
PTEN Overexpression Alters Autophagy Levels and Slows Sodium Arsenite-Induced Hepatic Stellate Cell Fibrosis


Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Treatments
2.2. Masson Staining for Detecting Liver Fibrosis
2.3. Cell Culture
2.4. Transmission Electronic Microscopy
2.5. Cell Transfection
2.6. Quantitative Real-Time PCR (qRT-PCR Analysis)
2.7. Western Blot
2.8. Statistical Analysis
3. Results
3.1. Autophagy and Fibrosis Were Established in the Liver Tissues of Mice Exposed to Different Concentrations of Sodium Arsenite
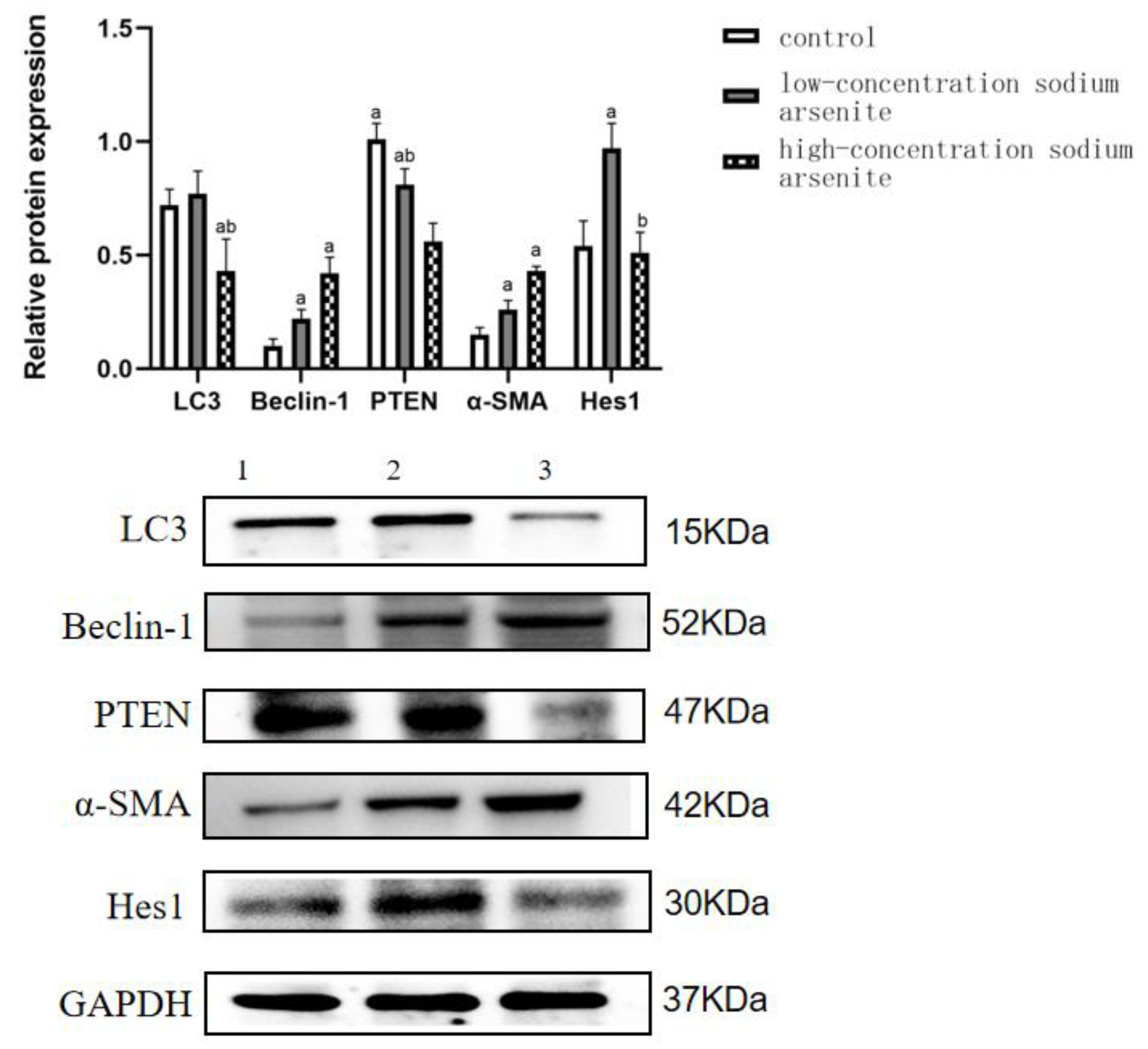
3.2. PTEN Is Involved in the Autophagy and Fibrosis in Mouse Liver Tissue Caused by Sodium Arsenite Treatment
3.3. PTEN Overexpression Can Reduce Autophagy Induced by Sodium Arsenite
3.4. PTEN Overexpression Can Reduce Fibrosis Induced by Sodium Arsenite in LX-2 Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Singh, R.; Singh, S.; Parihar, P.; Singh, V.P.; Prasad, S.M. Arsenic contamination, consequences and remediation techniques: A review. Ecotoxicol. Environ. Saf. 2015, 112, 247–270. [Google Scholar] [CrossRef]
- Rahaman, S.; Rahman, M.; Mise, N.; Sikder, T.; Ichihara, G.; Uddin, K.; Kurasaki, M.; Ichihara, S. Environmental arsenic exposure and its contribution to human diseases, toxicity mechanism and management. Environ. Pollut. 2021, 289, 117940. [Google Scholar] [CrossRef] [PubMed]
- Renu, K.; Saravanan, A.; Elangovan, A.; Ramesh, S.; Annamalai, S.; Namachivayam, A.; Gopalakrishnan, A.V.; Abel, P.; Madhyastha, H.; Madhyastha, R.; et al. An appraisal on molecular and biochemical signalling cascades during arsenic-induced hepatotoxicity. Life Sci. 2020, 260, 118438. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Lyu, M.; Yang, J.; Wang, K. Research progress on the mechanism of liver injury induced by arsenic. Chin. J. Endem. 2021, 12, 75–80. [Google Scholar]
- Chakraborty, A.; Ghosh, S.; Biswas, B.; Pramanik, S.; Nriagu, J.; Bhowmick, S. Epigenetic modifications from arsenic exposure: A comprehensive review. Sci. Total. Environ. 2022, 810, 151218. [Google Scholar] [CrossRef]
- Yao, M.; Zeng, Q.; Luo, P.; Sun, B.; Liang, B.; Wei, S.; Xu, Y.; Wang, Q.; Liu, Q.; Zhang, A. Assessing the risk of coal-burning arsenic-induced liver damage: A population-based study on hair arsenic and cumulative arsenic. Environ. Sci. Pollut. Res. Int. 2021, 28, 50489–50499. [Google Scholar] [CrossRef]
- Tao, Y.; Wang, N.; Qiu, T.; Sun, X. The role of autophagy and NLRP3 inflammasome in liver fibrosis. Biomed Res Int. 2020, 2020, 1–8. [Google Scholar] [CrossRef]
- Ghatak, S.; Biswas, A.; Dhali, G.K.; Chowdhury, A.; Boyer, J.L.; Santra, A. Oxidative stress and hepatic stellate cell activation are key events in arsenic induced liver fibrosis in mice. Toxicol. Appl. Pharmacol. 2011, 251, 59–69. [Google Scholar] [CrossRef]
- Tao, Y.; Qiu, T.; Yao, X.; Jiang, L.; Wang, N.; Jia, X.; Wei, S.; Wang, Z.; Pei, P.; Zhang, J.; et al. Autophagic-CTSB-inflammasome axis modulates hepatic stellate cells activation in arsenic-induced liver fibrosis. Chemosphere 2020, 242, 124959. [Google Scholar] [CrossRef]
- Tesseraud, S.; Avril, P.; Bonnet, M.; Bonnieu, A.; Cassar-Malek, I.; Chabi, B.; Dessauge, F.; Gabillard, J.-C.; Perruchot, M.-H.; Seiliez, I. Autophagy in farm animals: Current knowledge and future challenges. Autophagy 2021, 17, 1809–1827. [Google Scholar] [CrossRef]
- Yim, W.W.Y.; Mizushiman, N. Lysosome biology in autophagy. Cell Discov. 2020, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- Wadgaonkar, P.; Chen, F. Connections between endoplasmic reticulum stress-associated unfolded protein response, mitochondria, and autophagy in arsenic-induced carcinogenesis. Semin. Cancer Biol. 2021, 76, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Han, X.; Ou, D.; Liu, T.; Li, Z.; Jiang, G.; Liu, J.; Zhang, J. Targeting PI3K/AKT/mTOR-mediated autophagy for tumor therapy. Appl. Microbiol. Biotechnol. 2020, 104, 575–587. [Google Scholar] [CrossRef] [PubMed]
- Song, M.S.; Salmena, L.; Pandolfi, P.P. The functions and regulation of the PTEN tumour suppressor. Nat. Rev. Mol. Cell Biol. 2012, 13, 283–296. [Google Scholar] [CrossRef] [PubMed]
- He, T.; Zhang, X.; Hao, J.; Ding, S. Phosphatase and Tensin Homolog in Non-neoplastic Digestive Disease: More Than Just Tumor Suppressor. Front. Physiol. 2021, 12, 684529. [Google Scholar] [CrossRef]
- Cheng, Y.; Tian, Y.; Xia, J.; Wu, X.; Yang, Y.; Li, X.; Huang, C.; Meng, X.; Ma, T.; Li, J. The role of PTEN in regulation of hepatic macrophages activation and function in progression and reversal of liver fibrosis [published correction appears in Toxicol Appl Pharmacol. 2022 Aug 15; 449:116094]. Toxicol. Appl. Pharmacol. 2017, 317, 51–62. [Google Scholar] [CrossRef]
- Wang, A.; Bu, F.-T.; Li, J.-J.; Zhang, Y.-F.; Jia, P.-C.; You, H.-M.; Wu, S.; Wu, Y.-Y.; Zhu, S.; Huang, C.; et al. MicroRNA-195-3p promotes hepatic stellate cell activation and liver fibrosis by suppressing PTEN expression. Toxicol. Lett. 2022, 355, 88–99. [Google Scholar] [CrossRef]
- Guo, L.; Tan, K.; Luo, Q.; Bai, X. Dihydromyricetin promotes autophagy and attenuates renal interstitial fibrosis by regulating miR-155-5p/PTEN signaling in diabetic nephropathy. Bosn. J. Basic Med. Sci. 2020, 20, 372–380. [Google Scholar] [CrossRef]
- Zhu, L.; Mou, Q.; Wang, Y.; Zhu, Z.; Cheng, M. Resveratrol contributes to the inhibition of liver fibrosis by inducing autophagy via the microRNA-20a-mediated activation of the PTEN/PI3K/AKT signaling pathway. Int. J. Mol. Med. 2020, 46, 2035–2046. [Google Scholar] [CrossRef]
- Patel, A.S.; Lin, L.; Geyer, A.; Haspel, J.A.; An, C.H.; Cao, J.; Rosas, I.O.; Morse, D. Autophagy in idiopathic pulmonary fibrosis. PLoS ONE. 2012, 7, e41394. [Google Scholar] [CrossRef]
- Araya, J.; Kojima, J.; Takasaka, N.; Ito, S.; Fujii, S.; Hara, H.; Yanagisawa, H.; Kobayashi, K.; Tsurushige, C.; Kawaishi, M.; et al. Insufficient autophagy in idiopathic pulmonary fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2013, 304, L56–L69. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Xu, X.; Zhang, Q.; Yuan, Z.; Tan, B. Critical implication of the PTEN/PI3K/AKT pathway during BMP2-induced heterotopic ossification. Mol. Med. Rep. 2021, 23, 254. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhu, Z.; Guan, J.; Hu, Y.; Zhou, W.; Ye, W.; Lin, B.; Weng, S.; Chen, Y.; Zheng, C. Hes1 Controls Proliferation and Apoptosis in Chronic Lymphoblastic Leukemia Cells by Modulating PTEN Expression. Mol. Biotechnol. 2022, 64, 1419–1430. [Google Scholar] [CrossRef]
- GB 151933-2014; National Health and Family Planning Commission of the People’s Republic of China Acute Oral Toxicity Test. Beijing International Standard Press: Beijing, China, 2014.
- Minatel, B.C.; Sage, A.P.; Anderson, C.; Hubaux, R.; Marshall, E.A.; Lam, W.L.; Martinez, V.D. Environmental arsenic exposure: From genetic susceptibility to pathogenesis. Environ. Int. 2018, 112, 183–197. [Google Scholar] [CrossRef]
- Emadi, A.; Gore, S.D. Arsenic trioxide-An old drug rediscovered. Blood Rev. 2010, 24, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Palma-Lara, I.; Martínez-Castillo, M.; Quintana-Pérez, J.; Arellano-Mendoza, M.; Tamay-Cach, F.; Valenzuela-Limón, O.; García-Montalvo, E.; Hernández-Zavala, A. Arsenic exposure: A public health problem leading to several cancers. Regul. Toxicol. Pharm. 2020, 110, 104539. [Google Scholar] [CrossRef]
- Hu, Y.; Xiao, T.; Zhang, A. Associations between and risks of trace elements related to skin and liver damage induced by arsenic from coal burning. Ecotox. Environ. Saf. 2021, 208, 111719. [Google Scholar] [CrossRef]
- Zhang, C.Y.; Yuan, W.G.; He, P.; Lei, J.H.; Wang, C.X. Liver fibrosis and hepatic stellate cells: Etiology, pathological hallmarks and therapeutic targets. World J. Gastroenterol. 2016, 22, 10512–10522. [Google Scholar] [CrossRef]
- Wang, Y.; Qin, Z. Coordination of autophagy with other cellular activities. Acta Pharmacol. Sin. 2013, 34, 585–594. [Google Scholar] [CrossRef]
- Xue, F.; Cheng, J.; Liu, Y.; Cheng, C.; Zhang, M.; Sui, W.; Chen, W.; Hao, P.; Zhang, Y.; Zhang, C. Cardiomyocyte-specific knockout of ADAM17 ameliorates left ventricular remodeling and function in diabetic cardiomyopathy of mice. Signal Transduct. Target. Ther. 2022, 7, 259. [Google Scholar] [CrossRef]
- Zheng, L.; Chen, X.; Guo, J.; Sun, H.; Liu, L.; Shih, D.Q.; Zhang, X. Differential expression of PTEN in hepatic tissue and hepatic stellate cells during rat liver fibrosis and its reversal. Int. J. Mol. Med. 2012, 30, 1424–1430. [Google Scholar] [CrossRef] [PubMed]
- Soto-Blanco, B. Herbal Glycosides in Healthcare. In Herbal Biomolecules in Healthcare Applications; Academic Press: Cambridge, MA, USA, 2022; pp. 239–282. [Google Scholar]
- Bueno, M.; Lai, Y.-C.; Romero, Y.; Brands, J.; Croix, C.M.S.; Kamga, C.; Corey, C.; Herazo-Maya, J.D.; Sembrat, J.; Lee, J.; et al. PINK1 deficiency impairs mitochondrial homeostasis and promotes lung fibrosis. J. Clin. Investig. 2015, 125, 521–538. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Ma, Y.; He, H.W.; Wang, J.P.; Jiang, J.D.; Shao, R.G. SLC9A3R1 stimulates autophagy via BECN1 stabilization in breast cancer cells. Autophagy 2015, 11, 2323–2334. [Google Scholar] [CrossRef] [PubMed]
- Aquila, S.; Santoro, M.; Caputo, A.; Panno, M.L.; Pezzi, V.; De Amicis, F. The tumor suppressor PTEN as molecular switch node regulating cell metabolism and autophagy: Implications in immune system and tumor microenvironment. Cells 2020, 9, 1725. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, Y.; Shi, M.; Wang, Y.; Zhang, F.; Yan, R.; Liu, L.; Xiao, Y.; Guo, B. Notch1 regulates PTEN expression to exacerbate renal tubulointerstitial fibrosis in diabetic nephropathy by inhibiting autophagy via interactions with Hes1. Biochem. Biophys. Res. Commun. 2018, 497, 1110–1116. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Arsenic Concentration |
|---|---|
| High-dose iAs3+ group | 5.00 μmol/L |
| Middle-dose iAs3+ group | 0.50 μmol/L |
| Low-dose iAs3+ group | 0.05 μmol/L |
| Control group | 0.00 μmol/L |
| Gene | Forward (5′–3′) | Reverse (3′–5′) |
|---|---|---|
| α-SMA | GCGTGGCTATTCCTTCGTGACTAC | CGTCAGGCAGTTCGTAGCTCTTC |
| LC3 | GAACAGGAGAAGGATGAAGACGG | AATCCACTGGGGACTGAAATAGC |
| Beclin-1 | CCAGGAACTCACAGCTCCATTAC | GGATGAATCTTCGAGAGACACCA |
| PTEN | GCTGAGAGACATTATGACACCGC | TTACACCAGTCCGTCCCTTTC |
| HES1 | TGGAGAGGCTGCCAAGGTTT | ACTGGAAGGTGACACTGCGTT |
| GAPDH | CGTGTTCCTACCCCCAATGTG | TAGCCCAAGATGCCCTTCAGT |
| Group | LC3 | Beclin-1 | PTEN | α-SMA | HES1 |
|---|---|---|---|---|---|
| Control | 1.04 ± 0.05 | 1.03 ± 0.03 | 1.03 ± 0.04 | 1.02 ± 0.02 | 1.03 ± 0.08 |
| Low-dose iAs3+ group | 2.66 ± 0.37 a | 1.84 ± 0.17 a | 1.79 ± 0.17 a | 3.05 ± 0.52 a | 1.49 ± 0.22 a |
| High-dose iAs3+ group | 3.25 ± 0.29 ab | 1.09 ± 0.10 b | 0.84 ± 0.10 b | 1.12 ± 0.13 b | 1.09 ± 0.70 |
| F | 54.111 | 47.218 | 56.167 | 41.351 | 9.951 |
| p | <0.001 | <0.001 | <0.001 | <0.001 | 0.012 |
| Group | LC3 | Beclin-1 | PTEN | α-SMA | HES1 |
|---|---|---|---|---|---|
| Control | 0.72 ± 0.07 | 0.10 ± 0.03 | 1.01 ± 0.07 | 0.15 ± 0.03 | 0.54 ± 0.11 |
| Low-dose iAs3+ group | 0.77 ± 0.10 | 0.22 ± 0.04 a | 0.81 ± 0.07 a | 0.26 ± 0.04 a | 0.97 ± 0.11 a |
| High-dose iAs3+ group | 0.43 ± 0.14 ab | 0.42 ± 0.07 a | 0.56 ± 0.08 ab | 0.43 ± 0.02 a | 0.51 ± 0.09 b |
| F | 8.809 | 33.706 | 24.849 | 69.346 | 17.210 |
| p | 0.016 | 0.001 | 0.003 | <0.001 | 0.003 |
| Group | Notch1 | HES1 | PTEN | Beclin-1 | LC3 |
|---|---|---|---|---|---|
| Control | 0.27 ± 0.05 | 0.44 ± 0.09 | 0.99 ± 0.12 | 0.88 ± 0.04 | 0.60 ± 0.07 |
| Blank plasmid group | 0.19 ± 0.03 | 0.36 ± 0.05 | 0.88 ± 0.07 | 0.76 ± 0.04 | 0.52 ± 0.05 |
| High-dose iAs3+ + blank plasmid group | 0.42 ± 0.03 a | 0.96 ± 0.04 a | 0.66 ± 0.14 a | 1.17 ± 0.06 a | 1.10 ± 0.18 a |
| PTEN overexpression group | 0.12 ± 0.04 b | 0.44 ± 0.02 b | 1.26 ± 0.03 b | 0.71 ± 0.03 b | 0.52 ± 0.08 b |
| High-dose iAs3+ + PTEN overexpression group | 0.16 ± 0.03 | 0.48 ± 0.10 | 0.87 ± 0.11 a | 0.82 ± 0.06 | 0.54 ± 0.12 |
| F | 34.569 | 38.920 | 13.469 | 42.223 | 15.295 |
| p | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 |
| Group | α-SMA | Collagen Ⅰ |
|---|---|---|
| Control | 0.64 ± 0.06 | 0.09 ± 0.01 |
| Blank plasmid group | 0.59 ± 0.02 | 0.11 ± 0.01 |
| High-dose iAs3+ + blank plasmid group | 1.02 ± 0.11 a | 0.21 ± 0.00 a |
| PTEN overexpression group | 0.63 ± 0.06 b | 0.11 ± 0.00 b |
| High-dose iAs3+ + PTEN overexpression group | 0.60 ± 0.07 | 0.15 ± 0.01 |
| F | 20.115 | 47.257 |
| p | <0.001 | <0.001 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, F.; Ding, G.; Yuan, Y.; Zhao, L.; Ding, W.; Wu, S. PTEN Overexpression Alters Autophagy Levels and Slows Sodium Arsenite-Induced Hepatic Stellate Cell Fibrosis. Toxics 2023, 11, 578. https://doi.org/10.3390/toxics11070578
Huang F, Ding G, Yuan Y, Zhao L, Ding W, Wu S. PTEN Overexpression Alters Autophagy Levels and Slows Sodium Arsenite-Induced Hepatic Stellate Cell Fibrosis. Toxics. 2023; 11(7):578. https://doi.org/10.3390/toxics11070578
Chicago/Turabian StyleHuang, Fei, Guanxin Ding, Yanjie Yuan, Lijun Zhao, Wenmeng Ding, and Shunhua Wu. 2023. "PTEN Overexpression Alters Autophagy Levels and Slows Sodium Arsenite-Induced Hepatic Stellate Cell Fibrosis" Toxics 11, no. 7: 578. https://doi.org/10.3390/toxics11070578
APA StyleHuang, F., Ding, G., Yuan, Y., Zhao, L., Ding, W., & Wu, S. (2023). PTEN Overexpression Alters Autophagy Levels and Slows Sodium Arsenite-Induced Hepatic Stellate Cell Fibrosis. Toxics, 11(7), 578. https://doi.org/10.3390/toxics11070578