


Acute PM2.5 Exposure in Distinct NSCLC Cell Lines Reveals Strong Oxidative Stress and Therapy Resistance Signatures Through Transcriptomic Analysis


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
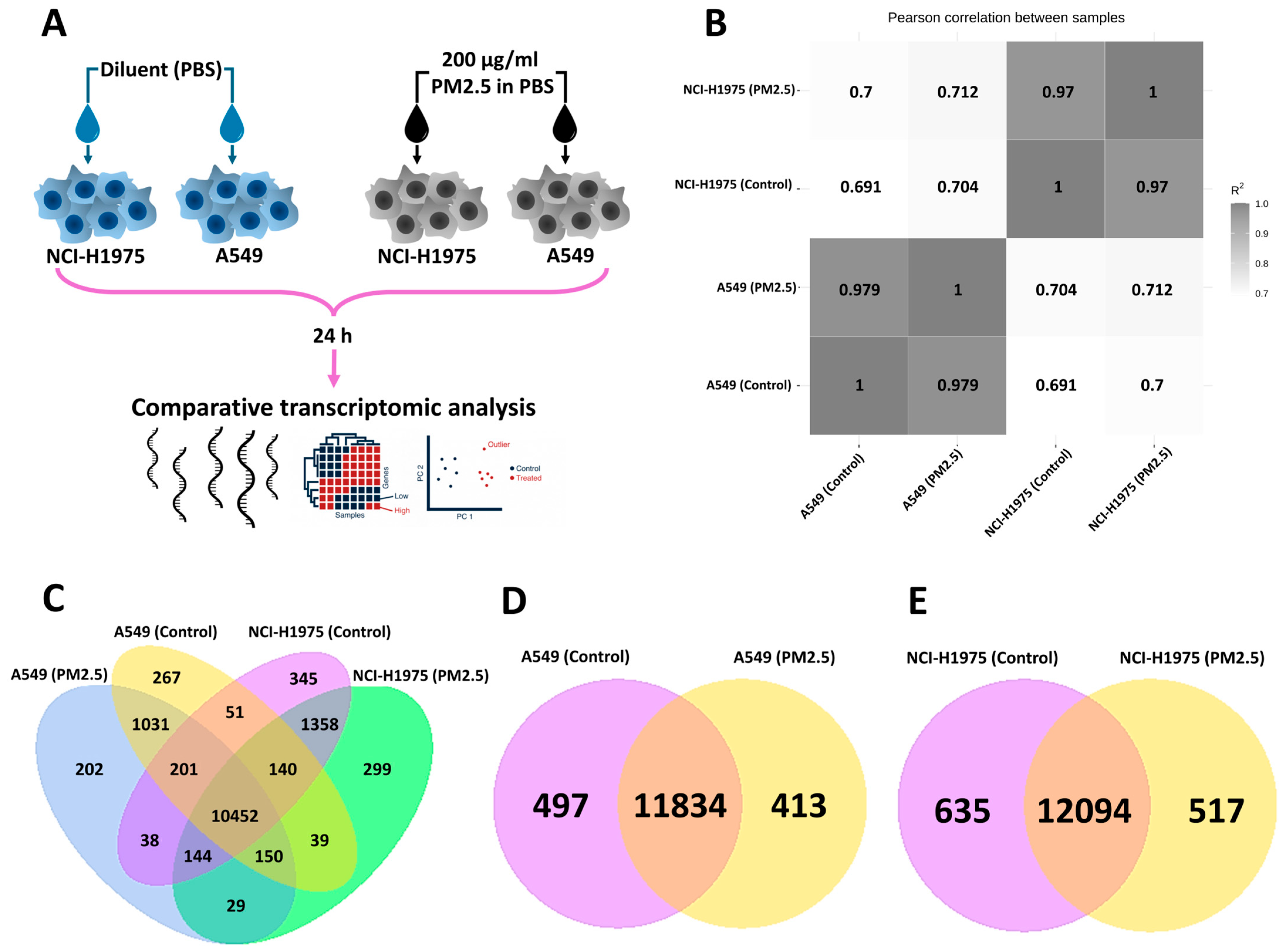
2.1. Cell Culture and PM2.5 Exposure
2.2. RNA Extraction, Quality Control, and Sequencing
2.3. Bioinformatics and Statistical Analysis
3. Results
3.1. Global Transcriptomic Profiling of NSCLC Cells Following PM2.5 Exposure
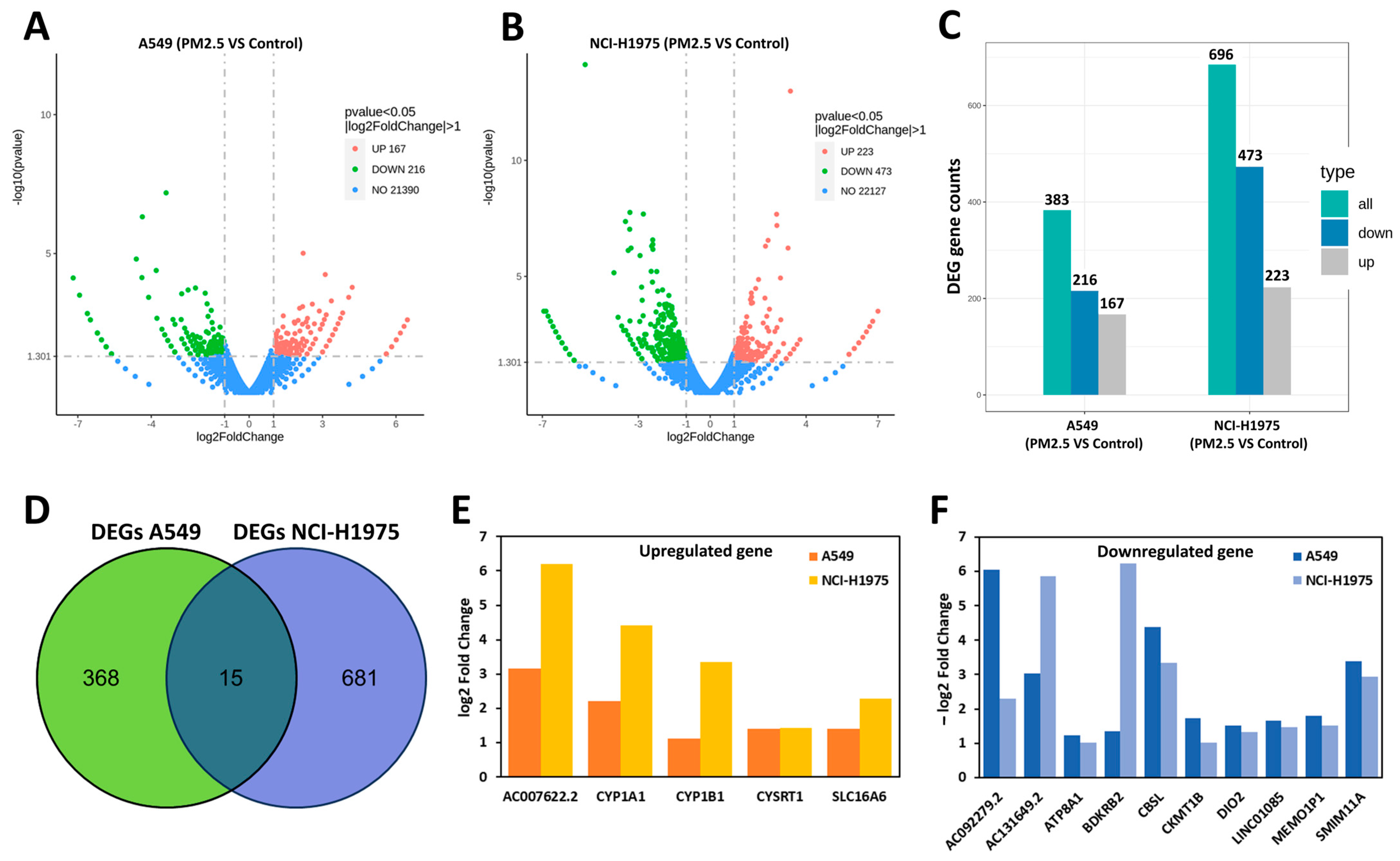
3.2. Differentially Expressed Genes (DEGs) in A549 and NCI-H1975 Cells Following PM2.5 Exposure
3.3. Pathway Enrichment Analysis of DEGs in A549 and NCI-H1975 Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Guo, J.; Chai, G.; Song, X.; Hui, X.; Li, Z.; Feng, X.; Yang, K. Long-term exposure to particulate matter on cardiovascular and respiratory diseases in low- and middle-income countries: A systematic review and meta-analysis. Front. Public Health 2023, 11, 1134341. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.-F.; Xu, Y.-H.; Shi, M.-H.; Lian, Y.-X. The impact of PM2.5 on the human respiratory system. J. Thorac. Dis. 2016, 8, E69–E74. [Google Scholar] [CrossRef] [PubMed]
- Pun, V.C.; Kazemiparkouhi, F.; Manjourides, J.; Suh, H.H. Long-Term PM2.5 Exposure and Respiratory, Cancer, and Cardiovascular Mortality in Older US Adults. Am. J. Epidemiol. 2017, 186, 961–969. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Pan, B.; Wu, J.; Chen, E.; Chen, L. Relationship between exposure to PM2.5 and lung cancer incidence and mortality: A meta-analysis. Oncotarget 2017, 8, 43322–43331. [Google Scholar] [CrossRef]
- Krishna, B.; Mandal, S.; Madhipatla, K.; Reddy, K.S.; Prabhakaran, D.; Schwartz, J.D. Daily nonaccidental mortality associated with short-term PM2.5 exposures in Delhi, India. Environ. Epidemiol. 2021, 5, e167. [Google Scholar] [CrossRef]
- Kosanpipat, B.; Wongwut, T.; Norrasan, N.; Watthanawongsa, P.; Phinyo, P.; Saeteng, S.; Siwachat, S.; Chewaskulyong, B.; Tantraworasin, A. Impact of PM2.5 exposure on mortality and tumor recurrence in resectable non-small cell lung carcinoma. Sci. Rep. 2024, 14, 24660. [Google Scholar] [CrossRef]
- Li, R.; Zhou, R.; Zhang, J. Function of PM2.5 in the pathogenesis of lung cancer and chronic airway inflammatory diseases. Oncol. Lett. 2018, 15, 7506–7514. [Google Scholar] [CrossRef]
- Chen, C.-Y.; Huang, K.-Y.; Chen, C.-C.; Chang, Y.-H.; Li, H.-J.; Wang, T.-H.; Yang, P.-C. The role of PM2.5 exposure in lung cancer: Mechanisms, genetic factors, and clinical implications. EMBO Mol. Med. 2025, 17, 31–40. [Google Scholar] [CrossRef]
- Wang, T.H.; Huang, K.Y.; Chen, C.C.; Chang, Y.H.; Chen, H.Y.; Hsueh, C.; Liu, Y.T.; Yang, S.C.; Yang, P.C.; Chen, C.Y. PM2.5 promotes lung cancer progression through activation of the AhR-TMPRSS2-IL18 pathway. EMBO Mol. Med. 2023, 15, e17014. [Google Scholar] [CrossRef]
- Hill, W.; Lim, E.L.; Weeden, C.E.; Lee, C.; Augustine, M.; Chen, K.; Kuan, F.-C.; Marongiu, F.; Evans, E.J.; Moore, D.A.; et al. Lung adenocarcinoma promotion by air pollutants. Nature 2023, 616, 159–167. [Google Scholar] [CrossRef]
- Chao, X.; Yi, L.; Lan, L.L.; Wei, H.Y.; Wei, D. Long-term PM2.5 exposure increases the risk of non-small cell lung cancer (NSCLC) progression by enhancing interleukin-17a (IL-17a)-regulated proliferation and metastasis. Aging 2020, 12, 11579–11602. [Google Scholar] [CrossRef] [PubMed]
- Ji, S.M.; Choi, J.-S.; Lee, J.Y.; Kim, S.; Bae, W.-Y.; Jang, Y.W.; Kim, J.-E.; Lee, S.H.; Nam, S.; Jeong, J.-W. Mild exposure to fine particulate matter promotes angiogenesis in non-small cell lung carcinoma. Environ. Pollut. 2023, 329, 121715. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Chen, D.; Zhao, H.; Xiao, C. The effects for PM2.5 exposure on non-small-cell lung cancer induced motility and proliferation. Springerplus 2016, 5, 2059. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, M.; Morais, S.; Delerue-Matos, C. SS07-06 OCCUPATIONAL EXPOSURE AS A FIREFIGHTER: CONTRIBUTION FROM BIOMONITORING ASSAYS. Occup. Med. 2024, 74 (Suppl. 1), 44–45. [Google Scholar] [CrossRef]
- Gili, J.; Maín, A.; van Drooge, B.L.; Viana, M. Source-resolved black carbon and PM2.5 exposures during wildfires and prescribed burns. Environ. Pollut. 2025, 368, 125660. [Google Scholar] [CrossRef]
- Panumasvivat, J.; Sapbamrer, R.; Sittitoon, N.; Khacha-ananda, S.; Kiratipaisarl, W.; Sirikul, W.; Insian, W.; Assavanopakun, P. Exploring the adverse effect of fine particulate matter (PM2.5) on wildland firefighters’ pulmonary function and DNA damage. Sci. Rep. 2024, 14, 7932. [Google Scholar] [CrossRef]
- Niu, B.-Y.; Li, W.-K.; Li, J.-S.; Hong, Q.-H.; Khodahemmati, S.; Gao, J.-F.; Zhou, Z.-X. Effects of DNA Damage and Oxidative Stress in Human Bronchial Epithelial Cells Exposed to PM2.5 from Beijing, China, in Winter. Int. J. Environ. Res. Public Health 2020, 17, 4874. [Google Scholar] [CrossRef]
- Liu, L.-L.; Yang, J.; Ye, Z.-L.; Tian, C.; Huang, X.-L.; Zhang, H.-Q.; Mo, X.-H. Effects of Long-Term Exposure to PM2.5 on Oxidative Stress Injury and Expression of Inflammatory Factors, NF-κB p65 and Cx43 in Bone Marrow of Mice. Front. Environ. Sci. 2022, 10, 747286. [Google Scholar] [CrossRef]
- Huggins, F.E.; Huffman, G.P.; Robertson, J.D. Speciation of elements in NIST particulate matter SRMs 1648 and 1650. J. Hazard. Mater. 2000, 74, 1–23. [Google Scholar] [CrossRef]
- Chansuebsri, S.; Kolar, P.; Kraisitnitikul, P.; Kantarawilawan, N.; Yabueng, N.; Wiriya, W.; Thepnuan, D.; Chantara, S. Chemical composition and origins of PM2.5 in Chiang Mai (Thailand) by integrated source apportionment and potential source areas. Atmos. Environ. 2024, 327, 120517. [Google Scholar] [CrossRef]
- Kong, L.; Feng, M.; Liu, Y.; Zhang, Y.; Zhang, C.; Li, C.; Qu, Y.; An, J.; Liu, X.; Tan, Q.; et al. Elucidating the pollution characteristics of nitrate, sulfate and ammonium in PM2.5 in Chengdu, southwest China, based on 3-year measurements. Atmos. Chem. Phys. 2020, 20, 11181–11199. [Google Scholar] [CrossRef]
- Li, Y.; Wang, X.; Xu, P.; Gui, J.; Guo, X.; Yan, G.; Fei, X.; Yang, A. Chemical characterization and source identification of PM2.5 in the Huaxi urban area of Guiyang. Sci. Rep. 2024, 14, 30451. [Google Scholar] [CrossRef] [PubMed]
- Berg, C.D.; Schiller, J.H.; Boffetta, P.; Cai, J.; Connolly, C.; Kerpel-Fronius, A.; Kitts, A.B.; Lam, D.C.L.; Mohan, A.; Myers, R.; et al. Air Pollution and Lung Cancer: A Review by International Association for the Study of Lung Cancer Early Detection and Screening Committee. J. Thorac. Oncol. 2023, 18, 1277–1289. [Google Scholar] [CrossRef]
- He, J.; Wang, T.; Li, H.; Zhou, Y.; Liu, Y.; Xu, A. Synergistic Toxicity of Fine Particulate Matter and Ozone and Their Underlying Mechanisms. Toxics 2025, 13, 236. [Google Scholar] [CrossRef]
- Shaath, H.; Vishnubalaji, R.; Elango, R.; Khattak, S.; Alajez, N.M. Single-cell long noncoding RNA (lncRNA) transcriptome implicates MALAT1 in triple-negative breast cancer (TNBC) resistance to neoadjuvant chemotherapy. Cell Death Discov. 2021, 7, 23. [Google Scholar] [CrossRef]
- Jiang, J.; Lu, Y.; Zhang, F.; Huang, J.; Ren, X.L.; Zhang, R. The Emerging Roles of Long Noncoding RNAs as Hallmarks of Lung Cancer. Front. Oncol. 2021, 11, 761582. [Google Scholar] [CrossRef]
- Li, L.; Wei, H.; Zhang, Y.W.; Zhao, S.; Che, G.; Wang, Y.; Chen, L. Differential expression of long non-coding RNAs as diagnostic markers for lung cancer and other malignant tumors. Aging 2021, 13, 23842–23867. [Google Scholar] [CrossRef]
- Liu, B.; Xiang, W.; Liu, J.; Tang, J.; Wang, J.; Liu, B.; Long, Z.; Wang, L.; Yin, G.; Liu, J. The regulatory role of antisense lncRNAs in cancer. Cancer Cell Int. 2021, 21, 459. [Google Scholar] [CrossRef]
- Androutsopoulos, V.P.; Tsatsakis, A.M.; Spandidos, D.A. Cytochrome P450 CYP1A1: Wider roles in cancer progression and prevention. BMC Cancer 2009, 9, 187. [Google Scholar] [CrossRef]
- Ezzeldin, N.; El-Lebedy, D.; Darwish, A.; El-Bastawisy, A.; Hassan, M.; Abd El-Aziz, S.; Abdel-Hamid, M.; Saad-Hussein, A. Genetic polymorphisms of human cytochrome P450 CYP1A1 in an Egyptian population and tobacco-induced lung cancer. Genes Environ. 2017, 39, 7. [Google Scholar] [CrossRef]
- Wenzlaff, A.S.; Cote, M.L.; Bock, C.H.; Land, S.J.; Santer, S.K.; Schwartz, D.R.; Schwartz, A.G. CYP1A1 and CYP1B1 polymorphisms and risk of lung cancer among never smokers: A population-based study. Carcinogenesis 2005, 26, 2207–2212. [Google Scholar] [CrossRef] [PubMed]
- Shimada, T.; Yun, C.H.; Yamazaki, H.; Gautier, J.C.; Beaune, P.H.; Guengerich, F.P. Characterization of human lung microsomal cytochrome P-450 1A1 and its role in the oxidation of chemical carcinogens. Mol. Pharmacol. 1992, 41, 856–864. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, U.T.N.; Hsieh, H.-Y.; Chin, T.-Y.; Wu, G.; Lin, Y.P.; Lee, C.-Y.; Hsu, Y.-C.; Fan, Y.-J. Evaluation of Pm2.5 Influence on Human Lung Cancer Cells Using a Microfluidic Platform. Int. J. Med. Sci. 2024, 21, 1117–1128. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Xie, J.; Wu, D.; Chen, L.; Gong, Z.; Wu, R.; Hu, Y.; Zhao, J.; Xu, Y. A pan-cancer analysis revealed the role of the SLC16 family in cancer. Channels 2021, 15, 528–540. [Google Scholar] [CrossRef]
- Yu, S.; Wu, Y.; Li, C.; Qu, Z.; Lou, G.; Guo, X.; Ji, J.; Li, N.; Guo, M.; Zhang, M.; et al. Comprehensive analysis of the SLC16A gene family in pancreatic cancer via integrated bioinformatics. Sci. Rep. 2020, 10, 7315. [Google Scholar] [CrossRef]
- Xue, L.; Liu, J.; Xie, J.; Luo, J. Prognostic Value of SLC16A3(MCT4) in Lung Adenocarcinoma and Its Clinical Significance. Int. J. Gen. Med. 2021, 14, 8413–8425. [Google Scholar] [CrossRef]
- Niehues, H.; Rikken, G.; Kersten, F.F.J.; Eeftens, J.M.; van Vlijmen-Willems, I.M.J.J.; Rodijk-Olthuis, D.; Jansen, P.A.M.; Hendriks, W.J.A.J.; Ederveen, T.H.A.; Schalkwijk, J.; et al. CYSRT1: An Antimicrobial Epidermal Protein that Can Interact with Late Cornified Envelope Proteins. J. Investig. Dermatol. 2023, 143, 1498–1508.e7. [Google Scholar] [CrossRef]
- Wang, G.; Guo, S.; Zhang, W.; Li, D.; Wang, Y.; Zhan, Q. Co-expression network analysis identifies key modules and hub genes implicated in esophageal squamous cell cancer progression. Med. Omics 2021, 1, 100003. [Google Scholar] [CrossRef]
- Wang, M.; Yu, F.; Li, P. Noncoding RNAs as an emerging resistance mechanism to immunotherapies in cancer: Basic evidence and therapeutic implications. Front. Immunol. 2023, 14, 1268745. [Google Scholar] [CrossRef]
- Zhang, J.; Li, S.; Zhang, J.; Zhang, W.; Jiang, J.; Wu, H.; Wu, E.; Feng, Y.; Yang, L.; Li, Z. Docetaxel resistance-derived LINC01085 contributes to the immunotherapy of hormone-independent prostate cancer by activating the STING/MAVS signaling pathway. Cancer Lett. 2022, 545, 215829. [Google Scholar] [CrossRef]
- Ascenção, K.; Szabo, C. Emerging roles of cystathionine β-synthase in various forms of cancer. Redox Biol. 2022, 53, 102331. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Akaike, T.; Hayashida, K.; Miyamoto, Y.; Nakagawa, T.; Miyakawa, K.; Müller-Esterl, W.; Maeda, H. Identification of bradykinin receptors in clinical cancer specimens and murine tumor tissues. Int. J. Cancer 2002, 98, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhou, Y.; Wei, R.; Jiang, G.; Li, F.; Chen, X.; Wang, X.; Ma, D.; Xi, L. Bradykinin promotes proliferation, migration, and invasion of cervical cancer cells through STAT3 signaling pathways. Oncol. Rep. 2019, 42, 2521–2527. [Google Scholar] [CrossRef] [PubMed]
- Ding, C.; Xi, G.; Wang, G.; Cui, D.; Zhang, B.; Wang, H.; Jiang, G.; Song, J.; Xu, G.; Wang, J. Exosomal Circ-MEMO1 Promotes the Progression and Aerobic Glycolysis of Non-small Cell Lung Cancer Through Targeting MiR-101-3p/KRAS Axis. Front. Genet. 2020, 11, 962. [Google Scholar] [CrossRef]
- Shi, H.; Song, Y.; Song, Z.; Huang, C. CKMT1B is a potential prognostic biomarker and associated with immune infiltration in Lower-grade glioma. PLoS ONE 2021, 16, e0245524. [Google Scholar] [CrossRef]
- Li, D.; Xu, T.; Wang, X.; Ma, X.; Liu, T.; Wang, Y.; Jiang, S. The role of ATP8A1 in non-small cell lung cancer. Int. J. Clin. Exp. Pathol. 2017, 10, 7760–7766. [Google Scholar]
- Canals, J.; Navarro, A.; Viñolas, N.; Díaz, T.; Marrades, R.M.; Moisés, J.; Acosta, M.; Cros, C.; Bing, H.; He, Y.; et al. 1167P DIO2 is implicated in the antitumor effect of the lung embryonic stem cell conditioned medium and impacts prognosis in non-small cell lung cancer. Ann. Oncol. 2021, 32, S937. [Google Scholar] [CrossRef]
- Chen, Q.; Wang, Y.; Yang, L.; Sun, L.; Wen, Y.; Huang, Y.; Gao, K.; Yang, W.; Bai, F.; Ling, L.; et al. PM2.5 promotes NSCLC carcinogenesis through translationally and transcriptionally activating DLAT-mediated glycolysis reprograming. J. Exp. Clin. Cancer Res. 2022, 41, 229. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panya, A.; Thongyim, S.; Sattayawat, P.; Inwongwan, S. Acute PM2.5 Exposure in Distinct NSCLC Cell Lines Reveals Strong Oxidative Stress and Therapy Resistance Signatures Through Transcriptomic Analysis. Toxics 2025, 13, 484. https://doi.org/10.3390/toxics13060484
Panya A, Thongyim S, Sattayawat P, Inwongwan S. Acute PM2.5 Exposure in Distinct NSCLC Cell Lines Reveals Strong Oxidative Stress and Therapy Resistance Signatures Through Transcriptomic Analysis. Toxics. 2025; 13(6):484. https://doi.org/10.3390/toxics13060484
Chicago/Turabian StylePanya, Aussara, Saruda Thongyim, Pachara Sattayawat, and Sahutchai Inwongwan. 2025. "Acute PM2.5 Exposure in Distinct NSCLC Cell Lines Reveals Strong Oxidative Stress and Therapy Resistance Signatures Through Transcriptomic Analysis" Toxics 13, no. 6: 484. https://doi.org/10.3390/toxics13060484
APA StylePanya, A., Thongyim, S., Sattayawat, P., & Inwongwan, S. (2025). Acute PM2.5 Exposure in Distinct NSCLC Cell Lines Reveals Strong Oxidative Stress and Therapy Resistance Signatures Through Transcriptomic Analysis. Toxics, 13(6), 484. https://doi.org/10.3390/toxics13060484