Abstract
Removing toxic formaldehyde (HCHO) from environmental water is crucial for human health and the ecosystem. Perovskite-type Lanthanum cobalt oxide (LaCoO3) has achieved great success in a wide range of catalytic processes; however, this concept has been rarely applied to the degradation of HCHO. Here, we prepared perovskite-type catalysts with different La/Co molar ratios, and the time for HCHO oxidation degradation at room temperature was shortened by 12 times (10 min vs. 119 min) compared to other heterogeneous catalysts. LaCoO3 exhibits superior catalytic activity for HCHO degradation at room temperature when the La/Co molar ratio is 1:1 compared to lanthanum cobalt oxides with other molar ratios. The X-ray photoelectron spectroscopy (XPS) test results show that increasing the La/Co molar ratio reduces the Co2+ content in the catalyst, while Co2+ plays the most important role in the catalyst. Quencher experiments indicated that sulfate radicals (SO4·−) and hydroxyl radicals (·OH) were the primary reactive species for the removal of HCHO. This finding suggests that the catalytic oxidation reaction involving HCHO operates as a heterogeneous Fenton-like oxidation reaction.
1. Introduction
Formaldehyde (HCHO) is a chemical raw material that has found application in various industries, including wood furniture processing, plastic synthesis, and skincare products [1,2]. Given the extensive utilization of HCHO, the inevitable consequence of its release into the environment is the production of HCHO wastewater. The World Health Organization (WHO) has classified HCHO as a suspected carcinogen and teratogen [3,4]. Degrading HCHO in wastewater is crucial for protecting aquatic environments and human health. Currently, methods for HCHO removal include adsorption [5,6], biological processes [7], photocatalysis [8], electrochemistry [9], Fenton oxidation [10], and Fenton-like oxidation [11], all widely applied for HCHO removal from wastewater.
Among these methods, adsorption is a simple and widely used method for removing HCHO from wastewater. The adsorption method has the advantages of simple adsorption material structure, convenient operation and high adsorption efficiency. There are many kinds of adsorbents, such as porous carbon [12,13], polymers [14], nano-powders [15], etc. These adsorption materials all have a good pore structure, high specific surface area, good thermal stability and chemical resistance to alkaline and acidic media. However, the adsorption process does not involve degradation, the adsorbed pollutants still exist and may continue to cause pollution. Given that the adsorption of HCHO occurs exclusively from wastewater to the adsorbent, with no subsequent breakdown into harmless environmental substances, the HCHO accumulated in the adsorbent remains a concern. Furthermore, the adsorption capacity of adsorbents is not unlimited. In most cases, adsorbents eventually reach a state of saturation, which leads to desorption. Consequently, alternative degradation methods currently under development for the removal of HCHO from wastewater include biological processes and advanced oxidation processes (AOPs). These methodologies have been demonstrated to achieve the mineralization of HCHO. In particular, biological methods have been shown to be more environmentally friendly, with a negligible propensity for secondary pollution. Nevertheless, the utilization of microorganisms in biotechnology necessitates prolonged domestication and screening processes. A prevalent challenge pertains to the microorganisms’ limited tolerance for HCHO wastewater, which often results in a low survival rate in high-concentration HCHO wastewater or acid–base environments [16]. AOPs are highly regarded due to their broad applicability, strong anti-interference capabilities, and the degradation of organic matter through mineralization. The photocatalytic method, electrochemical method, Fenton oxidation method, and Fenton-like oxidation method can all be considered as AOPs, and the goal of these methods is to generate highly oxidizing free radicals capable of mineralizing pollutants. The key factors in the process of treating organic pollutants by photocatalytic method, electrochemical method, Fenton oxidation method, and Fenton-like oxidation method are the generation of reactive species [17]. The rate of degradation of organic pollutants is contingent upon the number of free radicals produced and their molecular structure [18]. Photocatalysis suffers from low degradation efficiency, mainly due to the large difference in the absorption effect of the reaction system on light, and under high-power light radiation, the number of active free radicals produced is limited [19,20].
The hydroxyl radicals produced by the electrochemical method are predominantly concentrated in the proximity of the electrode. The probability of these radicals colliding and reacting with the HCHO molecules dispersed in the solution system is minimal due to the remarkably brief survival duration of the hydroxyl radicals (10−6–10−3 s) [21]. Fenton oxidation is defined as a specific oxidation system composed of H2O2, Fe2+, and acid. This system facilitates the decomposition of hydrogen peroxide through Fe2+, resulting in the production of hydroxyl radicals (·OH). These hydroxyl radicals subsequently attack and mineralize organic pollutants, thereby achieving deep oxidation of organic pollutants. Fenton oxidation boasts a number of advantages, including its rapid reaction rate and high organic removal rate, as well as its low energy requirements [14]. This makes it a widely used method for the treatment of organic pollutants in industrial wastewater [22,23]. For example, Fenton oxidation was employed to treat Octahydro-1,3,5,7-tetranitro-1,3,5,7-tetrazocine (HMX) wastewater. The results show that under the reaction conditions of pH = 3.0, H2O2 concentration of 30.0 mmol/L, and Fe2+ concentration of 0.7 mmol/L, the degradation performance of HMX reached its optimum after 180 min, with a removal rate as high as 90.8% [24]. Caiyi Jiang prepared Fe–N co-doped carbon catalyst (αFe@CTS-T) and used it to degrade RhB. αFe@CTS-T releases Fe2+ during the reaction process, which activates hydrogen peroxide to generate hydroxyl radicals. The maximum degradation rate of RhB was 99%. High catalytic activity was attributed to the high content of Fe2+ and Fe3+ in the catalyst [25]. Although the H2O2, Fe2+, and acid system is effective in degrading organic pollutants, this process also has disadvantages. As the reaction proceeds, Fe2+ will convert into Fe3+, and then form iron precipitation (Fe(OH)3), which affects the activation ability of hydrogen peroxide, and ultimately leads to the cessation of the oxidation reaction. In order to sustain the degradation reaction, it is imperative to add Fe2+ continuously, so this process consumes a large amount of catalyst Fe2+, and a secondary pollutant, iron mud (Fe(OH)3), was generated. Moreover, the Fenton oxidation process needs to be carried out under strong acid conditions to ensure a high degradation rate. Finally, when degrading high-concentration HCHO wastewater, the degradation efficiency is not ideal [26].
The heterogeneous Fenton catalyst has been demonstrated to exhibit characteristics such as high recyclability and ease of operation. In addition, it has been observed to reduce the amount of catalyst required, yet its catalytic activity is lower than that of the homogeneous Fenton catalyst [27,28,29]. A heterogeneous Fenton catalyst GF-PDA-FeOOH was prepared with glass fiber as the carrier, and glass fiber was modified through oxidative self-polymerization of dopamine, and then prepared by the impregnation method in an FeSO4 solution. Although the GF-PDA-FeOOH catalyst exhibits excellent cyclic performance, the degradation rate of methylene blue (MB), Congo red (CR) and crystal violet (CV) can still maintain above 80% after being used 6 cycles, When the degradation rates of MB, CR, and CV exceed 90%, the required temperature is 40 °C, and the time exceeds 100 min [30]. In order to improve the oxidation efficiency of the heterogeneous Fenton-like oxidation reaction, researchers introduced light to synergistically catalyze the formation of free radicals. Athikaphan P et al. [31] prepared a catalyst n-ZVI/TiO2 by wet impregnation and sodium borohydride reduction method. The degradation of HCHO was only 55.07% without UV light. The complete degradation of HCHO can only be achieved by adding the catalyst and then exposing it to UV light. Moreover, the degradation of HCHO under this catalyst condition is greatly affected by pH value. The degradation efficiency of HCHO rapidly decreased when pH value was greater than 3 because the catalyst can only catalyze the generation of hydroxyl radicals from hydrogen peroxide under acidic conditions. Bi F et al. [32] successfully prepared a new type of magnetic recyclable ZnO/NiFe2O4 composite nanofibrous photocatalyst by parallel electrospinning. When ZnO/NiFe2O4 is used as a catalyst, 0.05 mL of H2O2 is added to the reaction system and the degradation rate of RhB can reach 99.57% after 10 min of illumination. Without light irradiation, the degradation rate of RhB was only 10% after reacting in the dark for 30 min. In the absence of hydrogen peroxide and only with catalyst ZnO/NiFe2O4, the degradation rate of RhB was only 50.99% after 210 min of reaction. The degradation reaction is promoted in the presence of light irradiation. However, the implementation of light irradiation has been observed to result in an increase in the establishment and operational costs of the Fenton-like oxidation method. It is imperative to identify catalysts capable of facilitating the conversion of hydrogen peroxide and potassium peroxymonosulfate into ·OH and SO4·− during oxidation processes. The method of introducing transition metal-activated oxidants to generate free radicals has been demonstrated to be more economical and easier to operate than light irradiation [33].
Perovskite catalysts have been extensively studied in the catalytic treatment of wastewater and have been shown to have good degradation effects [34,35,36]. The chemical general formula of perovskite-type oxides is ABO3. Lanthanide-based perovskite has been reported to be a good candidate for heterogeneous catalysis. In addition, Co is the most effective transition metal for homogeneous catalysis, so heterogeneous Co-based catalysts exhibiting low Co2+ leaching rates have attracted the attention of researchers. Therefore, it is necessary to introduce Co into the B site of perovskite catalysts to prepare stable cobalt-containing perovskite-type catalysts [37].
Although the perovskite-type lanthanum cobalt oxide has been extensively studied in catalytic oxidation, its application in the degradation of HCHO has not been reported. Additionally, there is no literature on the effect of the La/Co molar ratio on the catalytic activity. In this study, we have prepared perovskite-type lanthanum cobalt oxide nanoparticle catalysts with varying La/Co molar ratios via the sol–gel method. A structural analysis of lanthanum cobalt oxides prepared with varying La/Co molar ratios reveals that they all exhibit a perovskite structure. However, their catalytic activity varies during HCHO degradation. The lanthanum cobalt oxides with a La/Co molar ratio of 1:1 exhibited the highest catalytic activity for HCHO degradation. As the La/Co molar ratio increased, the catalytic activity for HCHO degradation decreased. We investigated the effect of different process parameters (catalyst type, catalyst concentration, oxidant type, oxidant concentration, HCHO concentration, initial pH value) on the degradation efficiency of HCHO, and to determine the optimal experimental conditions. Our research found that although perovskite-type lanthanum cobalt oxides exhibit excellent catalytic performance in promoting PMS-mediated HCHO degradation, their efficacy in catalyzing hydrogen peroxide-mediated HCHO degradation is unsatisfactory.
2. Materials and Methods
2.1. Materials
Lanthanum nitrate (La(NO3)3·6H2O) with a purity of 99.5%, cobalt nitrate (Co(NO3)2·6H2O) with a purity of 98.5%, HCHO solution (37 wt%), sodium thiosulfate (Na2S2O3) with a purity of 99.0%, Potassium monopersulfate salt (KHSO5) with a purity of 42.0%, acetic acid (CH3COOH) with a purity of 99.5% and acetylacetone (CH3COCH2COCH3) with a purity of 99.0%. They were all purchased from Sinopharm Chemical Reagent Co., Ltd., Shanghai, China.
2.2. Methods
2.2.1. Preparation of Lanthanum Cobalt Oxide
The required reagent amounts were calculated based on a molar ratio of lanthanum nitrate, cobalt nitrate, and citric acid of 1:1:4. 3.523 g of lanthanum nitrate and 2.368 g of cobalt nitrate were weighed separately and dissolved in beakers each containing 5 mL of deionized water to prepare the corresponding solutions. Under stirring conditions, the lanthanum nitrate and cobalt nitrate solutions were combined. 6.252 g of citric acid was added slowly to this mixed solution, and after complete dissolution, the mixed solution was heated to 80 °C and the reaction is continued until the mixed solution is converted into a viscous sol. The gel was transferred to a drying oven and dried at 120 °C for 6 h. Finally, the dried solid was transferred to a crucible and calcined in a muffle furnace at 600 °C for 4 h to obtain a solid powder, denoted as LaCoO3-1. The molar ratios of lanthanum nitrate, cobalt nitrate, and citric acid were adjusted to 2:1:6 and 3:1:8, respectively. Lanthanum cobalt oxide was prepared following the same method as above and designated as LaCoO3-2 and LaCoO3-3.
2.2.2. Characterization of Lanthanum Cobalt Oxide
Thermogravimetric (TG, PerkinElmer, TGA4000, Waltham, MA, USA) analysis was performed on lanthanum cobalt oxides and their precursors. Samples were tested under an atmosphere with a flow rate of 20 mL/min. The temperature range was 50–800 °C, with a heating rate of 10 °C/min. The sample was stabilized at 100 °C for 5 min during the heating process.
The lanthanum cobalt oxide of LaCoO3-1, LaCoO3-2, and LaCoO3-3 was analyzed using X-ray powder diffraction (XRD, Rigaku, Smartlab, Tokyo, Japan) (copper target radiation source, step size 0.02°, 2θ = 5–80°).
Transmission electron microscopy (TEM, JEOL, JEM-F200, Tokyo, Japan) analyzed the grain size and interplanar spacing of LaCoO3-1.
The valence states of lanthanum and cobalt in LaCoO3-1 were analyzed using X-ray photoelectron spectroscopy (XPS, Thermo Fischer, ESCALAB 250Xi, Waltham, MA, USA). Cobalt measurement conditions: Al Kα radiation, total acquisition time 308.1 s, test energy 30.0 eV, step size 0.10 eV, 411 steps. Lanthanum testing conditions: Al Kα radiation, total acquisition time 200.4 s, test energy range 30.0 eV, step size 0.10 eV, 401 steps.
2.2.3. The Performance of HCHO Degradation
HCHO solution (approximately 1 mg/mL), sodium hydroxide solution, and iodine solution were added to the iodine bottle. After thoroughly mixing, sulfuric acid solution was added. The reaction was allowed to proceed for 15 min, followed by titration with standard sodium thiosulfate solution. A blank test was performed by replacing HCHO solution with deionized water and titrating with standard sodium thiosulfate solution using the same method. The concentration of the HCHO standard solution was calculated based on the volume of standard sodium thiosulfate solution consumed, resulting in 1.086 mg/mL [38].
5 mL of HCHO standard solution was added to a 10 mL reaction tube. After the temperature of the HCHO solution stabilized at 25 °C, 0.06 g of LaCoO3 and 0.13 g of PMS were added. The reaction was then started and allowed to proceed for 10 min. Every 2 min, 30 μL of reaction solution sample was taken, filtered through a filter membrane with a pore size of 0.45 μm, and reacted with sodium thiosulfate solution to terminate the reaction. The resulting solution was transferred to a colorimetric tube, followed by the addition of acetic acid and acetylacetone. Finally, the solution was diluted to the mark with deionized water. The colorimetric tubes were transferred to a boiling water bath to react for 3 min. The concentration of HCHO in the solution was determined by visible spectrophotometry. Finally, the degradation rate of HCHO was calculated based on the concentration of HCHO [38]. The effect of different process parameters (catalyst type, catalyst concentration, oxidant type, oxidant concentration, HCHO concentration, initial pH value) on the degradation efficiency of HCHO was investigated.
3. Results
3.1. TG Analysis
As shown in Figure 1, lanthanum cobalt oxide precursors showed three stages of weight loss. The first stage started at 200 °C, which was due to the decomposition of unreacted citric acid. Within 360(380)–400 °C, lanthanum cobalt oxide precursors underwent rapid weight loss, primarily due to the decomposition of citrates and nitrates. The decomposition temperature of LaCoO3-2 precursor and LaCoO3-3 precursor in the second stage was 20 °C lower than that of LaCoO3-1 precursor. This difference may be attributed to the observation that with an increase in the La/Co molar ratio, the decomposition temperatures of citrate and nitrate decrease. Lanthanum cobalt oxide precursors underwent a gradual weight loss within the temperature range of 400–600 °C, a phenomenon primarily attributed to the decomposition of residual citrate and nitrate. Above 600 °C, no significant weight loss was observed, indicating that LaCoO3 was formed at 600 °C. The solid powders obtained by calcining the lanthanum cobalt oxide precursor at 600 °C, namely LaCoO3-1, LaCoO3-2, and LaCoO3-3, exhibited almost no weight loss in the temperature range of 50–800 °C, which also indicates that the lanthanum cobalt oxide was formed at 600 °C. Consequently, 600 °C was selected as the final calcination temperature.
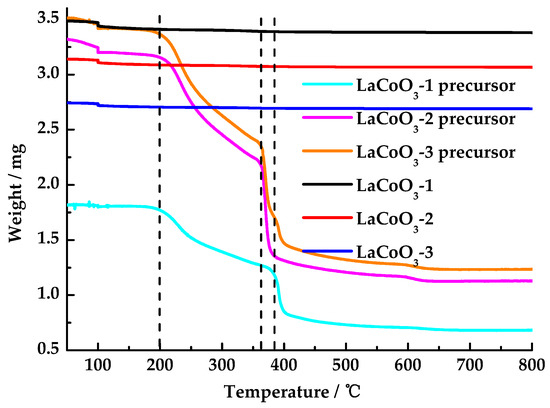
Figure 1.
TG curves of LaCoO3-1, LaCoO3-2, LaCoO3-3, LaCoO3-1 precursor, LaCoO3-2 precursor, and LaCoO3-3 precursor.
3.2. XRD Analysis
As shown in Figure 2, the diffraction peaks (23.22°, 32.88°, 40.66°, 47.46°, 53.6°, 58.92°, 69.82°, and 78.76°) correspond to those of perovskite-type LaCoO3 (JCPDS No. 48-0123) [36,39], and the diffraction peaks (15.72°, 27.96°, 39.58°, 48.58°, 55.36°, 64.3°, and 69.84°) correspond to those of La2O3 (JCPDS No. 02-0607) [40]. All the samples possess perovskite-structured LaCoO3 phases. No other peaks were detected except for the perovskite phase, indicating that the LaCoO3-1 has a single phase. In contrast, LaCoO3-2 and LaCoO3-3 contain both the perovskite phase and the La2O3 phase, indicating that the materials are two-phase. As the molar ratio of La/Co increases, the diffraction intensity of the perovskite-type LaCoO3 decreases, and a crystalline phase of La2O3 appears, whose diffraction intensity also increases with the molar ratio of La/Co. This indicates that when the molar ratio of La/Co is 1:1, a perovskite structure of La-O-Co is formed. As the molar ratio of La/Co increases, there is an excess of La atoms. The surplus La atoms form a La-O-La structure, resulting in the appearance of La2O3 crystalline phase in the spectrum. The XRD pattern of the recycled LaCoO3-1 indicates that the perovskite-type remains unaltered, suggesting that the perovskite-type LaCoO3-1 exhibits remarkable reusability.
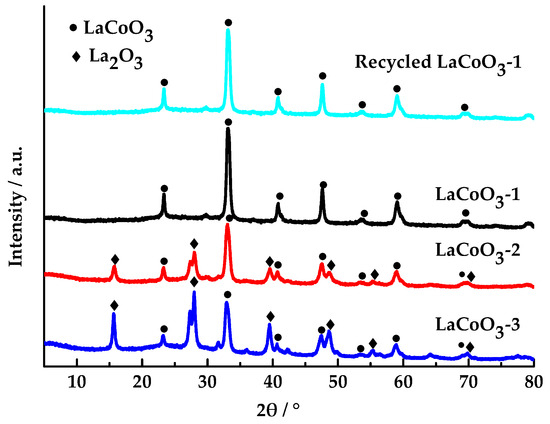
Figure 2.
XRD patterns of LaCoO3-1, LaCoO3-2, LaCoO3-3, and recycled LaCoO3-1.
3.3. TEM Analysis
As shown in Figure 3, the particle sizes of the LaCoO3-1, LaCoO3-2, and LaCoO3-3 all range from 20 to 50 nm. The particles of LaCoO3-3 are more uniformly dispersed than those of LaCoO3-1 and LaCoO3-2. The lattice fringe spacings of 0.19 nm and 0.30 nm correspond well to the (024) crystal plane of perovskite-structured LaCoO3 and the (002) crystal plane of La2O3, respectively [41]. LaCoO3-1 exhibited only the (024) crystal plane, while LaCoO3-2 and LaCoO3-3 displayed both crystal planes, indicating that both perovskite and lanthanum oxide crystal structures coexist in LaCoO3-2 and LaCoO3-3.
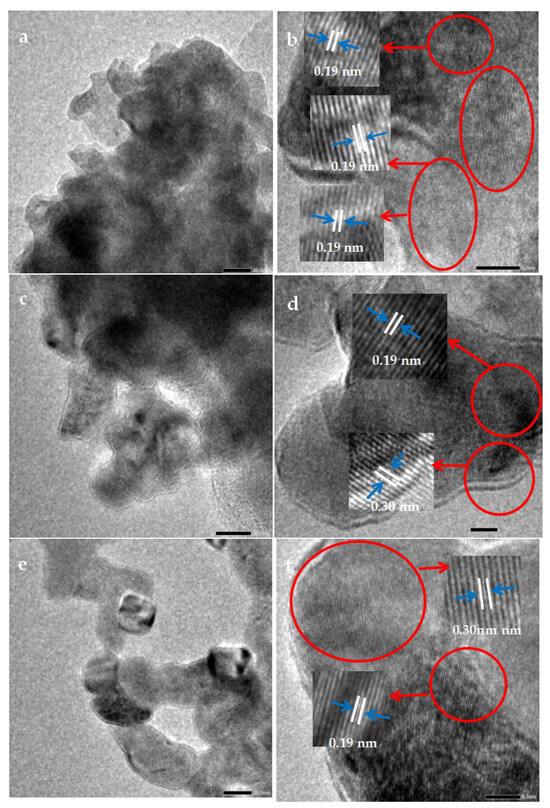
Figure 3.
TEM images of (a) LaCoO3-1; (c) LaCoO3-2; (e) LaCoO3-3 and HRTEM images of (b) LaCoO3-1; (d) LaCoO3-2; (f) LaCoO3-3.
3.4. XPS Analysis
Figure 4a shows the La 3d spectra of LaCoO3-1, LaCoO3-2 and LaCoO3-3, which all contained four peaks. The La3d XPS spectrum of LaCoO3-1 reveals a binding energy of 849.9 eV for La 3d3/2 and 833.1 eV for La 3d5/2, with satellite peaks appearing at 853.5 eV and 836.7 eV. The La 3d splitting energy is about 16.8 eV, close to the splitting energy of pure La2O3, indicating that the oxidation state of lanthanum element species in LaCoO3-1 is primarily +3 [42,43]. Since LaCoO3-2 and LaCoO3-3 have increased the La2O3 content, the electron binding energy of La 3d in LaCoO3-2 and LaCoO3-3 has increased.
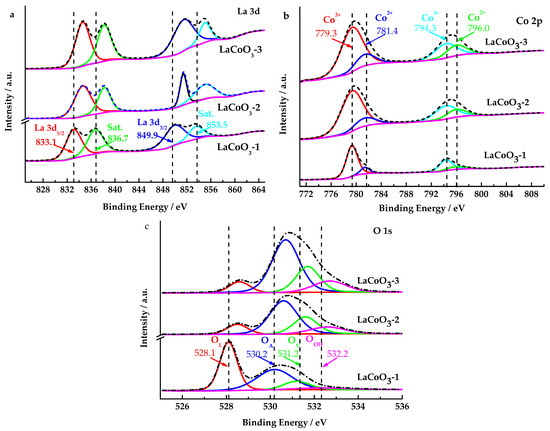
Figure 4.
XPS spectra of LaCoO3-1, LaCoO3-2 and LaCoO3-3 ((a): La 3d; (b): Co 2p; (c): O 1s).
As shown in Figure 4b, the Co 2p XPS spectrum of the LaCoO3-1, LaCoO3-2 and LaCoO3-3 could be fitted into four peaks. The two peaks located at 779.3 and 794.3 eV were close to the standard data for Co3+, while the other two peaks at 781.4 eV and 796.0 eV were related to Co2+ [43]. Although the four peaks of the three samples were at the same position, the proportion of Co2+ in the total Co was different. The proportions of Co2+ to total Co atom in the LaCoO3-1, LaCoO3-2 and LaCoO3-3 are 19.07%, 22.13%, and 25.25%, respectively. However, as the La/Co molar ratio increased, the content of Co atoms in the sample decreased. XPS data indicates that the Co at% proportion in LaCoO3-1, LaCoO3-2, and LaCoO3-3 are 10.39, 4.58, and 3.60, respectively, and the La at% proportion are 17.72, 19.82, and 19.16, respectively. Consequently, the atomic ratios of La atoms to Co atoms in LaCoO3-1, LaCoO3-2, and LaCoO3-3 are determined to be 1.7, 4.3, and 5.3, respectively. The molar ratios of La to Co in the raw materials used for preparing LaCoO3-1, LaCoO3-2, and LaCoO3-3 are 1, 2, and 3, respectively. Therefore, it can be deduced that the atomic ratios of La atoms to Co atoms in LaCoO3-1, LaCoO3-2, and LaCoO3-3 are 1, 2, and 3, respectively. As indicated by the aforementioned data, an increase in the molar ratio of La to Co results in an increased probability of La atoms being distributed on the catalyst surface.
Figure 4c displays that four oxygen species at 528.1, 530.2, 531.2 and 532.2 eV were observed in the O 1s spectra of LaCoO3-1, which were assigned to the lattice oxygen of metal oxides (O2−, denoted as OL), adsorbed oxygen species (O22−O−, denoted as OA), oxygen vacancy (Ov), and surface hydroxyl species (OH−, denoted as OOH), respectively [44].
Compared to LaCoO3-1, the electron binding energy of O 1s and La 3d in LaCoO3-2 and LaCoO3-3 has increased. The proportions of OL to total O atoms decrease sequentially in LaCoO3-1, LaCoO3-2 and LaCoO3-3, with proportions of 43.89%, 11.96%, and 8.83%, respectively. These results suggest that the increase in lanthanum oxide can promote the reduction of lattice oxygen, which is detrimental to catalytic oxidation reactions.
3.5. Investigation of Reaction Conditions for HCHO Degradation
3.5.1. Effects of Catalyst Types on HCHO Degradation
As shown in Figure 5, the HCHO degradation performance of LaCoO3 catalysts with different La/Co molar ratios was investigated. The study revealed that without a catalyst, the HCHO degradation rate reached only 40.1% after 10 min. The perovskite-type LaCoO3-1 exhibited the best catalytic effect, achieving a HCHO degradation rate of 95.7% within 4 min. As the lanthanum content increased, the HCHO degradation rate decreased significantly. When using LaCoO3-2 and LaCoO3-3 as catalysts for HCHO degradation, the degradation rates were 85.8% and 78.7%, respectively. The degradation of HCHO slowed after 2 min. When catalysts LaCoO3-1, LaCoO3-2 and LaCoO3-3 were used, respectively, the degradation rate of HCHO increased by only 2%, 3%, and 4% after 2 min. Under conditions where only catalysts LaCoO3-1, LaCoO3-2, LaCoO3-3 without PMS as the oxidant, HCHO degradation was negligible. Moreover, the removal rates of HCHO were not significantly different among the three types of catalysts (less than 5%), indicating that the prepared LaCoO3 catalyst exhibits weak adsorption capacity for HCHO.
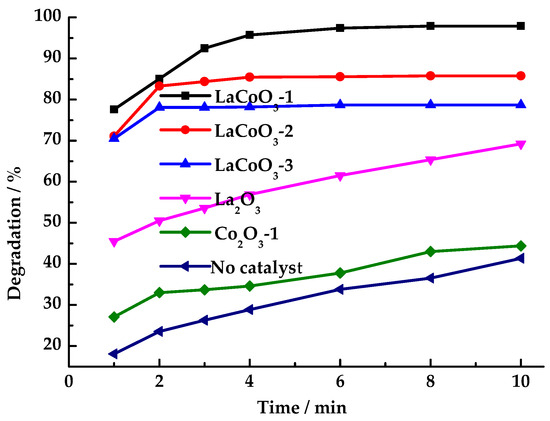
Figure 5.
Effects of catalyst types on degradation rate of HCHO (Reaction conditions: HCHO solution 1.086 mg/mL, PMS 26 mg/mL, catalyst 12 mg/mL, 25 °C).
Although the HCHO degradation performance of LaCoO3-1 is higher than LaCoO3-2 and LaCoO3-3, LaCoO3-2 and LaCoO3-3 exhibited greater catalytic activity toward PMS than LaCoO3-1 in the reaction system. Testing with starch-potassium iodide paper revealed that the reaction solution no longer turned the paper blue after 8 min, indicating that LaCoO3-2 and LaCoO3-3 catalyzed the production of SO4·− and ·OH from PMS at a faster rate. However, these species underwent quenching on the catalyst surface and did not fully participate in the HCHO oxidation reaction. Furthermore, the PMS concentration dropped to a low level after 2 min, reducing the efficiency of continued HCHO degradation. LaCoO3-1 catalyzed PMS to produce SO4·− and ·OH at a slower rate than LaCoO3-2 and LaCoO3-3. Moreover, the residence time of SO4·− and ·OH in the reaction system catalyzed by LaCoO3-1 is longer than that in systems catalyzed by LaCoO3-2 and LaCoO3-3, thus demonstrating higher catalytic efficiency than LaCoO3-2 and LaCoO3-3.
This phenomenon can be attributed to the fact that an increase in La content in LaCoO3 leads to enhanced adsorption of PMS on the surface of LaCoO3, thereby augmenting the catalytic activity of LaCoO3 for PMS. This, in turn, results in the acceleration of the rate of free radical generation by PMS, with a consequent quenching of a substantial number of active free radicals on the surface of LaCoO3.
When La2O3 and Co2O3 were used as catalysts, the degradation rates of HCHO were 69.2% and 44.4%, respectively. The reaction solution was detected by starch-potassium iodide paper, which was still blue after 120 min. This finding indicates that the presence of mixed Co3+/Co2+ oxidation states within the LaCoO3 perovskite structure is a significant contributing factor. Furthermore, the catalytic mechanism of LaCoO3 deviates from that of La2O3 and Co2O3. The instability of the O-O bond in PMS, which is prone to break after receiving electrons, is a key factor in this process. The electron transfer between Co2+ and Co3+ in LaCoO3 promotes the cleavage of O-O and generates SO4·− [45]. Subsequent studies selected LaCoO3-1 as the catalyst to investigate its catalytic performance in degrading HCHO by PMS.
As shown in Figure 6, the HCHO degradation rate peaked at 99.9% after 8 min when LaCoO3-1 was used at a dosage of 8 mg/mL. At a LaCoO3-1 dosage of 12 mg/mL, the HCHO degradation rate exceeded that of other dosages during the initial 4 min. However, after a period of 6 min, the observed degradation rate fell below that which was recorded at the 8 mg/mL dosage. This finding suggests that when the LaCoO3-1 dosage is less than 12 mg/mL, increasing the dosage accelerates the rate at which LaCoO3-1 activates PMS to produce SO4·− and ·OH, thereby enhancing HCHO degradation efficiency. However, as time progresses, the PMS concentration decreases, reducing the generation rate of SO4·− and ·OH and consequently lowering the HCHO degradation efficiency. When the LaCoO3-1 dosage exceeds1 2 mg/mL, LaCoO3-1 catalyzed PMS to produce SO4·− and ·OH more rapidly. Some SO4·− and ·OH fail to react with HCHO molecules before undergoing quenching [46]. In other words, the self-quenching reaction between the excess free radicals leads to a decrease in the HCHO degradation rate.
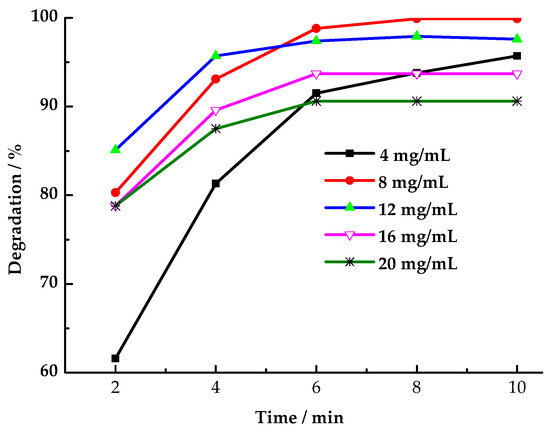
Figure 6.
Effect of LaCoO3-1 dosage on degradation rate of HCHO (Reaction conditions: HCHO solution 1.086 mg/mL, PMS 26 mg/mL, 25 °C).
3.5.2. Effect of PMS Dosage on HCHO Degradation
As shown in Figure 7, the HCHO degradation rate increases with the PMS dosage. This phenomenon can be attributed to the observation that increasing the dosage of PMS results in an elevation of the concentration of SO4·− and ·OH generated by LaCoO3-1 catalysis within the reaction system. Consequently, this increase in concentration leads to an enhancement in the probability of interaction between SO4·− and ·OH with HCHO molecules. After 6 min, the concentration of PMS in the reaction system becomes negligible, leading to the imminent cessation of the HCHO degradation reaction. It has been demonstrated that, at a constant PMS dosage, an extension of the reaction time beyond 6 min has a negligible effect on the degradation rate. When the oxidant PMS was replaced with an equivalent molar amount of H2O2 (43 μL) while maintaining other reaction conditions, the HCHO degradation rates at 2, 4, 6, 8, and 10 min were 14.5%, 27.6%, 30.1%, 30.8%, and 30.8%, respectively. These degradation rates were found to be considerably lower than those observed with PMS as the oxidant. Given the comparable oxidative capabilities of SO4·− and ·OH, the significant disparity in degradation rates suggests one of two possibilities. The catalyst LaCoO3-1 may exhibit no catalytic activity toward hydrogen peroxide, or the ·OH generated by the catalyst may not participate in the oxidation reaction for HCHO degradation. Monitoring hydrogen peroxide concentration changes in the solution using starch-potassium iodide paper revealed no hydrogen peroxide residue after 6 min. This indicates that LaCoO3-1 catalyzed the generation of ·OH and ·OOH from hydrogen peroxide [47]. However, these radicals persisted in the reaction system for an insufficient duration to participate in the oxidation of HCHO. Instead, they combined on the catalyst surface, forming water (lacking oxidizing capacity) and oxygen (with weaker oxidizing capacity), which rapidly escaped from the reaction system. This also demonstrates that SO4·− exhibits a longer lifetime than ·OH in this reaction system [48]. The long existence time of SO4·− in the reaction system increases its contact opportunity with HCHO molecules, thus improving the degradation rate.
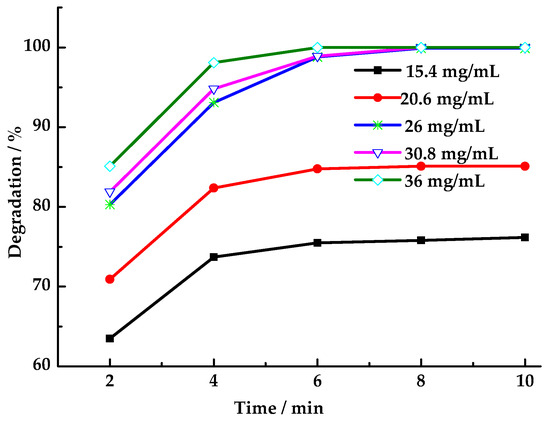
Figure 7.
Effect of PMS dosage on degradation rate of HCHO (Reaction conditions: HCHO solution 1.086 mg/mL, catalyst 8 mg/mL, 25 °C).
3.5.3. Effect of HCHO Concentration on HCHO Degradation
The applicable HCHO concentration of the catalyst and oxidant was investigated by changing the HCHO concentration with an optimized amount of catalyst and oxidant. As shown in Figure 8, when the HCHO concentration was 0.543 mg/mL, 1.086 mg/mL, 1.629 mg/mL, the degradation rates of HCHO were all above 90%. Consequently, at a constant catalyst concentration and molar ratio of PMS/HCHO, a decrease in HCHO concentration results in a reduction in PMS concentration within the system. While this enhancement of the catalyst’s efficiency in activating PMS to generate radicals is advantageous, it concomitantly diminishes the contact opportunities between PMS and HCHO, resulting in lower degradation rates. Conversely, elevated HCHO concentrations have been observed to concomitantly increase PMS concentrations within the system. However, when the amount of catalyst remains constant, the catalyst’s capacity to catalyze PMS activation and generate free radicals is limited, resulting in incomplete HCHO degradation. Consequently, to attain elevated HCHO degradation rates across a broad concentration range, the quantity of both the catalyst and oxidant in the reaction system must be adjusted concurrently.
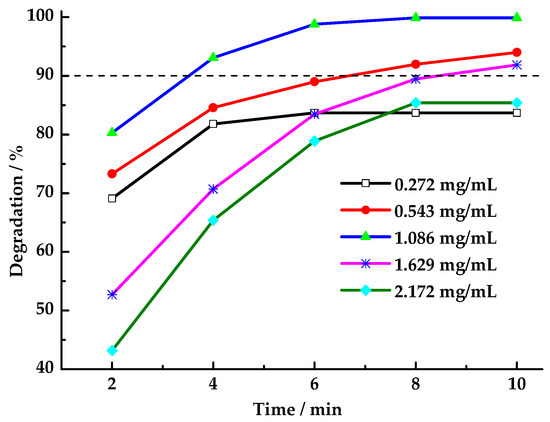
Figure 8.
Effect of HCHO concentration on degradation rate of HCHO (Reaction conditions: n(PMS)/n(HCHO) = 2.5, catalyst 8 mg/mL, 25 °C).
3.5.4. Effect of pH Value on HCHO Degradation
The acidity or alkalinity of HCHO wastewater is uncertain, and the pH value of the solution will affect the removal efficiency of pollutants. Thus, the catalytic oxidation system must perform effectively across a broad pH range. As shown in Figure 9, the performance of LaCoO3-1 in activating PMS for HCHO degradation was investigated across a range of solutions with different pH values. Results indicate that under near-neutral conditions (pH = 3–11), HCHO degradation rates exceeded 90% within 10 min, with neutral conditions yielding the highest degradation rate. As acidity or alkalinity increased, the HCHO degradation rate decreased. The presence of strong acids and bases in the reaction medium was found to be deleterious to the degradation process. Under alkaline conditions, the degradation rate declined because SO4·− reacted with OH− to form hydroxyl radicals (·OH), which have a relatively short lifetime in the solution system. Under acidic conditions, pH changes may cause varying amounts of lanthanum or cobalt ions to leach into the solution, disrupting the catalyst structure. Overall, LaCoO3-1-catalyzed PMS degradation of HCHO exhibits a broad pH tolerance range.
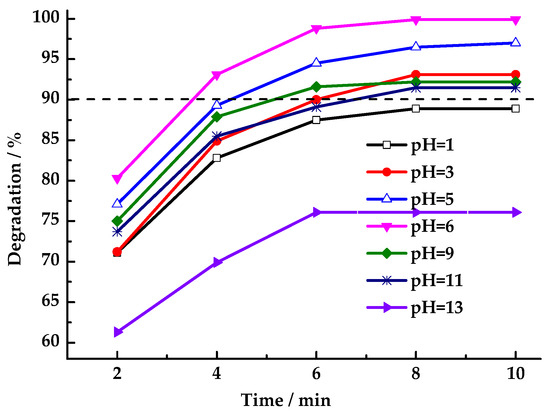
Figure 9.
Effect of pH value on degradation rate of HCHO (Reaction conditions: n(PMS)/n(HCHO) = 2.5, catalyst 8 mg/mL, 25 °C).
3.5.5. Reusability and Stability of the Perovskite LaCoO3-1
LaCoO3-1 was subjected to four repeated catalytic degradation experiments of HCHO, and the degradation rates of HCHO were 98%, 96%, 94%, and 91%, respectively. After four cycles, the degradation rate remains above 90%, and the XRD pattern of the recycled LaCoO3-1 shows that the perovskite structure has not changed, indicating that the perovskite LaCoO3-1 has excellent reusability. The gradual decrease in degradation rate may result from minor loss of active components within the catalyst. Since the highest stable oxidation state of cobalt is +3, it is possible that part of Co2+ in the LaCoO3-1 is converted to Co3+ as the catalyst is used, thus the catalytic efficiency is reduced [49].
4. Discussion
To further investigate the HCHO degradation mechanism in the LaCoO3-1/PMS system. A series of experiments were conducted with the use of scavengers, the objective of which was to ascertain whether the reaction in question was of a radical nature. Tert-butanol was considered in order to determine the contribution of radicals in the degradation of HCHO. After adding t-butanol (1 mL), the HCHO degradation efficiency decreased significantly (67.3%), whereas adding an equal volume of deionized water had almost no effect on the HCHO degradation efficiency (98.7%). A plausible mechanism is proposed to explain the catalytic degradation of HCHO by LaCoO3-1 and PMS system. The LaCoO3-1 continuously activates PMS through reversible redox reactions of Co2+/Co3+, efficiently producing SO4·− and ·OH, and acting on HCHO. PMS was adsorbed onto the LaCoO3-1 surface for reaction, but it also accelerated electron transfer and promoted Co2+ regeneration. The initial reaction of Co2+ and PMS results in the formation of SO4·−, OH− and Co3+, with the concomitant reduction of Co3+ to Co2+. Furthermore, SO4·−and OH− can react to form ·OH, thus forming a Co2+/Co3+ cycle, ensuring sufficient Co2+ to continuously catalyze PMS during HCHO degradation and improving degradation efficiency.
Perovskite-type LaCoO3-1 has been shown to catalyze the PMS reaction, thereby generating radicals. In addition, it has been observed to catalyze the decomposition of hydrogen peroxide. However, they cannot catalyze the oxidative degradation of HCHO by hydrogen peroxide. A substantial advancement would be achieved by adjusting the perovskite structure and elemental composition to achieve catalytic oxidative degradation of HCHO by hydrogen peroxide. The present state of affairs indicates that reaction systems employing hydrogen peroxide as an oxidant, photocatalytic systems, and electrocatalytic systems demonstrate inadequate degradation efficiency for HCHO (Table 1).

Table 1.
Comparison of the catalytic oxidative degradation of HCHO in water.
Although the catalytic activity of lanthanum cobalt oxide for HCHO degradation decreases with an increase in the La/Co molar ratio, its catalytic activity for PMS decomposition increases with an increase in the La/Co molar ratio. However, the catalytic effect of lanthanum oxide on PMS decomposition is extremely weak, and the structure and composition of lanthanum cobalt oxides still warrant in-depth investigation.
5. Conclusions
When the concentration of HCHO was 1.086 mg/mL (5 mL), the dosage of LaCoO3-1 was 8 mg/mL, and n(PMS)/n(HCHO) = 2.5, the removal rate of HCHO exceeded 90% across a pH range of 3 to 11 after 10 min at 25 °C. A comparison of the present report with previous reports concerning the catalytic oxidative degradation of HCHO in water (Table 1) reveals the superior catalytic activity of the perovskite-type LaCoO3-activated PMS system for HCHO degradation at room temperature. This reaction occurs without the need for light or electricity and results in a reduced reaction time.
XRD, XPS and TEM analysis confirmed the formation of perovskite structure. The catalytic HCHO degradation process of perovskite-type LaCoO3 is significantly different from that of La2O3 and Co2O3. The cooperation between La and Co significantly increased the degradation of HCHO. The complete degradation of HCHO in the research depended on the Co2+ on the catalyst surface. The introduction of La2O3 significantly increased the oxygen vacancy and Co2+ content.
Unfortunately, LaCoO3 can only catalyze the decomposition of hydrogen peroxide, but cannot provide free radicals for reaction with HCHO. This limits the use of hydrogen peroxide as a green oxidant.
Author Contributions
Conceptualization, Q.M.; methodology, Q.M. and Q.G.; validation, S.L., T.L., Z.F., B.M. and Y.Z.; formal analysis, Q.G.; investigation, S.L. and T.L.; resources, Q.M.; data curation, Q.G. and S.L.; writing—original draft preparation, Q.G. and S.L.; writing—review and editing, Q.M.; project administration, Q.M.; funding acquisition, Q.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by National Natural Science Fund of China, grant number 22072105.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The original contributions presented in this study are included in the article. Further inquiries can be directed to the corresponding author.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Ding, N.; Li, Z.; Hao, Y.; Yang, X. A new amine moiety-based near-infrared fluorescence probe for detection of formaldehyde in real food samples and mice. Food Chem. 2022, 384, 132426. [Google Scholar] [CrossRef]
- Xu, Q.; Sun, H.; Ren, M.; Kong, F. A novel cellulose-based fluorescent probe for the quantitative detection of HCHO in real food samples and living cells. Ind. Crops Prod. 2023, 204, 117406. [Google Scholar] [CrossRef]
- Yamada, M.; Funaki, S.; Miki, S. Formaldehyde interacts with RNA rather than DNA: Accumulation of formaldehyde by the RNA-inorganic hybrid material. Int. J. Biol. Macromol. 2019, 122, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Tong, Z.; Han, C.; Luo, W.; Wang, X.; Li, H.; Luo, H.; Zhou, J.; Qi, J.; He, R. Accumulated hippocampal formaldehyde induces age-dependent memory decline. Age 2013, 35, 583–596. [Google Scholar] [CrossRef]
- Suresh, S.; Kante, K.; Fini, E.H.; Bandosz, T.J. Combination of alkalinity and porosity enhances formaldehyde adsorption on pig manure -derived composite adsorbents. Micropor. Mesopor. Mat. 2019, 286, 155. [Google Scholar] [CrossRef]
- Yang, X.; Zhao, H.; Qu, Z.; He, M.; Tang, Z.; Lai, S.; Wang, Z. The effect of oxygen-containing functional groups on formaldehyde adsorption in solution on carbon surface: A density functional theory study. J. Environ. Chem. Eng. 2021, 9, 105987. [Google Scholar] [CrossRef]
- Yang, T.; Liu, Y.; Xia, G.; Zhu, X.; Zhao, Y. Degradation of formaldehyde and methylene blue using wood-templated biomimetic TiO2. J. Clean. Prod. 2021, 329, 129726. [Google Scholar] [CrossRef]
- Dai, M.; Li, F.; Zhang, J.; Shi, Q.; Wu, Y.; Kong, Q. Treatment of formaldehyde-containing wastewater and power generation by constructed wetland–microbial fuel cells enhanced by formaldehyde-degrading bacteria. J. Water Process Eng. 2024, 59, 104984. [Google Scholar] [CrossRef]
- Nisa, T.; Khokhar, W.A.; Imran, U.; Khokhar, S.A.; Soomro, N. Electrochemical treatment of wastewater containing urea-formaldehyde and melamine-formaldehyde. Chemosphere 2023, 338, 139587. [Google Scholar] [CrossRef] [PubMed]
- Bello, M.M.; Raman, A.A.; Asghar, A. A Review on Approaches for Addressing the Limitations of Fenton Oxidation for Recalcitrant Wastewater Treatment. Process Saf. Environ. Prot. 2019, 126, 119–140. [Google Scholar] [CrossRef]
- Ma, Q.; Hao, Y.; Xue, Y.; Niu, Y.; Chang, X. Removal of Formaldehyde from Aqueous Solution by Hydrogen Peroxide. J. Water Chem. Technol. 2022, 44, 297–303. [Google Scholar] [CrossRef]
- Mutia, A.S.; Ariyanto, T.; Prasetyo, I. Ciprofloxacin Removal from Simulated Wastewater Through a Combined Process of Adsorption and Oxidation Processes Using Fe/C Adsorbent. Water Air Soil Pollut. 2022, 233, 146. [Google Scholar] [CrossRef]
- Isinkaralar, K.; Gullu, G.; Turkyilmaz, A. Experimental study of formaldehyde and BTEX adsorption onto activated carbon from lignocellulosic biomass. Biomass Convers. Bior. 2023, 13, 4279–4289. [Google Scholar] [CrossRef]
- Henda, M.B.; Sadon, S.H.; Abdelmalek, Z.; Li, Z.; Le, Q.H. Removal of formaldehyde pollutant from petroleum industry wastewaters by polymers: A molecular dynamics simulation. Eng. Anal. Bound. Elem. 2023, 151, 400–405. [Google Scholar] [CrossRef]
- Vakylabad, A.B. Treatment of highly concentrated formaldehyde effluent using adsorption and ultrasonic dissociation on mesoporous copper iodide (CuI) nano-powder. J. Environ. Manag. 2021, 285, 112085. [Google Scholar] [CrossRef] [PubMed]
- Si, Z.; Song, X.; Wang, Y.; Cao, X.; Zhao, Y.; Ge, X.; Wang, Y. Assessment and Optimization of Formaldehyde Removal Using Tidal Flow Constructed Wetlands. Pol. J. Environ. Stud. 2021, 30, 987–992. [Google Scholar] [CrossRef]
- Chao, L.; Xiao, F.C.; Qin, Q.Z.; Chen, Y.Z.; Zai, X.L.; Jian, R.N.; Zhuang, H.; Qian, X.; Zhan, W.T.; Wei, T.M. Novel porous perovskite composite CeO2@LaMnO3/3 DOM SiO2 as an effective catalyst for activation of PMS toward oxidation of urotropine. Adv. Powder Technol. 2022, 33, 103802. [Google Scholar] [CrossRef]
- Isac-Gutul, T.; Tutovan, E.; Nika, D.L. Photo-Degradation of Dexamethasone through Radical-Based Advanced Oxidation Processes Using UV/H2O2 and Fe2+/UV/H2O2 Systems. Phys. Solid State 2025, 67, 292–301. [Google Scholar] [CrossRef]
- Santana, R.M.R.; Napoleao, D.C.; Dos Santos Júnior, S.G. Photo-Fenton process under sunlight irradiation for textile wastewater degradation: Monitoring of residual hydrogen peroxide by spectrophotometric method and modeling artificial neural network models to predict treatment. Chem. Pap. 2021, 75, 2305–2316. [Google Scholar] [CrossRef]
- Li, Y. Construction of WO3–visible light–H2O2 advanced oxidation system for degradation of organic pollutant. J. Mater. Sci. Mater. Electron. 2024, 35, 2070. [Google Scholar] [CrossRef]
- Wang, L.L.; Lan, X.; Peng, W.Y.; Wang, Z.H. Uncertainty and misinterpretation over identification, quantification and transformation of reactive species generated in catalytic oxidation processes: A review. J. Hazard Mater. 2021, 408, 124436. [Google Scholar] [CrossRef]
- Fama, Y.; Imae, T. Catalytic oxidation of formaldehyde in water by calcium phosphate-based Pt composites. RSC Adv. 2015, 5, 15944. [Google Scholar] [CrossRef]
- Zoh, K.-D.; Stenstrom, M.K. Fenton oxidation of hexahydro-1, 3, 5-trinitro-1, 3, 5-triazine (RDX) and octahydro-1, 3, 5, 7-tetrani-tro-1, 3, 5, 7-tetrazocine (HMX). Water Res. 2002, 36, 1331–1341. [Google Scholar]
- Chai, G.; Xi, H.; Qian, Y. Performance optimization and mechanism of HMX degradation by Fenton oxidation method. J Iran Chem. Soc. 2025, 22, 1009–1018. [Google Scholar] [CrossRef]
- Jiang, C.; Xing, B.; Yang, L. Fe–N co-doped carbon derived from one-step pyrolysis modified chitosan for activating H2O2 to degrade organic wastewater. J. Mater. Sci. 2024, 59, 9491–9501. [Google Scholar] [CrossRef]
- Ma, Q.; Shi, S.; Yang, F.; Zhang, X. Removal of formaldehyde in water with low concentration of hydrogen peroxide catalyzed by lanthanum-silicon oxide composite. Desalin. Water Treat. 2023, 300, 101–106. [Google Scholar] [CrossRef]
- Imohiosen, F.A.; Ofudje, E.A.; Al-Ahmary, K.M. Pharmaceutical effluent degradation using hydrogen peroxide-supported zerovalent iron nanoparticles catalyst. Sci. Rep. 2024, 14, 23957. [Google Scholar] [CrossRef]
- Xiong, Y.; Zhou, T.; Bao, J. Degradation mechanism of Bisphenol S via hydrogen peroxide/persulfate activated by sulfidated nanoscale zero valent iron. Environ. Sci. Pollut. Res. 2023, 30, 83545–83557. [Google Scholar] [CrossRef]
- Khan, A.; Yasin, S.; Mahmood, H. Mono-ethanolamine breakdown by UV/hydrogen peroxide via MEA photolysis: Kinetics, energy rate/order and degradation efficiency for mono-ethanolamine wastewater treatment. Braz. J. Chem. Eng. 2025, 42, 1079–1090. [Google Scholar] [CrossRef]
- Yu, C.; Liu, S.; Hu, W. Preparation of ferric hydroxide chelated glass fiber modified with dopamine chemistry for effective degradation of organic dyes by heterogeneous Fenton method. Res. Chem. Intermediat. 2025, 51, 2089–2104. [Google Scholar] [CrossRef]
- Athikaphan, P.; Wongsanga, K.; Klanghiran, S. Degradation of formaldehyde by photo-Fenton process over n-ZVI/TiO2 catalyst. Environ. Sci. Pollut. Res. 2023, 30, 90397–90409. [Google Scholar] [CrossRef]
- Bi, F.; Zhou, B.; Li, R. Synthesis of novel ZnO/NiFe2O4 heterostructures photocatalysts for photocatalytic and photo-Fenton removal of dyes. J. Mater. Sci. Mater. Electron. 2025, 36, 78. [Google Scholar] [CrossRef]
- Oh, W.D.; Dong, Z.; Ronn, G.; Lim, T.T. Surface-active bismuth ferrite as superior peroxymonosulfate activator for aqueous sulfamethoxazole removal: Performance, mechanism and quantification of sulfate radical. J. Hazard Mater. 2017, 325, 71–81. [Google Scholar] [CrossRef]
- Nie, Y.; Zhou, H.; Tian, S.; Tian, X.; Yang, C.; Li, Y.; Tian, Y. Anionic ligands driven efficient ofloxacin degradation over LaMnO3 suspended particles in water due to the enhanced peroxymonosulfate activation. Chem. Eng. J. 2022, 427, 130998. [Google Scholar] [CrossRef]
- Sojic Merkulov, D.; Vlazan, P.; Poienar, M.; Bognar, S.; Ianasi, C.; Sfirloaga, P. Sustainable removal of 17α-ethynylestradiol from aqueous environment using rare earth doped lanthanum manganite nanomaterials. Catal. Today 2023, 424, 113746. [Google Scholar] [CrossRef]
- Pang, X.; Guo, Y.; Zhang, Y.; Xu, B.; Qi, F. LaCoO3 perovskite oxide activation of peroxymonosulfate for aqueous 2-phenyl-5-sulfobenzimidazole degradation: Effect of synthetic method and the reaction mechanism. Chem. Eng. J. 2016, 304, 897–907. [Google Scholar] [CrossRef]
- Jain, A.; Pal, S.L.; Jaiswal, Y.; Srivastava, S. Designing a feasible phenol destruction process using LaM1−xCuxO3 (M = Co, Cr, Fe) perovskites as heterogeneous Fenton-like catalysts. Arab. J. Sci. Eng. 2022, 47, 5777–5796. [Google Scholar] [CrossRef]
- Ma, Q.G.; Huo, P.C.; Wang, K.S.; Yuan, Y.; Bai, S.J.; Zhao, C.T.; Li, W.Z. Preparation of Perovskite-Type LaMnO3 and Its Catalytic Degradation of Formaldehyde in Wastewater. Molecules 2024, 29, 3822. [Google Scholar] [CrossRef]
- Scelfo, S.; Geobaldo, F.; Pirone, R.; Russo, N. Catalytic wet air oxidation of D-glucose by perovskite type oxides (Fe, Co, Mn) for the synthesis of value-added chemicals. Carbohyd. Res. 2022, 514, 108529. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Peng, H. One-pot synthesis of La2O3-decorated Mg-Al oxides nanosheets for solar-light driven photocatalytic activity. Colloids Surf. A 2020, 604, 125316. [Google Scholar] [CrossRef]
- Li, C.; Li, Z.; Chen, X.; Zhang, Q.; Zhang, C.; Yue, X.; Xing, Q.; Tian, Z.; Ma, W.; Qi, H. Three-dimensional ordered macroporous eria-lanthanum cobaltate composite as efficient catalyst to activate peroxymonosulfate for N,N-dimethylformamide degradation. J. Mater. Sci. 2022, 57, 16280–16300. [Google Scholar]
- Zhong, Z.; Pan, J.; Li, M.; Wang, J.; Jiang, S.; Lin, J.; Xiong, Z.; Xie, A.; Luo, S. Enhanced electrocatalytic performance of LaCo1−xMnxO3 perovskite catalyst for glycerol oxidation. MRS Commun. 2022, 12, 786–793. [Google Scholar] [CrossRef]
- Wu, Y.H.; Liu, H.; Li, G.Y.; Jin, L.J.; Ou, X.M.; Dong, L.H.; Jin, G.Z.; Li, B. Tuning composition on B sites of LaM0.5Mn0.5O3 (M = Cu Co, Fe, Ni, Cr) perovskite catalysts in NOx efficient reduction. Appl. Surf. Sci. 2020, 508, 145158. [Google Scholar] [CrossRef]
- Chen, F.; Yan, H.; Wang, J.; Wang, H.; Sun, Y.; Chen, X.; Lu, W.; Chen, W. High-efficient catalytic ozonation for degradation of nitrobenzene in water with Ce-doped LaCoO3 catalyst. J. Mater. Sci. 2024, 59, 3406–3420. [Google Scholar] [CrossRef]
- Chen, X.; Qiao, X.; Wang, D. Kinetics of oxidative decolorization and mineralization of Acid Orange 7 by dark and photoassisted Co 2+ -catalyzed peroxymonosulfate system. Chemosphere 2007, 67, 802–808. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhao, Z.W.; Shao, P.H. Activation of peroxymonosulfate with magnetic Fe3O4 -MnO2 core-shell nanocomposites for 4-chlorophenol degradation. Chem. Eng. J. 2015, 262, 854–861. [Google Scholar] [CrossRef]
- Brillas, E.; Sires, I.; Oturan, M.A. Electro-Fenton process and related electrochemical technologies based on Fenton’s reaction chemistry. Chem. Rev. 2009, 109, 6570–6631. [Google Scholar] [CrossRef]
- Lin, K.Y.A.; Lin, T.Y. Degradation of Acid Azo Dyes Using Oxone Activated by Cobalt Titanate Perovskite. Water Air Soil Pollut. 2018, 229, 10. [Google Scholar] [CrossRef]
- Wei, Y.; Ni, L.; Li, M.; Zhao, J. Acid treated Sr-substituted LaCoO3 perovskite for toluene oxidation. Catal. Commun. 2021, 155, 106314. [Google Scholar] [CrossRef]
- Mohammadifard, Z.; Saboori, R.; Mirbagheri, N.S.; Sabbaghi, S. Heterogeneous photo-Fenton degradation of formaldehyde using MIL-100(Fe) under visible light irradiation. Environ. Pollut. 2019, 251, 783–791. [Google Scholar] [CrossRef]
- Moussavia, G.; Yazdanbakhshb, A.; Heidarizad, M. The removal of formaldehyde from concentrated synthetic wastewater using O3/MgO/H2O2 process integrated with the biological treatment. J. Hazard. Mater. 2009, 171, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Guimarães, J.R.; Farah, C.R.T.; Maniero, M.G.; Fadini, P.S. Degradation of formaldehyde by advanced oxidation processes. J. Environ. Manag. 2012, 107, 96–101. [Google Scholar] [CrossRef]
- Tan, Y.; Yin, C.; Zheng, S.; Di, Y.; Sun, Z.; Li, C. Design and controllable preparation of Bi2MoO6/attapulgite photocatalyst for the removal of tetracycline and formaldehyde. Appl. Clay Sci. 2021, 215, 106319. [Google Scholar] [CrossRef]
- Haddad, F.A.; Moussavi, G.; Moradi, M. Advanced oxidation of formaldehyde in aqueous solution using thechemical-less UVC/VUV process: Kinetics and mechanism evaluation. J. Water Process Eng. 2019, 27, 120–125. [Google Scholar] [CrossRef]
- Talaiekhozani, M.; Salari, M.; Talaei, M.; Bagheri, M.; Eskandari, Z. Formaldehyde removal from wastewater and air by using UV, ferrate(VI) and UV/ferrate(VI). J. Environ. Manag. 2016, 184, 204–209. [Google Scholar] [CrossRef] [PubMed]
- Yuan, D.; Tian, L.; Gu, D.; Shen, X.; Zhu, L.; Wu, H.; Wang, B. Fast and efficient oxidation of formaldehyde in wastewater via the Solar Thermal Electrochemical Process tuned by thermo electrochemistry. J. Clean. Prod. 2017, 156, 310–316. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).