
Antidotes in Clinical Toxicology—Critical Review

 ,
,  , , and
, , and
Abstract
1. Introduction
2. Twenty per Cent or Fat Emulsion (20%) (Lipid Rescue)
3. Acetylcholinesterase Reactivators
4. Antidotes for Metal Intoxications
4.1. BAL
4.2. Succimer
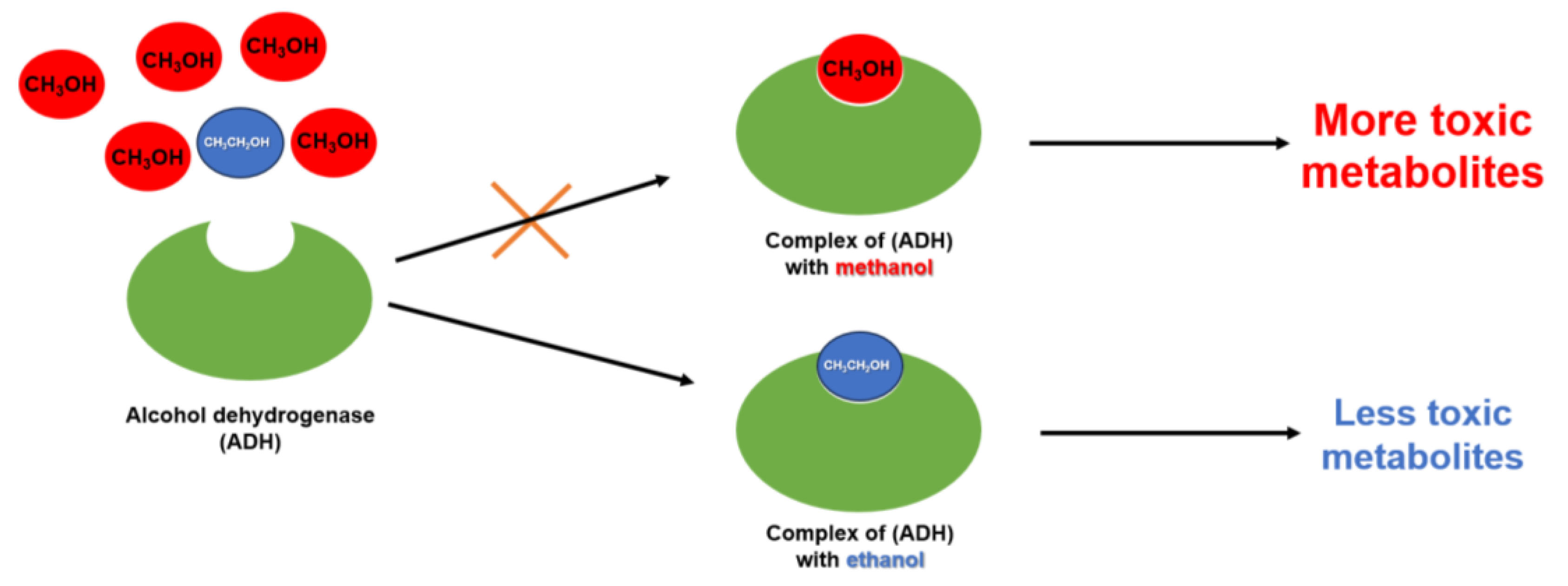
5. Antidotes for Methanol and Ethylene Glycol Poisoning
5.1. Ethanol
5.2. Fomepizole
5.3. Folinic Acid
6. Atropine
- Respiratory tract—relaxation of smooth muscles, increasing lumen of the bronchi, reducing mucus secretion.
- Heart—antivagal action on the heart; it increases heart rate and cardiac output and affects the sinoatrial node (to a lesser extent the atrioventricular node), accelerating nodal conduction and shortening the PQ interval.
- Digestive tract—reduces gastrointestinal motility and acts as an antiemetic.
- Premedication before or immediately during the procedure.
- During resuscitation until the heart rhythm is stabilised.
- In bradycardia or arrhythmias.
- Reversed neuromuscular block.
- Auxiliary in spastic conditions of smooth muscle in the abdominal cavity (biliary and renal colic).
- In radiological diagnostics, when it is desired to relax the smooth muscle and slow down the intestinal transit.
- In ophthalmology prior to endoscopy.
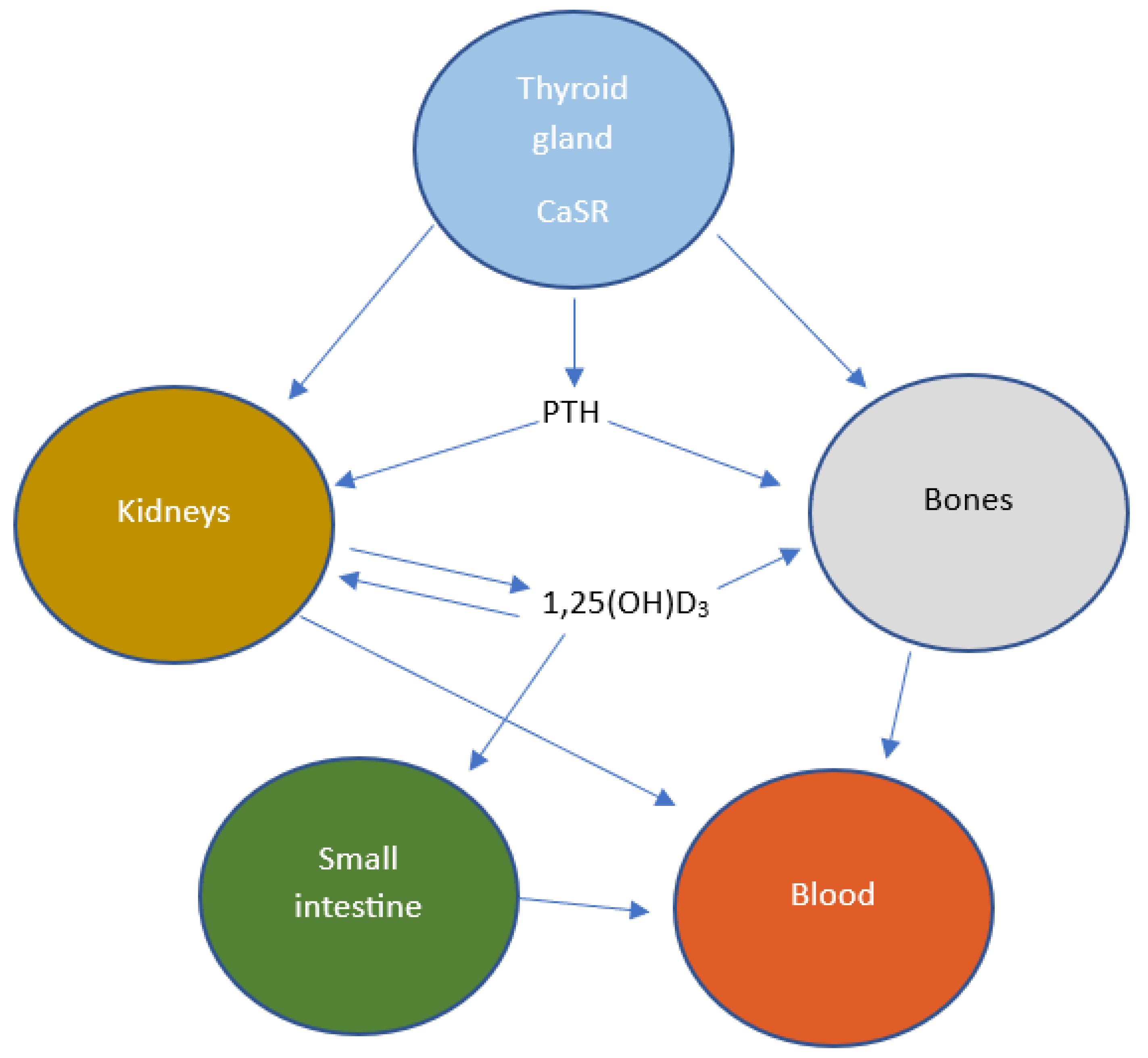
7. Calcium Chloride
8. Dantrolene
9. Diphenhydramine
10. Flumazenil
11. Glucagon
12. Hydroxocobalamin
13. Insulin, Glucose (HIET)
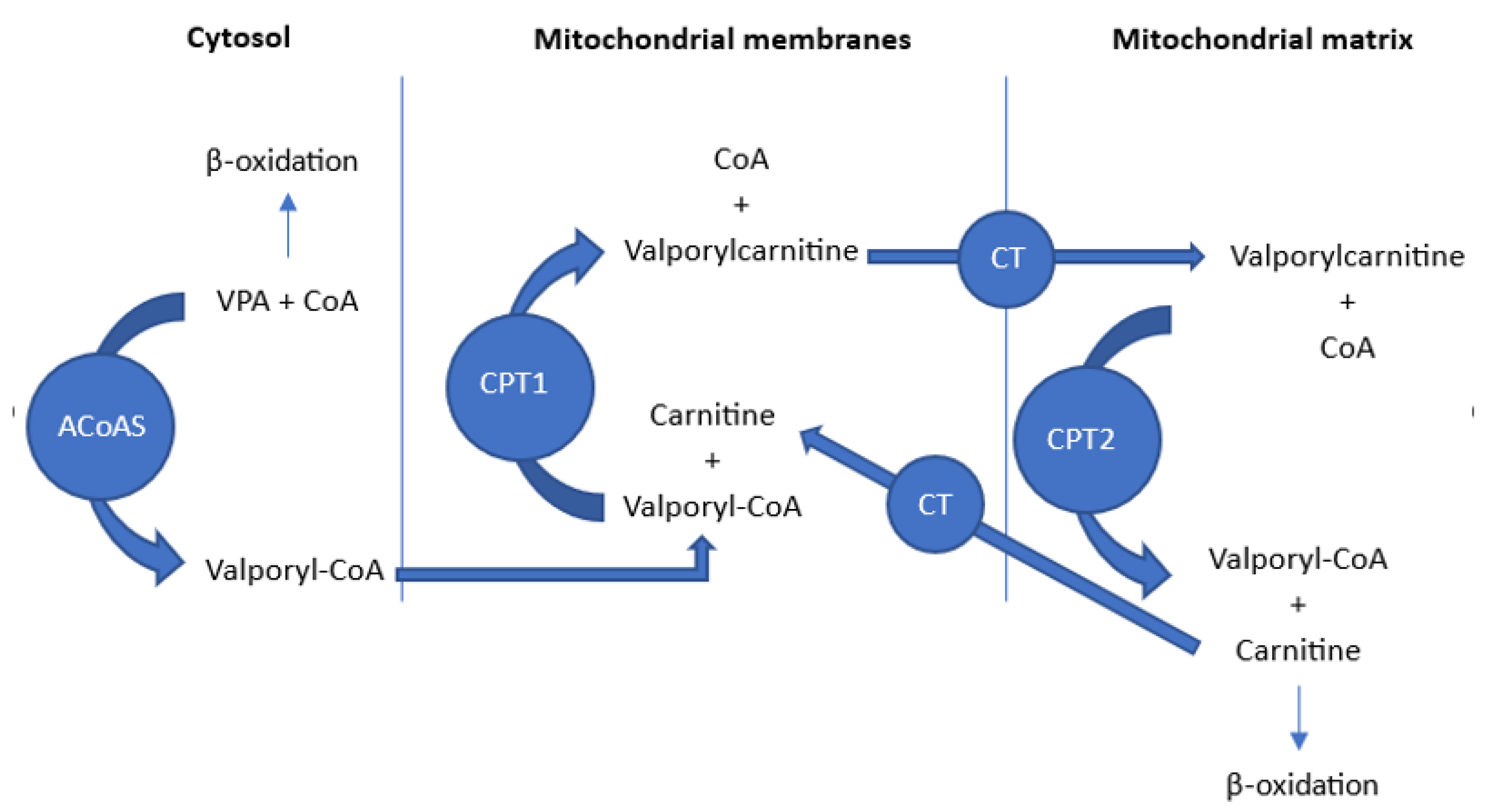
14. L-Carnitine
15. Methylene Blue
16. N-Acetylcysteine
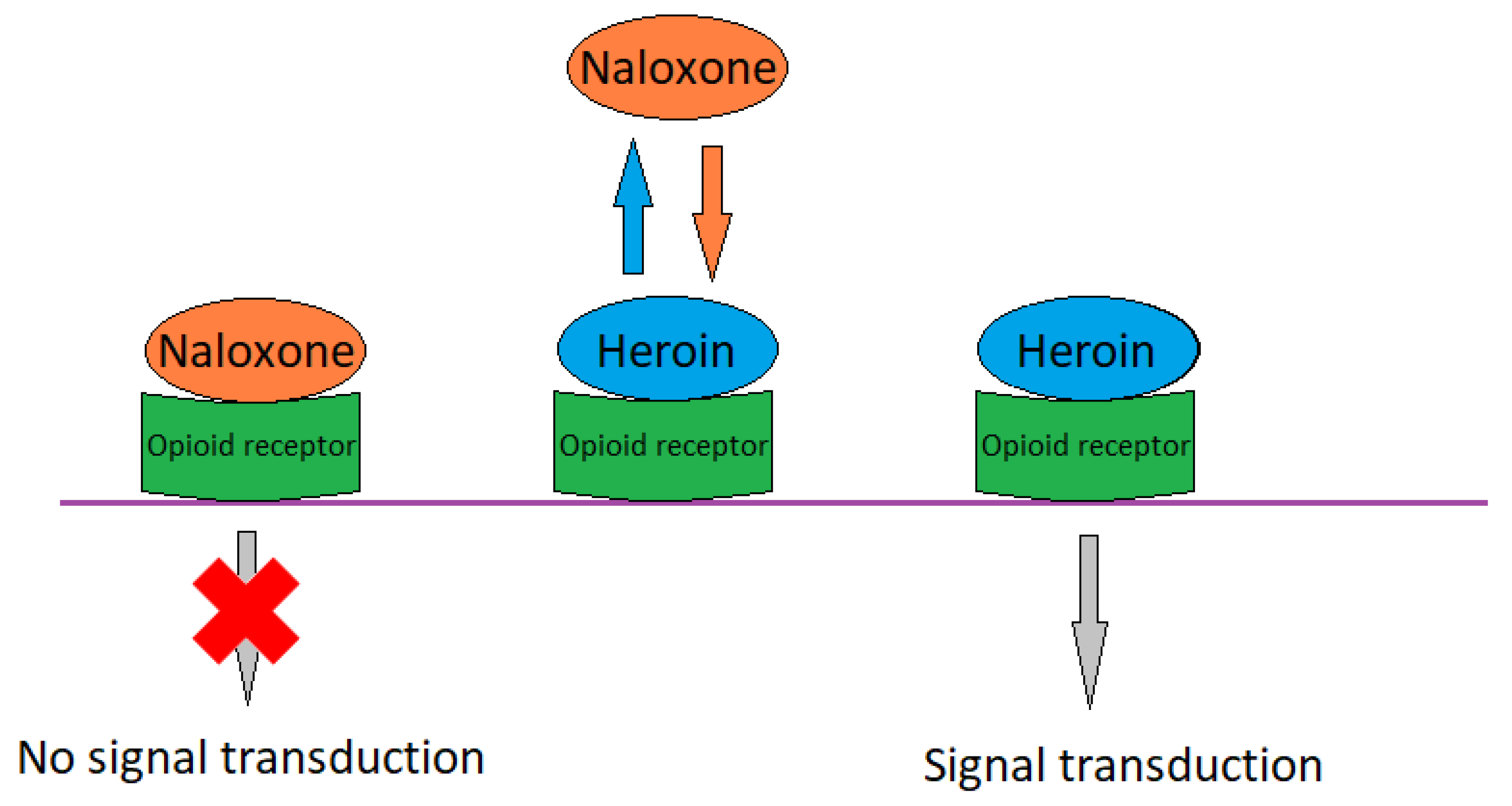
17. Naloxone
18. Octreotide
19. Oxygen
20. Physostigmine
21. Prussian Blue
22. Protamine Sulphate
23. Pyridoxine
24. Starch
25. Vitamin K
26. Conclusions
Funding
Conflicts of Interest
References
- World Health Organization. Guidelines for Establishing a Poison Centre; World Health Organization: Geneva, Switzerland, 2020; ISBN 978-92-4-000952-3. [Google Scholar]
- Smollin, C.G. Toxicology: Pearls and Pitfalls in the Use of Antidotes. Emerg. Med. Clin. N. Am. 2010, 28, 149–161. [Google Scholar] [CrossRef] [PubMed]
- De Garbino, J.P.; Haines, J.A.; Jacobsen, D.; Meredith, T. Evaluation of Antidotes: Activities of the International Programme on Chemical Safety. J. Toxicol. Clin. Toxicol. 1997, 35, 333–343. [Google Scholar] [CrossRef]
- Yin, H.; Zhang, X.; Wei, J.; Lu, S.; Bardelang, D.; Wang, R. Recent Advances in Supramolecular Antidotes. Theranostics 2021, 11, 1513–1526. [Google Scholar] [CrossRef] [PubMed]
- Anez-Bustillos, L.; Dao, D.T.; Baker, M.A.; Fell, G.L.; Puder, M.; Gura, K.M. Intravenous Fat Emulsion Formulations for the Adult and Pediatric Patient: Understanding the Differences. Nutr. Clin. Pract. 2016, 31, 596–609. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, G. Lipid Emulsion Infusion Rescues Dogs from Bupivacaine-Induced Cardiac Toxicity. Reg. Anesth. Pain Med. 2003, 28, 198–202. [Google Scholar] [CrossRef]
- Sepulveda, E.A.; Pak, A. Lipid Emulsion Therapy. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Neal, J.M.; Barrington, M.J.; Fettiplace, M.R.; Gitman, M.; Memtsoudis, S.G.; Mörwald, E.E.; Rubin, D.S.; Weinberg, G. The Third American Society of Regional Anesthesia and Pain Medicine Practice Advisory on Local Anesthetic Systemic Toxicity: Executive Summary 2017. Reg. Anesth. Pain Med. 2018, 43, 113–123. [Google Scholar] [CrossRef]
- Geyer, R.P. Parenteral Nutrition. Physiol. Rev. 1960, 40, 150–186. [Google Scholar] [CrossRef]
- Gold, V. (Ed.) The IUPAC Compendium of Chemical Terminology: The Gold Book, 4th ed.; International Union of Pure and Applied Chemistry (IUPAC): Research Triangle Park, NC, USA, 2019. [Google Scholar]
- Mercey, G.; Verdelet, T.; Renou, J.; Kliachyna, M.; Baati, R.; Nachon, F.; Jean, L.; Renard, P.-Y. Reactivators of Acetylcholinesterase Inhibited by Organophosphorus Nerve Agents. Acc. Chem. Res. 2012, 45, 756–766. [Google Scholar] [CrossRef]
- Lushington, G.; Guo, J.-X.; Hurley, M. Acetylcholinesterase: Molecular Modeling with the Whole Toolkit. Curr. Top. Med. Chem. 2006, 6, 57–73. [Google Scholar] [CrossRef]
- Marrs, T.C. Organophosphate Poisoning. Pharmacol. Ther. 1993, 58, 51–66. [Google Scholar] [CrossRef]
- Dvir, H.; Silman, I.; Harel, M.; Rosenberry, T.L.; Sussman, J.L. Acetylcholinesterase: From 3D Structure to Function. Chem. Biol. Interact. 2010, 187, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Pita, R.; Domingo, J. The Use of Chemical Weapons in the Syrian Conflict. Toxics 2014, 2, 391–402. [Google Scholar] [CrossRef]
- Hoenig, S.L. Compendium of Chemical Warfare Agents; Springer: New York, NY, USA, 2007; ISBN 978-0-387-34626-7. [Google Scholar]
- Korabecny, J.; Soukup, O.; Dolezal, R.; Spilovska, K.; Nepovimova, E.; Andrs, M.; Nguyen, T.; Jun, D.; Musilek, K.; Kucerova-Chlupacova, M.; et al. From Pyridinium-Based to Centrally Active Acetylcholinesterase Reactivators. Mini Rev. Med. Chem. 2014, 14, 215–221. [Google Scholar] [CrossRef]
- Sharma, R.; Gupta, B.; Singh, N.; Acharya, J.R.; Musilek, K.; Kuca, K.; Ghosh, K. Development and Structural Modifications of Cholinesterase Reactivators against Chemical Warfare Agents in Last Decade: A Review. Mini Rev. Med. Chem. 2015, 15, 58–72. [Google Scholar] [CrossRef] [PubMed]
- Sirin, G.S.; Zhou, Y.; Lior-Hoffmann, L.; Wang, S.; Zhang, Y. Aging Mechanism of Soman Inhibited Acetylcholinesterase. J. Phys. Chem. B 2012, 116, 12199–12207. [Google Scholar] [CrossRef]
- Jokanović, M. Structure-Activity Relationship and Efficacy of Pyridinium Oximes in the Treatment of Poisoning with Organophosphorus Compounds: A Review of Recent Data. Curr. Top. Med. Chem. 2012, 12, 1775–1789. [Google Scholar] [CrossRef]
- Kuča, K.; Pohanka, M. Chemical Warfare Agents. In Molecular, Clinical and Environmental Toxicology; Luch, A., Ed.; Experientia Supplementum; Birkhäuser Basel: Basel, Switzerland, 2010; Volume 100, pp. 543–558. ISBN 978-3-7643-8337-4. [Google Scholar]
- Wilson, I.B.; Ginsburg, S. A Powerful Reactivator of Alkylphosphate-Inhibited Acetylcholinesterase. Biochim. Biophys. Acta 1955, 18, 168–170. [Google Scholar] [CrossRef]
- Eyer, P. The Role of Oximes in the Management of Organophosphorus Pesticide Poisoning. Toxicol. Rev. 2003, 22, 165–190. [Google Scholar] [CrossRef]
- Worek, F.; Aurbek, N.; Thiermann, H. Reactivation of Organophosphate-Inhibited Human AChE by Combinations of Obidoxime and HI 6in Vitro. J. Appl. Toxicol. 2007, 27, 582–588. [Google Scholar] [CrossRef]
- Chai, P.R.; Hayes, B.D.; Erickson, T.B.; Boyer, E.W. Novichok Agents: A Historical, Current, and Toxicological Perspective. Toxicol. Commun. 2018, 2, 45–48. [Google Scholar] [CrossRef]
- Carletti, E.; Aurbek, N.; Gillon, E.; Loiodice, M.; Nicolet, Y.; Fontecilla-Camps, J.-C.; Masson, P.; Thiermann, H.; Nachon, F.; Worek, F. Structure–Activity Analysis of Aging and Reactivation of Human Butyrylcholinesterase Inhibited by Analogues of Tabun. Biochem. J. 2009, 421, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Dixon, H.; Clark, A.P.-Z.; Cabell, L.A.; McDonough, J.A. MMB4 DMS Nanoparticle Suspension Formulation with Enhanced Stability for the Treatment of Nerve Agent Intoxication. Int. J. Toxicol. 2013, 32, 18S–29S. [Google Scholar] [CrossRef] [PubMed]
- Katz, F.S.; Pecic, S.; Tran, T.H.; Trakht, I.; Schneider, L.; Zhu, Z.; Ton-That, L.; Luzac, M.; Zlatanic, V.; Damera, S.; et al. Discovery of New Classes of Compounds That Reactivate Acetylcholinesterase Inhibited by Organophosphates. ChemBioChem 2015, 16, 2205–2215. [Google Scholar] [CrossRef] [PubMed]
- de Koning, M.C.; Horn, G.; Worek, F.; van Grol, M. Synthesis and in Vitro Evaluation of Novel Non-Oximes for the Reactivation of Nerve Agent Inhibited Human Acetylcholinesterase. Chem. Biol. Interact. 2020, 326, 109139. [Google Scholar] [CrossRef]
- Nepovimova, E.; Kuca, K. Chemical Warfare Agent NOVICHOK—Mini-Review of Available Data. Food Chem. Toxicol. 2018, 121, 343–350. [Google Scholar] [CrossRef]
- Winter, M.; Wille, T.; Musilek, K.; Kuca, K.; Thiermann, H.; Worek, F. Investigation of the Reactivation Kinetics of a Large Series of Bispyridinium Oximes with Organophosphate-Inhibited Human Acetylcholinesterase. Toxicol. Lett. 2016, 244, 136–142. [Google Scholar] [CrossRef]
- Karasova, J.Z.; Chladek, J.; Hroch, M.; Josef, F.; Hnidkova, D.; Kuca, K. Pharmacokinetic Study of Two Acetylcholinesterase Reactivators, Trimedoxime and Newly Synthesized Oxime K027, in Rat Plasma: Pharmacokinetic Study of Oxime K027 and Trimedoxime. J. Appl. Toxicol. 2013, 33, 18–23. [Google Scholar] [CrossRef]
- Sepsova, V.; Krusek, J.; Zdarova Karasova, J.; Zemek, F.; Musilek, K.; Kuca, K.; Soukup, O. The Interaction of Quaternary Reversible Acetylcholinesterase Inhibitors with the Nicotinic Receptor. Physiol. Res. 2014, 63, 771–777. [Google Scholar] [CrossRef]
- Antonijevic, B.; Stojiljkovic, M.P. Unequal Efficacy of Pyridinium Oximes in Acute Organophosphate Poisoning. Clin. Med. Res. 2007, 5, 71–82. [Google Scholar] [CrossRef]
- Dimercaprol. Available online: https://go.drugbank.com/drugs/DB06782 (accessed on 27 April 2023).
- Williams, M. The Merck Index: An Encyclopedia of Chemicals, Drugs, and Biologicals, 15th ed.; $150 with 1-Year Free Access to The Merck Index Online; O’Neil, M.J., Ed.; Royal Society of Chemistry: Cambridge, UK, 2013; 2708p, ISBN 9781849736701. [Google Scholar] [CrossRef]
- PubChem Dimercaprol. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/3080 (accessed on 27 April 2023).
- National Institute of Diabetes and Digestive and Kidney Diseases. Wilson Disease Agents. In LiverTox: Clinical and Research Information on Drug-Induced Liver Injury; National Institute of Diabetes and Digestive and Kidney Diseases: Bethesda, MD, USA, 2012. [Google Scholar]
- Vilensky, J.A.; Redman, K. British Anti-Lewisite (Dimercaprol): An Amazing History. Ann. Emerg. Med. 2003, 41, 378–383. [Google Scholar] [CrossRef]
- Dawn, L.; Whited, L. Dimercaprol. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Scheinberg, I.H.; Sternlieb, I. Treatment of the Neurologic Manifestations of Wilson’s Disease. Arch. Neurol. 1995, 52, 339–340. [Google Scholar] [CrossRef] [PubMed]
- Mückter, H.; Lieb, B.; Reich, F.-X.; Hunder, G.; Walther, U.; Fichtl, B. Are We Ready to Replace Dimercaprol (BAL) as an Arsenic Antidote? Hum. Exp. Toxicol. 1997, 16, 460–465. [Google Scholar] [CrossRef] [PubMed]
- Flora, S.J.S.; Jain, K.; Panghal, A.; Patwa, J. Chemistry, Pharmacology, and Toxicology of Monoisoamyl Dimercaptosuccinic Acid: A Chelating Agent for Chronic Metal Poisoning. Chem. Res. Toxicol. 2022, 35, 1701–1719. [Google Scholar] [CrossRef] [PubMed]
- Haynes, W.M. (Ed.) CRC Handbook of Chemistry and Physics; CRC Press: Boca Raton, FL, USA, 2014; ISBN 978-0-429-17019-5. [Google Scholar]
- Cao, Y.; Skaug, M.A.; Andersen, O.; Aaseth, J. Chelation Therapy in Intoxications with Mercury, Lead and Copper. J. Trace Elem. Med. Biol. 2015, 31, 188–192. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.; Strupp, B.J. The Scientific Basis for Chelation: Animal Studies and Lead Chelation. J. Med. Toxicol. 2013, 9, 326–338. [Google Scholar] [CrossRef]
- Kosnett, M.J. Succimer. In Critical Care Toxicology; Brent, J., Burkhart, K., Dargan, P., Hatten, B., Megarbane, B., Palmer, R., White, J., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 2987–2991. ISBN 978-3-319-17899-8. [Google Scholar]
- Wang, S.C.; Ting, K.S.; Wu, C.C. Chelating Therapy with Na-DMS in Occupational Lead and Mercury Intoxications. Chin. Med. J. 1965, 84, 437–439. [Google Scholar]
- Yen, T.-H.; Yen, J.-S. Chelation Trial in Patients with Cardiovascular Disease. Am. Heart J. 2023, 256, 1. [Google Scholar] [CrossRef]
- Yen, J.-S.; Yen, T.-H. Chelation Therapy for Kidney Transplant Recipients with Lead Exposure. Am. J. Kidney Dis. 2023, 81, 118. [Google Scholar] [CrossRef]
- Patwa, J.; Thakur, A.; Flora, S.J.S. Alpha Lipoic Acid and Monoisoamyl-DMSA Combined Treatment Ameliorates Copper-Induced Neurobehavioral Deficits, Oxidative Stress, and Inflammation. Toxics 2022, 10, 718. [Google Scholar] [CrossRef]
- Aaseth, J.; Skaug, M.A.; Cao, Y.; Andersen, O. Chelation in Metal Intoxication—Principles and Paradigms. J. Trace Elem. Med. Biol. 2015, 31, 260–266. [Google Scholar] [CrossRef]
- Van Eijkeren, J.C.H.; Olie, J.D.N.; Bradberry, S.M.; Vale, J.A.; De Vries, I.; Clewell, H.J.; Meulenbelt, J.; Hunault, C.C. Modeling the Effect of Succimer (DMSA; Dimercaptosuccinic Acid) Chelation Therapy in Patients Poisoned by Lead. Clin. Toxicol. 2017, 55, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Domingo, J.L.; Ortega, A.; Paternain, J.L.; Llobet, J.M.; Corbella, J. Oral Meso-2, 3-dimercaptosuccinic Acid in Pregnant Sprague-dawley Rats: Teratogenicity and Alterations in Mineral Metabolism. I. Teratological Evaluation. J. Toxicol. Environ. Health 1990, 30, 181–190. [Google Scholar] [CrossRef] [PubMed]
- James, S.; Stevenson, S.W.; Silove, N.; Williams, K. Chelation for Autism Spectrum Disorder (ASD). Cochrane Database Syst. Rev. 2015. [Google Scholar] [CrossRef] [PubMed]
- PubChem Ethanol. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/702 (accessed on 28 July 2023).
- Le Daré, B.; Gicquel, T. Therapeutic Applications of Ethanol: A Review. J. Pharm. Pharm. Sci. 2019, 22, 525–535. [Google Scholar] [CrossRef] [PubMed]
- McMartin, K.; Jacobsen, D.; Hovda, K.E. Antidotes for Poisoning by Alcohols That Form Toxic Metabolites: Toxic Alcohol Antidotes. Br. J. Clin. Pharmacol. 2016, 81, 505–515. [Google Scholar] [CrossRef]
- Nekoukar, Z.; Zakariaei, Z.; Taghizadeh, F.; Musavi, F.; Banimostafavi, E.S.; Sharifpour, A.; Ghuchi, N.E.; Fakhar, M.; Tabaripour, R.; Safanavaei, S. Methanol Poisoning as a New World Challenge: A Review. Ann. Med. Surg. 2021, 66, 102445. [Google Scholar] [CrossRef]
- Bestic, M.; Blackford, M.; Reed, M. Fomepizole: A Critical Assessment of Current Dosing Recommendations. J. Clin. Pharmacol. 2009, 49, 130–137. [Google Scholar] [CrossRef]
- PubChem Fomepizole. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/3406 (accessed on 26 April 2023).
- Jones, A.W. Clinical and Forensic Toxicology of Methanol. Forensic Sci. Rev. 2021, 33, 117–143. [Google Scholar]
- Mégarbane, B. Treatment of Patients with Ethylene Glycol or Methanol Poisoning: Focus on Fomepizole. Open Access Emerg. Med. OAEM 2010, 2, 67. [Google Scholar] [CrossRef]
- PubChem Folinic Acid. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/135402009 (accessed on 26 April 2023).
- Kelly, G.S. Folates: Supplemental Forms and Therapeutic Applications. Altern. Med. Rev. 1998, 3, 208–220. [Google Scholar]
- Howard, S.C.; McCormick, J.; Pui, C.-H.; Buddington, R.K.; Harvey, R.D. Preventing and Managing Toxicities of High-Dose Methotrexate. Oncologist 2016, 21, 1471–1482. [Google Scholar] [CrossRef] [PubMed]
- Gristan, Y.D.; Moosavi, L. Folinic Acid. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Tjong, E.; Dimri, M.; Mohiuddin, S.S. Biochemistry, Tetrahydrofolate. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Menezo, Y.; Elder, K.; Clement, A.; Clement, P. Folic Acid, Folinic Acid, 5 Methyl TetraHydroFolate Supplementation for Mutations That Affect Epigenesis through the Folate and One-Carbon Cycles. Biomolecules 2022, 12, 197. [Google Scholar] [CrossRef] [PubMed]
- Roberts, D.M.; Yates, C.; Megarbane, B.; Winchester, J.F.; Maclaren, R.; Gosselin, S.; Nolin, T.D.; Lavergne, V.; Hoffman, R.S.; Ghannoum, M. Recommendations for the Role of Extracorporeal Treatments in the Management of Acute Methanol Poisoning: A Systematic Review and Consensus Statement. Crit. Care Med. 2015, 43, 461–472. [Google Scholar] [CrossRef] [PubMed]
- American Academy of Clinical Toxicology Ad Hoc Committee on the Treatment Guidelines for Methanol Poisoning; Barceloux, D.G.; Randall Bond, G.; Krenzelok, E.P.; Cooper, H.; Allister Vale, J. American Academy of Clinical Toxicology Practice Guidelines on the Treatment of Methanol Poisoning. J. Toxicol. Clin. Toxicol. 2002, 40, 415–446. [Google Scholar] [CrossRef]
- Benedek, T.G. Methotrexate: From Its Introduction to Non-Oncologic Therapeutics to Anti-TNF-α. Clin. Exp. Rheumatol. 2010, 28, S3–S8. [Google Scholar]
- Atropine and Cardiac Arrhythmia. N. Engl. J. Med. 1973, 288, 635. [CrossRef]
- Marrs, T.T.; Maynard, R.L.; Sidell, F. Chemical Warfare Agents: Toxicology and Treatment; John Wiley & Sons: Hoboken, NJ, USA, 2007; ISBN 978-0-470-06002-5. [Google Scholar]
- Szczeklik, A.; Gajewski, P. Interna Szczeklika 2022; MP: Gdańsk, Poland, 2023; ISBN 978-83-7430-669-0. [Google Scholar]
- Atropine. Available online: https://handbook.bcehs.ca/drug-monographs/atropine/ (accessed on 27 July 2023).
- Calcium Chloride. Available online: https://go.drugbank.com/drugs/DB01164 (accessed on 26 April 2023).
- PubChem Calcium Chloride. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/5284359 (accessed on 26 April 2023).
- Marraffa, J.M.; Cohen, V.; Howland, M.A. Antidotes for Toxicological Emergencies: A Practical Review. Am. J. Health-Syst. Pharm. 2012, 69, 199–212. [Google Scholar] [CrossRef]
- Pu, F.; Chen, N.; Xue, S. Calcium Intake, Calcium Homeostasis and Health. Food Sci. Hum. Wellness 2016, 5, 8–16. [Google Scholar] [CrossRef]
- Ratto, D.; Joyner, R.W. Dantrolene. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Liang, L.; Wei, H. Dantrolene, A Treatment for Alzheimer Disease? Alzheimer Dis. Assoc. Disord. 2015, 29, 1–5. [Google Scholar] [CrossRef]
- Kobayashi, S.; Bannister, M.L.; Gangopadhyay, J.P.; Hamada, T.; Parness, J.; Ikemoto, N. Dantrolene Stabilizes Domain Interactions within the Ryanodine Receptor. J. Biol. Chem. 2005, 280, 6580–6587. [Google Scholar] [CrossRef]
- Mosiołek, A.; Galanty, D. Wszystko co powinniśmy wiedzieć o złośliwym zespole neuroleptycznym. Psychiatria 2017, 14, 28–34. [Google Scholar]
- Sabouri, M.; Momeni, M.; Khorvash, F.; Rezvani, M.; Tabesh, H. The Effect of a Single Dose Dantrolene in Patients with Vasospasm Following Aneurysmal Subarachnoid Hemorrhage. Adv. Biomed. Res. 2017, 6, 83. [Google Scholar] [CrossRef]
- PubChem Diphenhydramine. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/3100 (accessed on 27 July 2023).
- Cherukuri, S.V.; Purvis, A.W.; Tosto, S.T.; Velayati, A. IV Lipid Emulsion Infusion in the Treatment of Severe Diphenhydramine Overdose. Am. J. Case Rep. 2019, 20, 758–763. [Google Scholar] [CrossRef]
- Hamano, H.; Ikeda, Y.; Goda, M.; Fukushima, K.; Kishi, S.; Chuma, M.; Yamashita, M.; Niimura, T.; Takechi, K.; Imanishi, M.; et al. Diphenhydramine May Be a Preventive Medicine against Cisplatin-Induced Kidney Toxicity. Kidney Int. 2021, 99, 885–899. [Google Scholar] [CrossRef] [PubMed]
- Sicari, V.; Zabbo, C.P. Diphenhydramine. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- DiphenhydrAMINE Monograph for Professionals. Available online: https://www.drugs.com/monograph/diphenhydramine.html (accessed on 27 April 2023).
- Bolser, D.C. Older-Generation Antihistamines and Cough Due to Upper Airway Cough Syndrome (UACS): Efficacy and Mechanism. Lung 2008, 186, 74–77. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Huynh, D.A.; Abbas, M.; Dabaja, A. Diphenhydramine Toxicity. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Scharman, E.J.; Erdman, A.R.; Wax, P.M.; Chyka, P.A.; Martin Caravati, E.; Nelson, L.S.; Manoguerra, A.S.; Christianson, G.; Olson, K.R.; Woolf, A.D.; et al. Diphenhydramine and Dimenhydrinate Poisoning: An Evidence-Based Consensus Guideline for Out-of-Hospital Management. Clin. Toxicol. 2006, 44, 205–223. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.-H.; Chen, D.-Y.; Chen, H.-Y.; Lee, K.-H. Intravenous Lipid-Emulsion Therapy in a Patient with Cardiac Arrest after Overdose of Diphenhydramine. J. Formos. Med. Assoc. 2016, 115, 1017–1018. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kamijo, Y.; Soma, K.; Sato, C.; Kurihara, K. Fatal Diphenhydramine Poisoning with Increased Vascular Permeability Including Late Pulmonary Congestion Refractory to Percutaneous Cardiovascular Support. Clin. Toxicol. 2008, 46, 864–868. [Google Scholar] [CrossRef]
- PubChem Flumazenil. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/3373 (accessed on 26 April 2023).
- Flumazenil. Available online: https://go.drugbank.com/drugs/DB01205 (accessed on 26 April 2023).
- Chacko, B.; Peter, J.V. Antidotes in poisoning. Indian J. Crit. Care Med. Peer-Rev. Off. Publ. Indian Soc. Crit. Care Med. 2019, 23, S241. [Google Scholar] [CrossRef]
- Sharbaf Shoar, N.; Bistas, K.G.; Saadabadi, A. Flumazenil. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Ghit, A.; Assal, D.; Al-Shami, A.S.; Hussein, D.E.E. GABAA Receptors: Structure, Function, Pharmacology, and Related Disorders. J. Genet. Eng. Biotechnol. 2021, 19, 123. [Google Scholar] [CrossRef]
- Zhu, S.; Noviello, C.M.; Teng, J.; Walsh, R.M.; Kim, J.J.; Hibbs, R.E. Structure of a Human Synaptic GABAA Receptor. Nature 2018, 559, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Haskali, M.B.; Roselt, P.D.; O’Brien, T.J.; Hutton, C.A.; Ali, I.; Vivash, L.; Jupp, B. Effective Preparation of [18F]Flumazenil Using Copper-Mediated Late-Stage Radiofluorination of a Stannyl Precursor. Molecules 2022, 27, 5931. [Google Scholar] [CrossRef]
- Wong, M. Reversal Agents in Sedation and Anesthesia Practice for Dentistry. Anesth. Prog. 2022, 69, 49–58. [Google Scholar] [CrossRef]
- Masui, K. Caution!! Reappearance of Remimazolam Effect after a Flumazenil Bolus: A Larger Bolus of Flumazenil and a Lower Total Remimazolam Clearance Are Higher Risks. J. Anesth. 2023, 37, 1–5. [Google Scholar] [CrossRef]
- Gajewski, P. (Ed.) Interna Szczeklika 2017; Medycyna Praktyczna: Kraków, Poland, 2017; ISBN 978-83-7430-517-4. [Google Scholar]
- Hood, S.D.; Norman, A.; Hince, D.A.; Melichar, J.K.; Hulse, G.K. Benzodiazepine Dependence and Its Treatment with Low Dose Flumazenil: Benzodiazepine Dependence and Its Treatment. Br. J. Clin. Pharmacol. 2014, 77, 285–294. [Google Scholar] [CrossRef]
- Chen, X.; Sang, N.; Song, K.; Zhong, W.; Wang, H.; Jiang, J.; Huang, Y.; Hu, P. Psychomotor Recovery Following Remimazolam-Induced Sedation and the Effectiveness of Flumazenil as an Antidote. Clin. Ther. 2020, 42, 614–624. [Google Scholar] [CrossRef] [PubMed]
- Trotti, L.M.; Arnulf, I. Idiopathic Hypersomnia and Other Hypersomnia Syndromes. Neurotherapeutics 2021, 18, 20–31. [Google Scholar] [CrossRef]
- Goh, E.T.; Andersen, M.L.; Morgan, M.Y.; Gluud, L.L. Flumazenil versus Placebo or No Intervention for People with Cirrhosis and Hepatic Encephalopathy. Cochrane Database Syst. Rev. 2017. [Google Scholar] [CrossRef] [PubMed]
- PubChem Glucagon. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/16132283 (accessed on 26 April 2023).
- Al-Tikrity, N.Y.; Sh-Abdulrazzaq, F.; Beyatli, A. Preparation of Nitrile Derivative and Study Its Effect as a Possible Novel Drug for Diabetes. Ann. Trop. Med. Public Health 2020, 23, 516–542. [Google Scholar] [CrossRef]
- Rix, I.; Nexøe-Larsen, C.; Bergmann, N.C.; Lund, A.; Knop, F.K. Glucagon Physiology. In Endotext; Feingold, K.R., Anawalt, B., Blackman, M.R., Boyce, A., Chrousos, G., Corpas, E., de Herder, W.W., Dhatariya, K., Dungan, K., Hofland, J., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- White, C.M. A Review of Potential Cardiovascular Uses of Intravenous Glucagon Administration. J. Clin. Pharmacol. 1999, 39, 442–447. [Google Scholar] [CrossRef]
- Morris, C.H.; Baker, J. Glucagon. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Rotella, J.-A.; Greene, S.L.; Koutsogiannis, Z.; Graudins, A.; Hung Leang, Y.; Kuan, K.; Baxter, H.; Bourke, E.; Wong, A. Treatment for Beta-Blocker Poisoning: A Systematic Review. Clin. Toxicol. 2020, 58, 943–983. [Google Scholar] [CrossRef] [PubMed]
- Graudins, A.; Lee, H.M.; Druda, D. Calcium Channel Antagonist and Beta-Blocker Overdose: Antidotes and Adjunct Therapies: Management of CCB and Beta-Blocker Toxicity. Br. J. Clin. Pharmacol. 2016, 81, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, G. Treatment of Poisoning Caused by β-Adrenergic and Calcium-Channel Blockers. Am. J. Health-Syst. Pharm. 2006, 63, 1828–1835. [Google Scholar] [CrossRef]
- Ramezanpour Ahangar, E.; Annamaraju, P. Hydroxocobalamin. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- PubChem Hydroxocobalamin Acetate. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/70689311 (accessed on 27 April 2023).
- Shapeton, A.D.; Mahmood, F.; Ortoleva, J.P. Hydroxocobalamin for the Treatment of Vasoplegia: A Review of Current Literature and Considerations for Use. J. Cardiothorac. Vasc. Anesth. 2019, 33, 894–901. [Google Scholar] [CrossRef] [PubMed]
- Thakkar, K.; Billa, G. Treatment of Vitamin B12 Deficiency–Methylcobalamine? Cyancobalamine? Hydroxocobalamin?—Clearing the Confusion. Eur. J. Clin. Nutr. 2015, 69, 1–2. [Google Scholar] [CrossRef]
- Thompson, J.P.; Marrs, T.C. Hydroxocobalamin in Cyanide Poisoning. Clin. Toxicol. 2012, 50, 875–885. [Google Scholar] [CrossRef]
- Elsevier—Drug Monograph|Hydroxocobalamin. Available online: https://elsevier.health/en-US/preview/hydroxocobalamin#indicationsdosage (accessed on 27 April 2023).
- Mayer, J.P.; Zhang, F.; DiMarchi, R.D. Insulin Structure and Function. Biopolymers 2007, 88, 687–713. [Google Scholar] [CrossRef]
- PubChem Insulin Human. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/118984375 (accessed on 28 July 2023).
- Lheureux, P.E.R.; Zahir, S.; Gris, M.; Derrey, A.-S.; Penaloza, A. Bench-to-Bedside Review: Hyperinsulinaemia/Euglycaemia Therapy in the Management of Overdose of Calcium-Channel Blockers. Crit. Care 2006, 10, 212. [Google Scholar] [CrossRef]
- Alshaya, O.A.; Alhamed, A.; Althewaibi, S.; Fetyani, L.; Alshehri, S.; Alnashmi, F.; Alharbi, S.; Alrashed, M.; Alqifari, S.F.; Alshaya, A.I. Calcium Channel Blocker Toxicity: A Practical Approach. J. Multidiscip. Healthc. 2022, 15, 1851–1862. [Google Scholar] [CrossRef]
- Krenz, J.R.; Kaakeh, Y. An Overview of Hyperinsulinemic-Euglycemic Therapy in Calcium Channel Blocker and β-Blocker Overdose. Pharmacotherapy 2018, 38, 1130–1142. [Google Scholar] [CrossRef]
- Azendour, H.; Belyamani, L.; Atmani, M.; Balkhi, H.; Haimeur, C. Severe Amlodipine Intoxication Treated by Hyperinsulinemia Euglycemia Therapy. J. Emerg. Med. 2010, 38, 33–35. [Google Scholar] [CrossRef] [PubMed]
- PubChem Levocarnitine. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/10917 (accessed on 27 April 2023).
- Lheureux, P.E.R.; Penaloza, A.; Zahir, S.; Gris, M. Science Review: Carnitine in the Treatment of Valproic Acid-Induced Toxicity—What Is the Evidence? Crit. Care 2005, 9, 431–440. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Dahash, B.A.; Sankararaman, S. Carnitine deficiency. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Vázquez, M.; Fagiolino, P.; Maldonado, C.; Olmos, I.; Ibarra, M.; Alvariza, S.; Guevara, N.; Magallanes, L.; Olano, I. Hyperammonemia Associated with Valproic Acid Concentrations. BioMed Res. Int. 2014, 2014, 217269. [Google Scholar] [CrossRef] [PubMed]
- Longo, N.; Frigeni, M.; Pasquali, M. Carnitine transport and fatty acid oxidation. Biochim. Biophys. Acta 2016, 1863, 2422–2435. [Google Scholar] [CrossRef]
- Farkas, V.; Bock, I.; Cseko, J.; Sandor, A. Inhibition of Carnitine Biosynthesis by Valproic Acid in Rats—The Biochemical Mechanism of Inhibition. Biochem. Pharmacol. 1996, 52, 1429–1433. [Google Scholar] [CrossRef]
- L-Carnitine Uses, Benefits & Dosage—Drugs.Com Herbal Database. Available online: https://www.drugs.com/npp/l-carnitine.html (accessed on 27 April 2023).
- PubChem Methylene Blue. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/6099 (accessed on 27 April 2023).
- Xue, H.; Thaivalappil, A.; Cao, K. The Potentials of Methylene Blue as an Anti-Aging Drug. Cells 2021, 10, 3379. [Google Scholar] [CrossRef]
- Pushparajah Mak, R.S.; Liebelt, E.L. Methylene Blue: An Antidote for Methemoglobinemia and Beyond. Pediatr. Emer. Care 2021, 37, 474–477. [Google Scholar] [CrossRef]
- Bistas, E.; Sanghavi, D. Methylene Blue. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Lim, D.-J. Methylene Blue-Based Nano and Microparticles: Fabrication and Applications in Photodynamic Therapy. Polymers 2021, 13, 3955. [Google Scholar] [CrossRef]
- PubChem Acetylcysteine. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/12035 (accessed on 28 July 2023).
- Guidance for Industry: Policy Regarding N-Acetyl-L-Cysteine. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/guidance-industry-policy-regarding-n-acetyl-l-cysteine (accessed on 27 April 2023).
- Pedre, B.; Barayeu, U.; Ezeriņa, D.; Dick, T.P. The Mechanism of Action of N-Acetylcysteine (NAC): The Emerging Role of H2S and Sulfane Sulfur Species. Pharmacol. Ther. 2021, 228, 107916. [Google Scholar] [CrossRef]
- Aldini, G.; Altomare, A.; Baron, G.; Vistoli, G.; Carini, M.; Borsani, L.; Sergio, F. N-Acetylcysteine as an Antioxidant and Disulphide Breaking Agent: The Reasons Why. Free Radic. Res. 2018, 52, 751–762. [Google Scholar] [CrossRef]
- Hinz-Brylew, N.; Rajewski, P.; Rajewski, P. Ostre Zatrucie Paracetamolem—Opis Trzech Przypadków Acute Paracetamol Poisoning—Three Cases Description. Forum Med. Rodz. 2011, 5, 429–433. [Google Scholar]
- Schwalfenberg, G.K. N-Acetylcysteine: A Review of Clinical Usefulness (an Old Drug with New Tricks). J. Nutr. Metab. 2021, 2021, 9949453. [Google Scholar] [CrossRef] [PubMed]
- De Flora, S.; Balansky, R.; La Maestra, S. Rationale for the Use of N-acetylcysteine in Both Prevention and Adjuvant Therapy of COVID-19. FASEB J. 2020, 34, 13185–13193. [Google Scholar] [CrossRef] [PubMed]
- Hariharan, A.; Hakeem, A.R.; Radhakrishnan, S.; Reddy, M.S.; Rela, M. The Role and Therapeutic Potential of NF-Kappa-B Pathway in Severe COVID-19 Patients. Inflammopharmacology 2021, 29, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Geiler, J.; Michaelis, M.; Naczk, P.; Leutz, A.; Langer, K.; Doerr, H.-W.; Cinatl, J. N-Acetyl-l-Cysteine (NAC) Inhibits Virus Replication and Expression of pro-Inflammatory Molecules in A549 Cells Infected with Highly Pathogenic H5N1 Influenza A Virus. Biochem. Pharmacol. 2010, 79, 413–420. [Google Scholar] [CrossRef]
- Shi, Z.; Puyo, C.A. N-Acetylcysteine to Combat COVID-19: An Evidence Review. Ther. Clin. Risk Manag. 2020, 16, 1047–1055. [Google Scholar] [CrossRef]
- Poe, F.L.; Corn, J. N-Acetylcysteine: A Potential Therapeutic Agent for SARS-CoV-2. Med. Hypotheses 2020, 143, 109862. [Google Scholar] [CrossRef]
- Van Dorp, E.L.; Yassen, A.; Dahan, A. Naloxone Treatment in Opioid Addiction: The Risks and Benefits. Expert. Opin. Drug Saf. 2007, 6, 125–132. [Google Scholar] [CrossRef]
- Sivilotti, M.L.A. Flumazenil, Naloxone and the ‘Coma Cocktail’: Flumazenil, Naloxone and the ‘Coma Cocktail’. Br. J. Clin. Pharmacol. 2016, 81, 428–436. [Google Scholar] [CrossRef]
- Wermeling, D.P. A Response to the Opioid Overdose Epidemic: Naloxone Nasal Spray. Drug Deliv. Transl. Res. 2013, 3, 63–74. [Google Scholar] [CrossRef]
- Straus, M.; Ghitza, U.; Tai, B. Preventing Deaths from Rising Opioid Overdose in the US–the Promise of Naloxone Antidote in Community-Based Naloxone Take-Home Programs. Subst. Abus. Rehabil. 2013, 4, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Morgan, J.; Jones, A.L. The Role of Naloxone in the Opioid Crisis. Toxicol. Commun. 2018, 2, 15–18. [Google Scholar] [CrossRef]
- Jordan, M.R.; Morrisonponce, D. Naloxone. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- PubChem Octreotide. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/448601 (accessed on 28 July 2023).
- Glatstein, M.; Scolnik, D.; Bentur, Y. Octreotide for the Treatment of Sulfonylurea Poisoning. Clin. Toxicol. 2012, 50, 795–804. [Google Scholar] [CrossRef] [PubMed]
- Baumgartner, K.; Devgun, J. Toxicology of Medications for Diabetes Mellitus. Crit. Care Clin. 2021, 37, 577–589. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, S.A.; Crandall, C.S.; McKinney, P.E. Octreotide: An Antidote for Sulfonylurea-Induced Hypoglycemia. Ann. Emerg. Med. 2000, 36, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Carr, R.; Zed, P.J. Octreotide for Sulfonylurea-Induced Hypoglycemia Following Overdose. Ann. Pharmacother. 2002, 36, 1727–1732. [Google Scholar] [CrossRef]
- Dougherty, P.P.; Klein-Schwartz, W. Octreotide’s Role in the Management of Sulfonylurea-Induced Hypoglycemia. J. Med. Toxicol. 2010, 6, 199–206. [Google Scholar] [CrossRef]
- Lamberts, S.W.J.; Hofland, L.J. ANNIVERSARY REVIEW: Octreotide, 40 Years Later. Eur. J. Endocrinol. 2019, 181, R173–R183. [Google Scholar] [CrossRef]
- Moore, D.C. Oxygen: The Antidote for Systemic Toxic Reactions from Local Anesthetic Drugs. JAMA 1960, 174, 842. [Google Scholar] [CrossRef]
- Oxygen|O (Element)—PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/element/Oxygen (accessed on 27 April 2023).
- Eichhorn, L.; Thudium, M.; Jüttner, B. The Diagnosis and Treatment of Carbon Monoxide Poisoning. Dtsch. Ärzteblatt Int. 2018. [Google Scholar] [CrossRef]
- Oxygen. Available online: https://go.drugbank.com/drugs/DB09140 (accessed on 27 April 2023).
- Triggle, D.J.; Mitchell, J.M.; Filler, R. The Pharmacology of Physostigmine. CNS Drug Rev. 1998, 4, 87–136. [Google Scholar] [CrossRef]
- PubChem Physostigmine. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/5983 (accessed on 28 July 2023).
- Muhtadi, F.J.; El-Hawary, S.S. Analytical Profile of Physostigmine Salicylate. In Analytical Profiles of Drug Substances; Elsevier: Amsterdam, The Netherlands, 1990; Volume 18, pp. 289–350. ISBN 978-0-12-260818-6. [Google Scholar]
- Burns, M.J.; Linden, C.H.; Graudins, A.; Brown, R.M.; Fletcher, K.E. A Comparison of Physostigmine and Benzodiazepines for the Treatment of Anticholinergic Poisoning. Ann. Emerg. Med. 2000, 35, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Dawson, A.H.; Buckley, N.A. Pharmacological Management of Anticholinergic Delirium—Theory, Evidence and Practice. Br. J. Clin. Pharmacol. 2016, 81, 516–524. [Google Scholar] [CrossRef] [PubMed]
- Ritter, J.; Flower, R.; Henderson, G.; Loke, Y.K.; MacEwan, D.; Rang, H.P. Rang & Dale’s Pharmacology, 9th ed.; Elsevier: Edinburgh, UK; London, UK; New York, NY, USA, 2020; ISBN 978-0-7020-7448-6. [Google Scholar]
- Larionova, J.; Guari, Y.; Sangregorio, C.; Guérin, C. Cyano-Bridged Coordination Polymer Nanoparticles. New J. Chem. 2009, 33, 1177. [Google Scholar] [CrossRef]
- Itaya, K.; Ataka, T.; Toshima, S. Spectroelectrochemistry and Electrochemical Preparation Method of Prussian Blue Modified Electrodes. J. Am. Chem. Soc. 1982, 104, 4767–4772. [Google Scholar] [CrossRef]
- Thompson, D.F.; Church, C.O. Prussian Blue for Treatment of Radiocesium Poisoning. Pharmacotherapy 2001, 21, 1364–1367. [Google Scholar] [CrossRef]
- Prussian Blue. Available online: https://go.drugbank.com/drugs/DB06783 (accessed on 28 July 2023).
- Richelmi, P.; Bono, F.; Guardia, L.; Ferrini, B.; Manzo, L. Salivary Levels of Thallium in Acute Human Poisoning. Arch. Toxicol. 1980, 43, 321–325. [Google Scholar] [CrossRef]
- Zhao, J.; Cai, X.; Gao, W.; Zhang, L.; Zou, D.; Zheng, Y.; Li, Z.; Chen, H. Prussian Blue Nanozyme with Multienzyme Activity Reduces Colitis in Mice. ACS Appl. Mater. Interfaces 2018, 10, 26108–26117. [Google Scholar] [CrossRef]
- Yang, H.S.; Shin, J.; Bhang, S.H.; Shin, J.-Y.; Park, J.; Im, G.-I.; Kim, C.-S.; Kim, B.-S. Enhanced Skin Wound Healing by a Sustained Release of Growth Factors Contained in Platelet-Rich Plasma. Exp. Mol. Med. 2011, 43, 622. [Google Scholar] [CrossRef]
- Applefield, D.; Krishnan, S. Protamine. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Sokolowska, E.; Kalaska, B.; Miklosz, J.; Mogielnicki, A. The Toxicology of Heparin Reversal with Protamine: Past, Present and Future. Expert. Opin. Drug Metab. Toxicol. 2016, 12, 897–909. [Google Scholar] [CrossRef]
- Ranasinghe, T.; Mays, T.; Quedado, J.; Adcock, A. Thrombolysis Following Heparin Reversal with Protamine Sulfate in Acute Ischemic Stroke: Case Series and Literature Review. J. Stroke Cerebrovasc. Dis. 2019, 28, 104283. [Google Scholar] [CrossRef]
- Protamine Sulfate. Available online: https://go.drugbank.com/drugs/DB09141 (accessed on 27 April 2023).
- Butterworth, J.; Lin, Y.A.; Prielipp, R.C.; Bennett, J.; Hammon, J.W.; James, R.L. Rapid Disappearance of Protamine in Adults Undergoing Cardiac Operation with Cardiopulmonary Bypass. Ann. Thorac. Surg. 2002, 74, 1589–1595. [Google Scholar] [CrossRef] [PubMed]
- DailyMed—PROTAMINE SULFATE-Protamine Sulfate Injection, Solution. Available online: https://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=e1964129-33f4-4e4e-86e3-8e6a4e65bd83 (accessed on 27 April 2023).
- Calderón-Ospina, C.A.; Nava-Mesa, M.O. B Vitamins in the Nervous System: Current Knowledge of the Biochemical Modes of Action and Synergies of Thiamine, Pyridoxine, and Cobalamin. CNS Neurosci. Ther. 2020, 26, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Badrinath, M.; John, S. Isoniazid Toxicity. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Abosamak, N.R.; Gupta, V. Vitamin B6 (Pyridoxine). In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- O’Connor, C.; Brady, M.F. Isoniazid. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Lheureux, P.; Penaloza, A.; Gris, M. Pyridoxine in Clinical Toxicology: A Review. Eur. J. Emerg. Med. 2005, 12, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Pecriaux, C. Place de la vitamine b6 dans le traitement des nausees et vomissements gravidiques. Gynécologie Obs. Fertil. Sénologie 2020, 48, 840–843. [Google Scholar] [CrossRef]
- Brown, W.H.; Iverson, B.L.; Anslyn, E.V.; Foote, C.S. Organic Chemistry, 7th ed.; Wadsworth Cengage Learning: Belmont, CA, Australia, 2014; ISBN 978-1-133-95284-8. [Google Scholar]
- Jane, J. Starch Properties, Modifications, and Applications. J. Macromol. Sci. Part. A 1995, 32, 751–757. [Google Scholar] [CrossRef]
- Edwards, N.A.; Quigley, P.; Hackett, L.P.; James, R. Death by Oral Ingestion of Iodine. Emerg. Med. Australas. 2005, 17, 173–177. [Google Scholar] [CrossRef]
- Seager, S.L.; Slabaugh, M.R.; Hansen, M.S. Chemistry for Today: General, Organic, and Biochemistry, 9th ed.; Cengage Learning: Boston, MA, USA, 2018; ISBN 978-1-305-96006-0.
- Haddad, L.M.; Shannon, M.W.; Winchester, J.F. (Eds.) Clinical Management of Poisoning and Drug Overdose, 3rd ed.; Saunders: Philadelphia, PA, USA, 1998; ISBN 978-0-7216-6409-5. [Google Scholar]
- Goedecke, C. Why Does Iodine Turn Starch Blue? ChemViews 2016. [Google Scholar] [CrossRef]
- Mladěnka, P.; Macáková, K.; Kujovská Krčmová, L.; Javorská, L.; Mrštná, K.; Carazo, A.; Protti, M.; Remião, F.; Nováková, L.; The OEMONOM Researchers and Collaborators. Vitamin K—Sources, Physiological Role, Kinetics, Deficiency, Detection, Therapeutic Use, and Toxicity. Nutr. Rev. 2022, 80, 677–698. [Google Scholar] [CrossRef]
- Violi, F.; Lip, G.Y.; Pignatelli, P.; Pastori, D. Interaction between Dietary Vitamin K Intake and Anticoagulation by Vitamin K Antagonists: Is It Really True? A Systematic Review. Medicine 2016, 95, e2895. [Google Scholar] [CrossRef]
- Webb, G. Modern Nutrition in Health and Disease, 9th ed.; $105.00; Shils, M.E., Olsen, J.A., Shike, M., Ross, A.C., Eds.; Williams & Wilkins: Baltimore, MD, USA, 1999; ISBN 0-683-30769-X. [Google Scholar] [CrossRef]
- Simpson, W.M.; Schuman, S.H. Recognition and Management of Acute Pesticide Poisoning. Am. Fam. Physician 2002, 65, 1599–1604. [Google Scholar] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | Specific Parameter Value |
|---|---|
| Mechanism of action | The compound binds to somatostatin-2 receptors that are located on pancreatic β cells, and this prevents the influx of calcium that is required for insulin secretion. |
| Peak effect | Half hour |
| Preferred route | Subcutaneous |
| Dosing schedule—adults | 50–100 μg every 6–12 h |
| Adverse effects | Hyperglycaemia, injection site pain, nausea, abdominal pain, flatulence, diarrhoea |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kobylarz, D.; Noga, M.; Frydrych, A.; Milan, J.; Morawiec, A.; Glaca, A.; Kucab, E.; Jastrzębska, J.; Jabłońska, K.; Łuc, K.; et al. Antidotes in Clinical Toxicology—Critical Review. Toxics 2023, 11, 723. https://doi.org/10.3390/toxics11090723
Kobylarz D, Noga M, Frydrych A, Milan J, Morawiec A, Glaca A, Kucab E, Jastrzębska J, Jabłońska K, Łuc K, et al. Antidotes in Clinical Toxicology—Critical Review. Toxics. 2023; 11(9):723. https://doi.org/10.3390/toxics11090723
Chicago/Turabian StyleKobylarz, Damian, Maciej Noga, Adrian Frydrych, Justyna Milan, Adrian Morawiec, Agata Glaca, Emilia Kucab, Julia Jastrzębska, Karolina Jabłońska, Klaudia Łuc, and et al. 2023. "Antidotes in Clinical Toxicology—Critical Review" Toxics 11, no. 9: 723. https://doi.org/10.3390/toxics11090723
APA StyleKobylarz, D., Noga, M., Frydrych, A., Milan, J., Morawiec, A., Glaca, A., Kucab, E., Jastrzębska, J., Jabłońska, K., Łuc, K., Zdeb, G., Pasierb, J., Toporowska-Kaźmierak, J., Półchłopek, S., Słoma, P., Adamik, M., Banasik, M., Bartoszek, M., Adamczyk, A., ... Jurowski, K. (2023). Antidotes in Clinical Toxicology—Critical Review. Toxics, 11(9), 723. https://doi.org/10.3390/toxics11090723