elegans Model. Comp. Biochem. Physiol. Part C Toxicol. Pharmacol. 2024, 283, 109963. [Google Scholar] [CrossRef]
- Kiper, K.; Freeman, J.L. Joint Action Toxicity of Arsenic (As) and Lead (Pb) Mixtures in Developing Zebrafish. Biomolecules 2022, 12, 1833. [Google Scholar] [CrossRef]
- Lee, J.; Freeman, J.L. Exposure to the Heavy-Metal Lead Induces DNA Copy Number Alterations in Zebrafish Cells. Chem. Res. Toxicol. 2020, 33, 2047–2053. [Google Scholar] [CrossRef] [PubMed]
- Luo, T.; Shen, M.; Zhou, J.; Wang, X.; Xia, J.; Fu, Z.; Jin, Y. Chronic Exposure to Low Doses of Pb Induces Hepatotoxicity at the Physiological, Biochemical, and Transcriptomic Levels of Mice. Environ. Toxicol. 2019, 34, 521–529. [Google Scholar] [CrossRef]
- Zhai, Q.; Qu, D.; Feng, S.; Yu, Y.; Yu, L.; Tian, F.; Zhao, J.; Zhang, H.; Chen, W. Oral Supplementation of Lead-Intolerant Intestinal Microbes Protects against Lead (Pb) Toxicity in Mice. Front. Microbiol. 2020, 10, 3161. [Google Scholar] [CrossRef]
- Bihaqi, S.W.; Zawia, N.H. Enhanced Taupathy and AD-like Pathology in Aged Primate Brains Decades after Infantile Exposure to Lead (Pb). Neurotoxicology 2013, 39, 95–101. [Google Scholar] [CrossRef]
- Mason, L.H.; Harp, J.P.; Han, D.Y. Pb Neurotoxicity: Neuropsychological Effects of Lead Toxicity. BioMed Res. Int. 2014, 2014, 840547. [Google Scholar] [CrossRef]
- Santa Maria, M.P.; Hill, B.D.; Kline, J. Lead (Pb) Neurotoxicology and Cognition. Appl. Neuropsychol. Child 2019, 8, 272–293. [Google Scholar] [CrossRef]
- Wani, A.L.; Ara, A.; Usmani, J.A. Lead Toxicity: A Review. Interdiscip. Toxicol. 2015, 8, 55–64. [Google Scholar] [CrossRef]
- Labban, M. Deterritorializing Extraction: Bioaccumulation and the Planetary Mine. Ann. Assoc. Am. Geogr. 2014, 104, 560–576. [Google Scholar] [CrossRef]
- Boldyrev, M. Lead: Properties, History, and Applications. WikiJ. Sci. 2018, 1, 1–23. [Google Scholar] [CrossRef]
- Kamenov, G.D.; Gulson, B.L. The Pb Isotopic Record of Historical to Modern Human Lead Exposure. Sci. Total Environ. 2014, 490, 861–870. [Google Scholar] [CrossRef] [PubMed]
- Levin, R.; Vieira, C.L.Z.; Rosenbaum, M.H.; Bischoff, K.; Mordarski, D.C.; Brown, M.J. The Urban Lead (Pb) Burden in Humans, Animals and the Natural Environment. Environ. Res. 2021, 193, 110377. [Google Scholar] [CrossRef]
- Lopes, P.P.; Stamenkovic, V.R. Past, Present, and Future of Lead–Acid Batteries. Science 2020, 369, 923–924. [Google Scholar] [CrossRef]
- Otieno, J.; Kowal, P.; Mąkinia, J. Monitoring Lead Concentration in the Surrounding Environmental Components of a Lead Battery Company: Plants, Air and Effluents—Case Study, Kenya. Int. J. Environ. Res. Public Health 2022, 19, 5195. [Google Scholar] [CrossRef]
- Hadi, F.; Aziz, T. A Mini Review on Lead (Pb) Toxicity in Plants. J. Biol. Life Sci. 2015, 6, 91–101. [Google Scholar] [CrossRef]
- Gorkhali, R.; Huang, K.; Kirberger, M.; Yang, J.J. Defining Potential Roles of Pb2+ in Neurotoxicity from a Calciomics Approach. Metallomics 2016, 8, 563–578. [Google Scholar] [CrossRef]
- Kirberger, M.; Yang, J.J. Structural Differences between Pb2+-and Ca2+-Binding Sites in Proteins: Implications with Respect to Toxicity. J. Inorg. Biochem. 2008, 102, 1901–1909. [Google Scholar] [CrossRef]
- Mitra, P.; Sharma, S.; Purohit, P.; Sharma, P. Clinical and Molecular Aspects of Lead Toxicity: An Update. Crit. Rev. Clin. Lab. Sci. 2017, 54, 506–528. [Google Scholar] [CrossRef] [PubMed]
- Riva, M.A.; Lafranconi, A.; D’orso, M.I.; Cesana, G. Lead Poisoning: Historical Aspects of a Paradigmatic “Occupational and Environmental Disease”. Saf. Health Work 2012, 3, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Egan, K.B.; Cornwell, C.R.; Courtney, J.G.; Ettinger, A.S. Blood Lead Levels in U.S. Children Ages 1–11 Years, 1976–2016. Environ. Health Perspect 2021, 129, 037003. [Google Scholar] [CrossRef]
- Desai, G.; Anzman-Frasca, S.; Vernarelli, J.A.; Ravenscroft, J.; Yang, J.; Burstein, G.; Kordas, K. Examining Links between Diet and Lead Exposure in Young Children: 2009 to 2014 National Health and Nutrition Examination Survey. Acad. Pediatr. 2021, 21, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Swaringen, B.F.; Gawlik, E.; Kamenov, G.D.; McTigue, N.E.; Cornwell, D.A.; Bonzongo, J.-C.J. Children’s Exposure to Environmental Lead: A Review of Potential Sources, Blood Levels, and Methods Used to Reduce Exposure. Environ. Res. 2022, 204, 112025. [Google Scholar] [CrossRef]
- Schwartz, J. Low-Level Lead Exposure and Children′ s IQ: A Metaanalysis and Search for a Threshold. Environ. Res. 1994, 65, 42–55. [Google Scholar] [CrossRef]
- Poudel, K.; Ikeda, A.; Fukunaga, H.; Brune Drisse, M.-N.; Onyon, L.J.; Gorman, J.; Laborde, A.; Kishi, R. How Does Formal and Informal Industry Contribute to Lead Exposure? A Narrative Review from Vietnam, Uruguay, and Malaysia. Rev. Environ. Health 2024, 39, 371–388. [Google Scholar] [CrossRef]
- Fortune, T.; Lurie, D.I. Chronic Low-Level Lead Exposure Affects the Monoaminergic System in the Mouse Superior Olivary Complex. J. Comp. Neurol. 2009, 513, 542–558. [Google Scholar] [CrossRef]
- Qu, X.; Xu, C.; Wang, H.; Xu, J.; Liu, W.; Wang, Y.; Jia, X.; Xie, Z.; Xu, Z.; Ji, C.; et al. Hippocampal Glutamate Level and Glutamate Aspartate Transporter (GLAST) Are Up-Regulated in Senior Rat Associated with Isoflurane-Induced Spatial Learning/Memory Impairment. Neurochem. Res. 2013, 38, 59–73. [Google Scholar] [CrossRef]
- Niciu, M.J.; Kelmendi, B.; Sanacora, G. Overview of Glutamatergic Neurotransmission in the Nervous System. Pharmacol. Biochem. Behav. 2012, 100, 656–664. [Google Scholar] [CrossRef]
- Gulyaeva, N.V. Neuroendocrine Control of Hyperglutamatergic States in Brain Pathologies: The Effects of Glucocorticoids. J. Evol. Biochem. Phys. 2022, 58, 1425–1438. [Google Scholar] [CrossRef]
- Roberts, R.C.; McCollum, L.A.; Schoonover, K.E.; Mabry, S.J.; Roche, J.K.; Lahti, A.C. Ultrastructural Evidence for Glutamatergic Dysregulation in Schizophrenia. Schizophr. Res. 2022, 249, 4–15. [Google Scholar] [CrossRef] [PubMed]
- González-Estecha, M.; Trasobares, E.M.; Tajima, K.; Cano, S.; Fernández, C.; López, J.L.; Unzeta, B.; Arroyo, M.; Fuentenebro, F. Trace Elements in Bipolar Disorder. J. Trace Elem. Med. Biol. 2011, 25, S78–S83. [Google Scholar] [CrossRef]
- Amadi, C.N.; Orish, C.N.; Frazzoli, C.; Orisakwe, O.E. Association of Autism with Toxic Metals: A Systematic Review of Case-Control Studies. Pharmacol. Biochem. Behav. 2022, 212, 173313. [Google Scholar] [CrossRef]
- Yassa, H.A. Autism: A Form of Lead and Mercury Toxicity. Environ. Toxicol. Pharmacol. 2014, 38, 1016–1024. [Google Scholar] [CrossRef]
- Benarroch, E.E. Glutamatergic Synaptic Plasticity and Dysfunction in Alzheimer Disease: Emerging Mechanisms. Neurology 2018, 91, 125–132. [Google Scholar] [CrossRef]
- Iovino, L.; Tremblay, M.E.; Civiero, L. Glutamate-Induced Excitotoxicity in Parkinson’s Disease: The Role of Glial Cells. J. Pharmacol. Sci. 2020, 144, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Arnold, F.J.; Putka, A.F.; Raychaudhuri, U.; Hsu, S.; Bedlack, R.S.; Bennett, C.L.; La Spada, A.R. Revisiting Glutamate Excitotoxicity in Amyotrophic Lateral Sclerosis and Age-Related Neurodegeneration. Int. J. Mol. Sci. 2024, 25, 5587. [Google Scholar] [CrossRef]
- Willard, S.S.; Koochekpour, S. Glutamate, Glutamate Receptors, and Downstream Signaling Pathways. Int. J. Biol. Sci. 2013, 9, 948. [Google Scholar] [CrossRef]
- Kessels, H.W.; Malinow, R. Synaptic AMPA Receptor Plasticity and Behavior. Neuron 2009, 61, 340–350. [Google Scholar] [CrossRef]
- Nicoletti, F.; Di Menna, L.; Iacovelli, L.; Orlando, R.; Zuena, A.R.; Conn, P.J.; Dogra, S.; Joffe, M.E. GPCR Interactions Involving Metabotropic Glutamate Receptors and Their Relevance to the Pathophysiology and Treatment of CNS Disorders. Neuropharmacology 2023, 235, 109569. [Google Scholar] [CrossRef]
- Alva, S.; Parithathvi, A.; Harshitha, P.; Dsouza, H.S. Influence of Lead on cAMP-Response Element Binding Protein (CREB) and Its Implications in Neurodegenerative Disorders. Toxicol. Lett. 2024, 400, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Ishida, K.; Kotake, Y.; Miyara, M.; Aoki, K.; Sanoh, S.; Kanda, Y.; Ohta, S. Involvement of Decreased Glutamate Receptor Subunit GluR2 Expression in Lead-Induced Neuronal Cell Death. J. Toxicol. Sci. 2013, 38, 513–521. [Google Scholar] [CrossRef]
- Ishida, K.; Kotake, Y.; Sanoh, S.; Ohta, S. Lead-Induced ERK Activation Is Mediated by GluR2 Non-Containing AMPA Receptor in Cortical Neurons. Biol. Pharm. Bull. 2017, 40, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Alleva, C.; Machtens, J.-P.; Kortzak, D.; Weyand, I.; Fahlke, C. Molecular Basis of Coupled Transport and Anion Conduction in Excitatory Amino Acid Transporters. Neurochem. Res. 2022, 47, 9–22. [Google Scholar] [CrossRef]
- Sidoryk-Węgrzynowicz, M.; Adamiak, K.; Strużyńska, L. Astrocyte–Neuron Interaction via the Glutamate–Glutamine Cycle and Its Dysfunction in Tau-Dependent Neurodegeneration. Int. J. Mol. Sci. 2024, 25, 3050. [Google Scholar] [CrossRef]
- Sood, A.; Preeti, K.; Fernandes, V.; Khatri, D.K.; Singh, S.B. Glia: A Major Player in Glutamate–GABA Dysregulation-mediated Neurodegeneration. J. Neurosci. Res. 2021, 99, 3148–3189. [Google Scholar] [CrossRef] [PubMed]
- Weidacker, K.; Johnston, S.J.; Mullins, P.G.; Boy, F.; Dymond, S. Impulsive Decision-Making and Gambling Severity: The Influence of γ-Amino-Butyric Acid (GABA) and Glutamate-Glutamine (Glx). Eur. Neuropsychopharmacol. 2020, 32, 36–46. [Google Scholar] [CrossRef]
- Limón, I.D.; Angulo-Cruz, I.; Sánchez-Abdon, L.; Patricio-Martínez, A. Disturbance of the Glutamate-Glutamine Cycle, Secondary to Hepatic Damage, Compromises Memory Function. Front. Neurosci. 2021, 15, 578922. [Google Scholar] [CrossRef]
- Olajide, O.J.; Gbadamosi, I.T.; Yawson, E.O.; Arogundade, T.; Lewu, F.S.; Ogunrinola, K.Y.; Adigun, O.O.; Bamisi, O.; Lambe, E.; Arietarhire, L.O. Hippocampal Degeneration and Behavioral Impairment during Alzheimer-like Pathogenesis Involves Glutamate Excitotoxicity. J. Mol. Neurosci. 2021, 71, 1205–1220. [Google Scholar] [CrossRef]
- Galineau, L.; Arlicot, N.; Dupont, A.-C.; Briend, F.; Houy-Durand, E.; Tauber, C.; Gomot, M.; Gissot, V.; Barantin, L.; Lefevre, A. Glutamatergic Synapse in Autism: A Complex Story for a Complex Disorder. Mol. Psychiatry 2023, 28, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Montanari, M.; Martella, G.; Bonsi, P.; Meringolo, M. Autism Spectrum Disorder: Focus on Glutamatergic Neurotransmission. Int. J. Mol. Sci. 2022, 23, 3861. [Google Scholar] [CrossRef] [PubMed]
- Da Silva Lemos, I.; Wessler, L.B.; Duarte, M.B.; da Silva, G.L.; Bernardo, H.T.; Candiotto, G.; Torres, C.A.; Petronilho, F.; Rico, E.P.; Streck, E.L. Exposure to Leucine Alters Glutamate Levels and Leads to Memory and Social Impairment in Zebrafish. Metab. Brain Dis. 2022, 37, 2925–2935. [Google Scholar] [CrossRef]
- Martins, M.L.; Pinheiro, E.F.; Saito, G.A.; Lima, C.A.C.D.; Leão, L.K.R.; Batista, E.d.J.O.; Passos, A.d.C.F.; Gouveia, A.; Oliveira, K.R.H.M., Jr.; Herculano, A.M. Distinct Acute Stressors Produce Different Intensity of Anxiety-like Behavior and Differential Glutamate Release in Zebrafish Brain. Front. Behav. Neurosci. 2024, 18, 1464992. [Google Scholar] [CrossRef] [PubMed]
- Kruse, A.O.; Bustillo, J.R. Glutamatergic Dysfunction in Schizophrenia. Transl. Psychiatry 2022, 12, 500. [Google Scholar] [CrossRef]
- Der Walt, E.V.; Brink, C.B.; van Vuren, E.J. Changes in Schizophrenia Symptoms, Tryptophan Metabolism, Neuroinflammation and the GABA-Glutamate Loop: A Pilot Study. S. Afr. J. Psychiatry 2025, 31, 6. [Google Scholar] [CrossRef]
- Clever, H.L.; Johnston, F.J. The Solubility of Some Sparingly Soluble Lead Salts: An Evaluation of the Solubility in Water and Aqueous Electrolyte Solution. J. Phys. Chem. Ref. Data 1980, 9, 751–784. [Google Scholar] [CrossRef]
- Mayer, R.A.; Godwin, H.A. Preparation of Media and Buffers with Soluble Lead. Anal. Biochem. 2006, 356, 142–144. [Google Scholar] [CrossRef]
- Tang, W.; Peng, J.; Chen, L.; Yu, C.; Wang, Y.; Zou, F.; Zheng, G.; Meng, X. Lead Inhibits Microglial Cell Migration via Suppression of Store-Operated Calcium Entry. Toxicol. Lett. 2024, 393, 69–77. [Google Scholar] [CrossRef]
- Büsselberg, D. Calcium Channels as Target Sites of Heavy Metals. Toxicol. Lett. 1995, 82, 255–261. [Google Scholar] [CrossRef]
- Lindsley, C.W.; Wisnoski, D.D.; Leister, W.H.; O’Brien, J.A.; Lemaire, W.; Williams, D.L.; Burno, M.; Sur, C.; Kinney, G.G.; Pettibone, D.J.; et al. Discovery of Positive Allosteric Modulators for the Metabotropic Glutamate Receptor Subtype 5 from a Series of N-(1,3-Diphenyl-1 H-Pyrazol-5-Yl)Benzamides That Potentiate Receptor Function in Vivo. J. Med. Chem. 2004, 47, 5825–5828. [Google Scholar] [CrossRef] [PubMed]
- Han, Q.; Zhang, W.; Guo, J.; Zhu, Q.; Chen, H.; Xia, Y.; Zhu, G. Mitochondrion: A Sensitive Target for Pb Exposure. J. Toxicol. Sci. 2021, 46, 345–358. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Liu, J.-Y.; Dong, J.-X.; Xiao, Q.; Zhao, J.; Jiang, F.-L. Toxicity of Pb2+ on Rat Liver Mitochondria Induced by Oxidative Stress and Mitochondrial Permeability Transition. Toxicol. Res. 2017, 6, 822–830. [Google Scholar] [CrossRef] [PubMed]
- Neuwirth, L.S.; El Idrissi, A. Developmental Pb2+-Exposure Alters KCC₂ and VSCC-Β3 Subunit Expression Patterns in the Postnatal Rat Brain and Cerebellar Granule Cell Cultures: Implications for Disrupted GABA-Shifts Resulting from Neurotoxicant Exposures. Psychol. Neurosci. 2021, 14, 49–72. [Google Scholar] [CrossRef]
- Iliyasu, M.O.; Ajayi, A. The Extent of Lead-Induced Pathogenesis of Alzheimer’s Disease, Synaptic Dysfunction and Cognitive Impairment in Rats. Alzheimer’s Dement. 2023, 19, e082971. [Google Scholar] [CrossRef]
- Rahman, A.; Rao, M.S.; Khan, K.M. Intraventricular Infusion of Quinolinic Acid Impairs Spatial Learning and Memory in Young Rats: A Novel Mechanism of Lead-Induced Neurotoxicity. J. Neuroinflamm. 2018, 15, 263. [Google Scholar] [CrossRef]
- Chlubek, M.; Baranowska-Bosiacka, I. Selected Biochemical Mechanisms of Lead Neurotoxicity. Pomeranian J. Life Sci. 2023, 69, 33–39. [Google Scholar] [CrossRef]
- Neal, A.P.; Guilarte, T.R. Molecular Neurobiology of Lead (Pb2+): Effects on Synaptic Function. Mol. Neurobiol. 2010, 42, 151–160. [Google Scholar] [CrossRef]
- Sadiq, S.; Ghazala, Z.; Chowdhury, A.; Büsselberg, D. Metal Toxicity at the Synapse: Presynaptic, Postsynaptic, and Long-Term Effects. J. Toxicol. 2012, 2012, 132671. [Google Scholar] [CrossRef]
- Florea, A.-M.; Taban, J.; Varghese, E.; Alost, B.T.; Moreno, S.; Büsselberg, D. Lead (Pb2+) Neurotoxicity: Ion-Mimicry with Calcium (Ca2+) Impairs Synaptic Transmission. A Review with Animated Illustrations of the Pre- and Post-Synaptic Effects of Lead. J. Local Glob. Health Sci. 2013, 2013, 4. [Google Scholar] [CrossRef]
- Xiao, C.; Gu, Y.; Zhou, C.-Y.; Wang, L.; Zhang, M.-M.; Ruan, D.-Y. Pb2+ Impairs GABAergic Synaptic Transmission in Rat Hippocampal Slices: A Possible Involvement of Presynaptic Calcium Channels. Brain Res. 2006, 1088, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Dudev, T.; Grauffel, C.; Lim, C. How Pb2+ Binds and Modulates Properties of Ca2+-Signaling Proteins. Inorg. Chem. 2018, 57, 14798–14809. [Google Scholar] [CrossRef] [PubMed]
- Alkondon, M.; Costa, A.C.S.; Radhakrishnan, V.; Aronstam, R.S.; Albuquerque, E.X. Selective Blockade of NMDA-Activated Channel Currents May Be Implicated in Learning Deficits Caused by Lead. FEBS Lett. 1990, 261, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Büsselberg, D.; Michael, D.; Platt, B. Pb2+ Reduces Voltage-and N-Methyl-D-Aspartate (NMDA)-Activated Calcium Channel Currents. Cell. Mol. Neurobiol. 1994, 14, 711–722. [Google Scholar] [CrossRef]
- Liu, J.; Guan, A.; Huo, Z.; Li, X.; Zhu, Y.; Liang, H.; Liu, W.; Zhou, H.; Lin, Z.; Yan, B. Distinct Neurotoxic Mechanisms of Thallium and Lead: Calcium-Mediated Apoptosis and Iron-Induced Ferroptosis in Zebrafish at Environmental Concentrations. J. Hazard. Mater. 2025, 492, 138288. [Google Scholar] [CrossRef]
- Marino, V.; Dal Cortivo, G.; Dell’Orco, D. Ionic Displacement of Ca2+ by Pb2+ in Calmodulin Is Affected by Arrhythmia-Associated Mutations. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2023, 1870, 119490. [Google Scholar] [CrossRef]
- Gavazzo, P.; Zanardi, I.; Baranowska-Bosiacka, I.; Marchetti, C. Molecular Determinants of Pb2+ Interaction with NMDA Receptor Channels. Neurochem. Int. 2008, 52, 329–337. [Google Scholar] [CrossRef]
- Ordemann, J.M.; Austin, R.N. Lead Neurotoxicity: Exploring the Potential Impact of Lead Substitution in Zinc-Finger Proteins on Mental Health. Metallomics 2016, 8, 579–588. [Google Scholar] [CrossRef]
- Guilarte, T.R.; Miceli, R.C.; Jett, D.A. Biochemical Evidence of an Interaction of Lead at the Zinc Allosteric Sites of the NMDA Receptor Complex: Effects of Neuronal Development. Neurotoxicology 1995, 16, 63–71. [Google Scholar]
- Guilarte, T.R.; McGlothan, J.L. Hippocampal NMDA Receptor mRNA Undergoes Subunit Specific Changes during Developmental Lead Exposure. Brain Res. 1998, 790, 98–107. [Google Scholar] [CrossRef]
- Neal, A.P.; Worley, P.F.; Guilarte, T.R. Lead Exposure during Synaptogenesis Alters NMDA Receptor Targeting via NMDA Receptor Inhibition. Neurotoxicology 2011, 32, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Gundacker, C.; Forsthuber, M.; Szigeti, T.; Kakucs, R.; Mustieles, V.; Fernandez, M.F.; Bengtsen, E.; Vogel, U.; Hougaard, K.S.; Saber, A.T. Lead (Pb) and Neurodevelopment: A Review on Exposure and Biomarkers of Effect (BDNF, HDL) and Susceptibility. Int. J. Hyg. Environ. Health 2021, 238, 113855. [Google Scholar] [CrossRef]
- Qneibi, M.; Bdir, S.; Bdair, M.; Aldwaik, S.A.; Sandouka, D.; Heeh, M.; Idais, T.I. AMPA Receptor Neurotransmission and Therapeutic Applications: A Comprehensive Review of Their Multifaceted Modulation. Eur. J. Med. Chem. 2024, 266, 116151. [Google Scholar] [CrossRef]
- Shen, Z.; Haragopal, H.; Li, Y.V. Zinc Modulates Synaptic Transmission by Differentially Regulating Synaptic Glutamate Homeostasis in Hippocampus. Eur. J. Neurosci. 2020, 52, 3710–3722. [Google Scholar] [CrossRef]
- Ding, J.-J.; Zou, R.-X.; He, H.-M.; Tang, Y.-Q.; Wang, H.-L. Effect of Pb Exposure on Synaptic Scaling through Regulation of AMPA Receptor Surface Trafficking. Toxicol. Sci. 2018, 165, 224–231. [Google Scholar] [CrossRef]
- Abreu, N.; Acosta-Ruiz, A.; Xiang, G.; Levitz, J. Mechanisms of Differential Desensitization of Metabotropic Glutamate Receptors. Cell Rep. 2021, 35, 109050. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Yan, C.; Yang, B.; Xie, H.; Zou, X.; Zhong, L.; Gao, Y.; Tian, Y.; Shen, X. The Role of Metabotropic Glutamate Receptor 5 in Developmental Lead Neurotoxicity. Toxicol. Lett. 2009, 191, 223–230. [Google Scholar] [CrossRef]
- Kasten-Jolly, J.; Lawrence, D.A. The Cationic (Calcium and Lead) and Enzyme Conundrum. J. Toxicol. Environ. Health Part B 2018, 21, 400–413. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Coro, A.; Sánchez-Hernández, B.E.; Montes, S.; Martínez-Lazcano, J.C.; González-Guevara, E.; Pérez-Severiano, F. Alterations in Gene Expression Due to Chronic Lead Exposure Induce Behavioral Changes. Neurosci. Biobehav. Rev. 2021, 126, 361–367. [Google Scholar] [CrossRef]
- Ramírez Ortega, D.; González Esquivel, D.F.; Blanco Ayala, T.; Pineda, B.; Gómez Manzo, S.; Marcial Quino, J.; Carrillo Mora, P.; de la Cruz, P.V. Cognitive Impairment Induced by Lead Exposure during Lifespan: Mechanisms of Lead Neurotoxicity. Toxics 2021, 9, 23. [Google Scholar] [CrossRef]
- Wang, T.; Guan, R.-L.; Liu, M.-C.; Shen, X.-F.; Chen, J.Y.; Zhao, M.-G.; Luo, W.-J. Lead Exposure Impairs Hippocampus Related Learning and Memory by Altering Synaptic Plasticity and Morphology During Juvenile Period. Mol. Neurobiol. 2016, 53, 3740–3752. [Google Scholar] [CrossRef] [PubMed]
- Strużyńska, L.; Chalimoniuk, M.; Sulkowski, G. Changes in Expression of Neuronal and Glial Glutamate Transporters in Lead-Exposed Adult Rat Brain. Neurochem. Int. 2005, 47, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Rahman, A.; Guillemin, G.J. Lead and Excitotoxicity. In Handbook of Neurotoxicity; Kostrzewa, R.M., Ed.; Springer International Publishing: Cham, Switzerland, 2021; pp. 1–39. ISBN 978-3-030-71519-9. [Google Scholar]
- Khalid, M.; Hodjat, M.; Abdollahi, M. Environmental Exposure to Heavy Metals Contributes to Diseases Via Deregulated Wnt Signaling Pathways. Iran. J. Pharm. Res. 2021, 20, 370–382. [Google Scholar] [CrossRef]
- Garza-Lombó, C.; Posadas, Y.; Quintanar, L.; Gonsebatt, M.E.; Franco, R. Neurotoxicity Linked to Dysfunctional Metal Ion Homeostasis and Xenobiotic Metal Exposure: Redox Signaling and Oxidative Stress. Antioxid. Redox Signal. 2018, 28, 1669–1703. [Google Scholar] [CrossRef]
- Karki, P.; Smith, K.; Johnson, J.; Aschner, M.; Lee, E.Y. Genetic Dys-Regulation of Astrocytic Glutamate Transporter EAAT2 and Its Implications in Neurological Disorders and Manganese Toxicity. Neurochem. Res. 2015, 40, 380–388. [Google Scholar] [CrossRef] [PubMed]
- Chlubek, M.; Baranowska-Bosiacka, I. Selected Functions and Disorders of Mitochondrial Metabolism under Lead Exposure. Cells 2024, 13, 1182. [Google Scholar] [CrossRef]
- Zhou, F.; Ouyang, L.; Xie, J.; Liu, S.; Li, Q.; Yang, S.; Li, J.; Su, R.; Rao, S.; Yan, L.; et al. Co-Exposure to Low-Dose Lead, Cadmium, and Mercury Promotes Memory Deficits in Rats: Insights from the Dynamics of Dendritic Spine Pruning in Brain Development. Ecotoxicol. Environ. Saf. 2023, 264, 115425. [Google Scholar] [CrossRef]
- Li, N.; Liu, X.; Zhang, P.; Qiao, M.; Li, H.; Li, X.; Zhang, H.; Yu, Z. The Effects of Early Life Lead Exposure on the Expression of Interleukin (IL) 1β, IL-6, and Glial Fibrillary Acidic Protein in the Hippocampus of Mouse Pups. Hum. Exp. Toxicol. 2015, 34, 357–363. [Google Scholar] [CrossRef]
- Wang, L.; Li, X.; Ying, M.; Hu, M.; Liu, Z. Amino Acid Transporters. In Oral Bioavailability and Drug Delivery; Hu, M., Li, X., Eds.; Wiley: Hoboken, NJ, USA, 2023; pp. 361–392. ISBN 978-1-119-66065-1. [Google Scholar]
- Huang, Y.; Liao, Y.; Zhang, H.; Li, S. Lead Exposure Induces Cell Autophagy via Blocking the Akt/mTOR Signaling in Rat Astrocytes. J. Toxicol. Sci. 2020, 45, 559–567. [Google Scholar] [CrossRef]
- Neal, A.P.; Stansfield, K.H.; Worley, P.F.; Thompson, R.E.; Guilarte, T.R. Lead Exposure during Synaptogenesis Alters Vesicular Proteins and Impairs Vesicular Release: Potential Role of NMDA Receptor–Dependent BDNF Signaling. Toxicol. Sci. 2010, 116, 249–263. [Google Scholar] [CrossRef]
- Ding, J.-J.; Zou, R.-X.; He, H.-M.; Lou, Z.-Y.; Xu, Y.; Wang, H.-L. Pb Inhibits Hippocampal Synaptic Transmission via Cyclin-Dependent Kinase-5 Dependent Synapsin 1 Phosphorylation. Toxicol. Lett. 2018, 296, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Mochida, S. Mechanisms of Synaptic Vesicle Exo- and Endocytosis. Biomedicines 2022, 10, 1593. [Google Scholar] [CrossRef] [PubMed]
- Bouton, C.M.L.S.; Frelin, L.P.; Forde, C.E.; Godwin, H.A.; Pevsner, J. Synaptotagmin I Is a Molecular Target for Lead. J. Neurochem. 2001, 76, 1724–1735. [Google Scholar] [CrossRef]
- Gąssowska, M.; Baranowska-Bosiacka, I.; Moczydłowska, J.; Frontczak-Baniewicz, M.; Gewartowska, M.; Strużyńska, L.; Gutowska, I.; Chlubek, D.; Adamczyk, A. Perinatal Exposure to Lead (Pb) Induces Ultrastructural and Molecular Alterations in Synapses of Rat Offspring. Toxicology 2016, 373, 13–29. [Google Scholar] [CrossRef] [PubMed]
- Jahn, R.; Cafiso, D.C.; Tamm, L.K. Mechanisms of SNARE Proteins in Membrane Fusion. Nat. Rev. Mol. Cell Biol. 2024, 25, 101–118. [Google Scholar] [CrossRef]
- Yang, C.; Kang, B.; Cao, Z.; Zhang, J.; Zhao, F.; Wang, D.; Su, P.; Chen, J. Early-Life Pb Exposure Might Exert Synapse-Toxic Effects Via Inhibiting Synapse-Associated Membrane Protein 2 (VAMP2) Mediated by Upregulation of miR-34b. JAD 2022, 87, 619–633. [Google Scholar] [CrossRef]
- Kurhaluk, N.; Kamiński, P.; Lukash, O.; Tkaczenko, H. Nitric Oxide-Dependent Regulation of Oxygen-Related Processes in a Rat Model of Lead Neurotoxicity: Influence of the Hypoxia Resistance Factor. Cell. Physiol. Biochem. 2024, 58, 597–629. [Google Scholar] [CrossRef]
- Gąssowska-Dobrowolska, M.; Chlubek, M.; Kolasa, A.; Tomasiak, P.; Korbecki, J.; Skowrońska, K.; Tarnowski, M.; Masztalewicz, M.; Baranowska-Bosiacka, I. Microglia and Astroglia—The Potential Role in Neuroinflammation Induced by Pre- and Neonatal Exposure to Lead (Pb). Int. J. Mol. Sci. 2023, 24, 9903. [Google Scholar] [CrossRef]
- Stansfield, K.H.; Ruby, K.N.; Soares, B.D.; McGlothan, J.L.; Liu, X.; Guilarte, T.R. Early-Life Lead Exposure Recapitulates the Selective Loss of Parvalbumin-Positive GABAergic Interneurons and Subcortical Dopamine System Hyperactivity Present in Schizophrenia. Transl. Psychiatry 2015, 5, e522. [Google Scholar] [CrossRef]
- Dason, J.S.; Romero-Pozuelo, J.; Marin, L.; Iyengar, B.G.; Klose, M.K.; Ferrús, A.; Atwood, H.L. Frequenin/NCS-1 and the Ca2+-Channel A1-Subunit Co-Regulate Synaptic Transmission and Nerve-Terminal Growth. J. Cell Sci. 2009, 122, 4109–4121. [Google Scholar] [CrossRef]
- Lasley, S.M.; Gilbert, M.E. Rat Hippocampal Glutamate and GABA Release Exhibit Biphasic Effects as a Function of Chronic Lead Exposure Level. Toxicol. Sci. 2002, 66, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Richetti, S.K.; Rosemberg, D.B.; Ventura-Lima, J.; Monserrat, J.M.; Bogo, M.R.; Bonan, C.D. Acetylcholinesterase Activity and Antioxidant Capacity of Zebrafish Brain Is Altered by Heavy Metal Exposure. Neurotoxicology 2011, 32, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Devóz, P.P.; Reis, M.B.d.; Gomes, W.R.; Maraslis, F.T.; Ribeiro, D.L.; Antunes, L.M.G.; Batista, B.L.; Grotto, D.; Reis, R.M.; Barbosa, F., Jr.; et al. Adaptive Epigenetic Response of Glutathione (GSH)-Related Genes against Lead (Pb)-Induced Toxicity, in Individuals Chronically Exposed to the Metal. Chemosphere 2021, 269, 128758. [Google Scholar] [CrossRef] [PubMed]
- Perez, R.R.; Sousa, C.A.; Vankeersbilck, T.; Machado, M.D.; Soares, E.V. Evaluation of the Role of Glutathione in the Lead-Induced Toxicity in Saccharomyces Cerevisiae. Curr. Microbiol. 2013, 67, 300–305. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Glutamatergic Target | Primary Molecular Effect | Corresponding Behavioral Phenotype |
|---|---|---|
| Voltage-gated calcium channels (VGCCs) | Inhibition of calcium influx at presynaptic terminals, reducing vesicle fusion and glutamate release | Decreased synaptic transmission, impaired learning and memory |
| Vesicular glutamate transporters (VGLUTs) | Disruption of proton gradient and vesicular packaging, leading to reduced glutamate availability in vesicles | Impaired excitatory signaling, deficits in cognitive performance |
| NMDA receptors (ionotropic) | Inhibition of receptor function and altered subunit expression; dysregulated Ca2+ signaling and synaptic plasticity | Reduced long-term potentiation (LTP), memory impairments, increased anxiety-like behaviors |
| AMPA receptors (ionotropic) | Altered receptor trafficking and expression; imbalance in fast excitatory transmission | Impaired learning, increased impulsivity, reduced cognitive flexibility |
| Metabotropic glutamate receptors (mGluRs) | Dysregulation of Group I/II/III receptors; altered intracellular signaling cascades | Social deficits, affective disturbances, behavioral rigidity |
| Excitatory amino acid transporters (EAATs) | Impaired astrocytic reuptake of glutamate; extracellular glutamate accumulation and excitotoxicity | Neurodegeneration, hyperexcitability, anxiety-like and seizure-like behaviors |
| Glutamine synthetase (GS) in astrocytes | Reduced enzymatic activity, impairing glutamate detoxification and recycling | Long-term neurotransmitter imbalance, chronic excitotoxic effects, neurocognitive decline |
| Glutamate dehydrogenase (GDH) and GOGAT | Interference in metabolic conversion between glutamate and α-ketoglutarate | Energetic stress, altered synaptic efficacy, cognitive dysfunction |
| Glutamate–GABA conversion (via GAD) | Disrupted inhibitory–excitatory balance due to altered GABA synthesis from glutamate | Increased anxiety-like behavior, seizure susceptibility, altered stress reactivity |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tamagno, W.A.; Freeman, J.L. Glutamate-Mediated Neural Alterations in Lead Exposure: Mechanisms, Pathways, and Phenotypes. Toxics 2025, 13, 519. https://doi.org/10.3390/toxics13070519
Tamagno WA, Freeman JL. Glutamate-Mediated Neural Alterations in Lead Exposure: Mechanisms, Pathways, and Phenotypes. Toxics. 2025; 13(7):519. https://doi.org/10.3390/toxics13070519
Chicago/Turabian StyleTamagno, Wagner A., and Jennifer L. Freeman. 2025. "Glutamate-Mediated Neural Alterations in Lead Exposure: Mechanisms, Pathways, and Phenotypes" Toxics 13, no. 7: 519. https://doi.org/10.3390/toxics13070519
APA StyleTamagno, W. A., & Freeman, J. L. (2025). Glutamate-Mediated Neural Alterations in Lead Exposure: Mechanisms, Pathways, and Phenotypes. Toxics, 13(7), 519. https://doi.org/10.3390/toxics13070519