
miR-153-3p via PIK3R1 Is Involved in Cigarette Smoke-Induced Neurotoxicity in the Brain

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Subjects
2.2. Assessment of Behavior
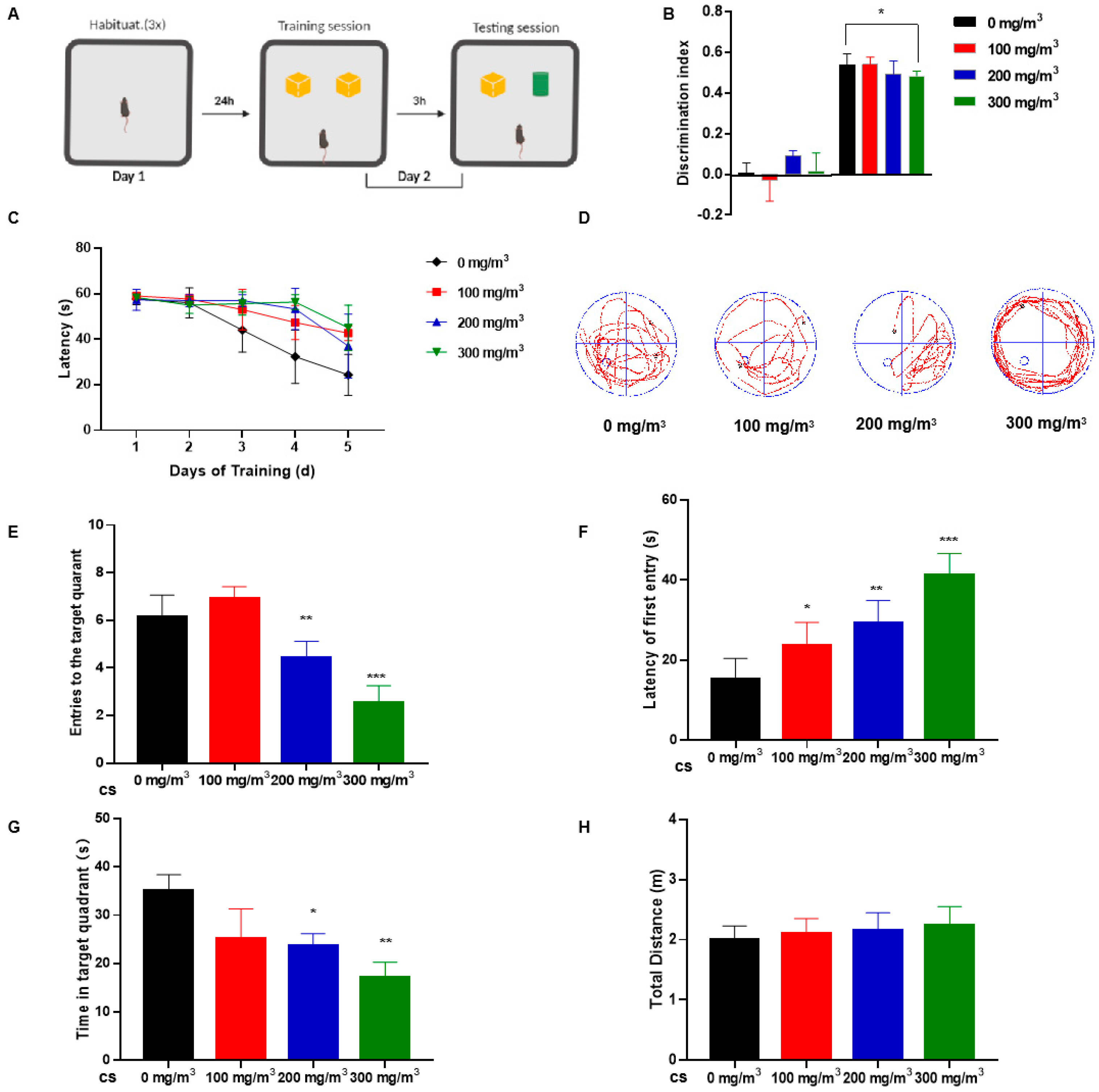
2.2.1. The Novel Object Recognition (NOR) Test Procedure
2.2.2. Conducting the Morris Water Maze (MWM) Test
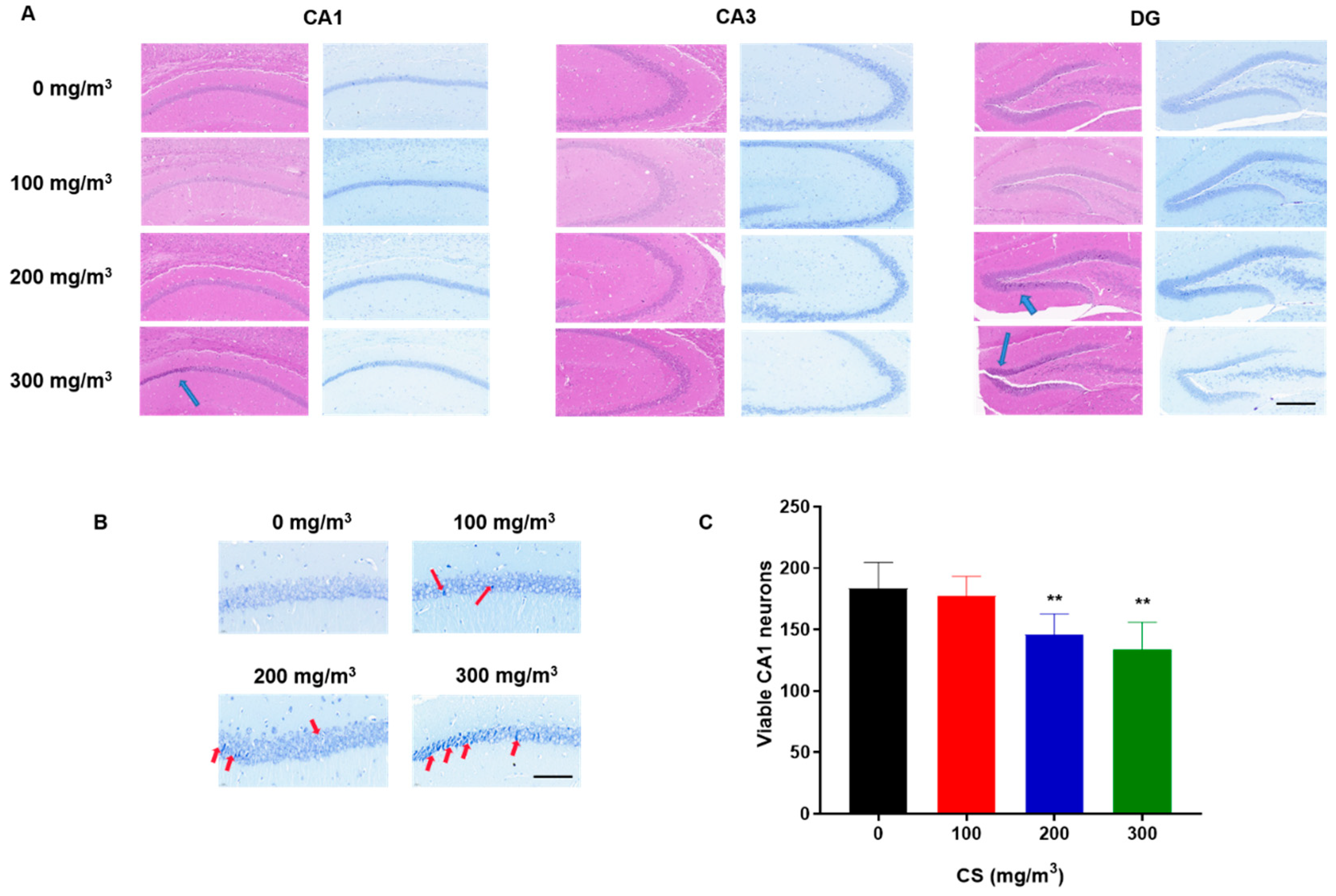
2.3. Hematoxylin and Eosin (H&E) Staining
2.4. Nissl Staining
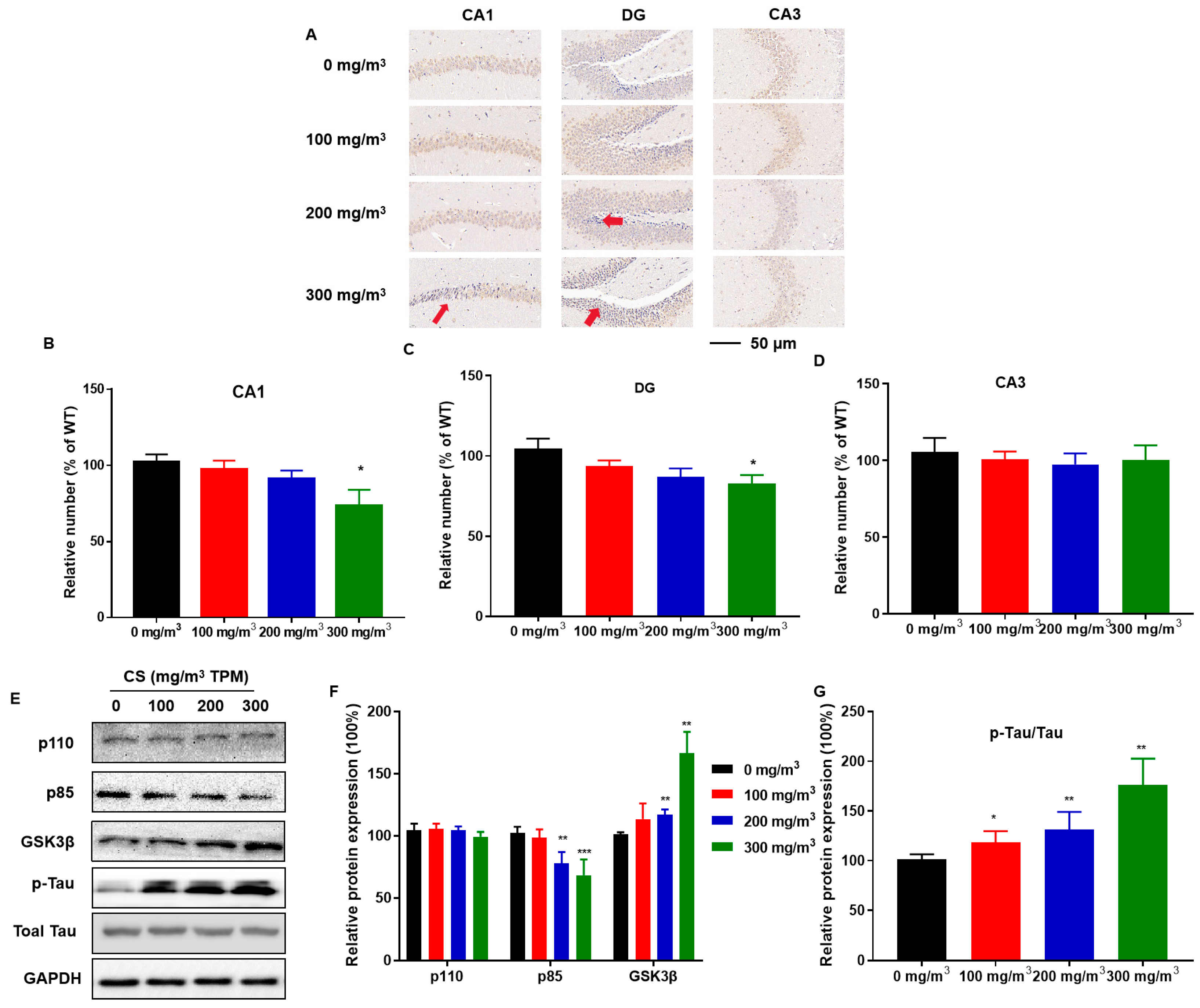
2.5. Neuron-Specific Nuclear Protein (NeuN) Immunohistochemistry
2.6. Preparation of Cigarette Smoke Extract (CSE)
2.7. Cell Culture and Treatment
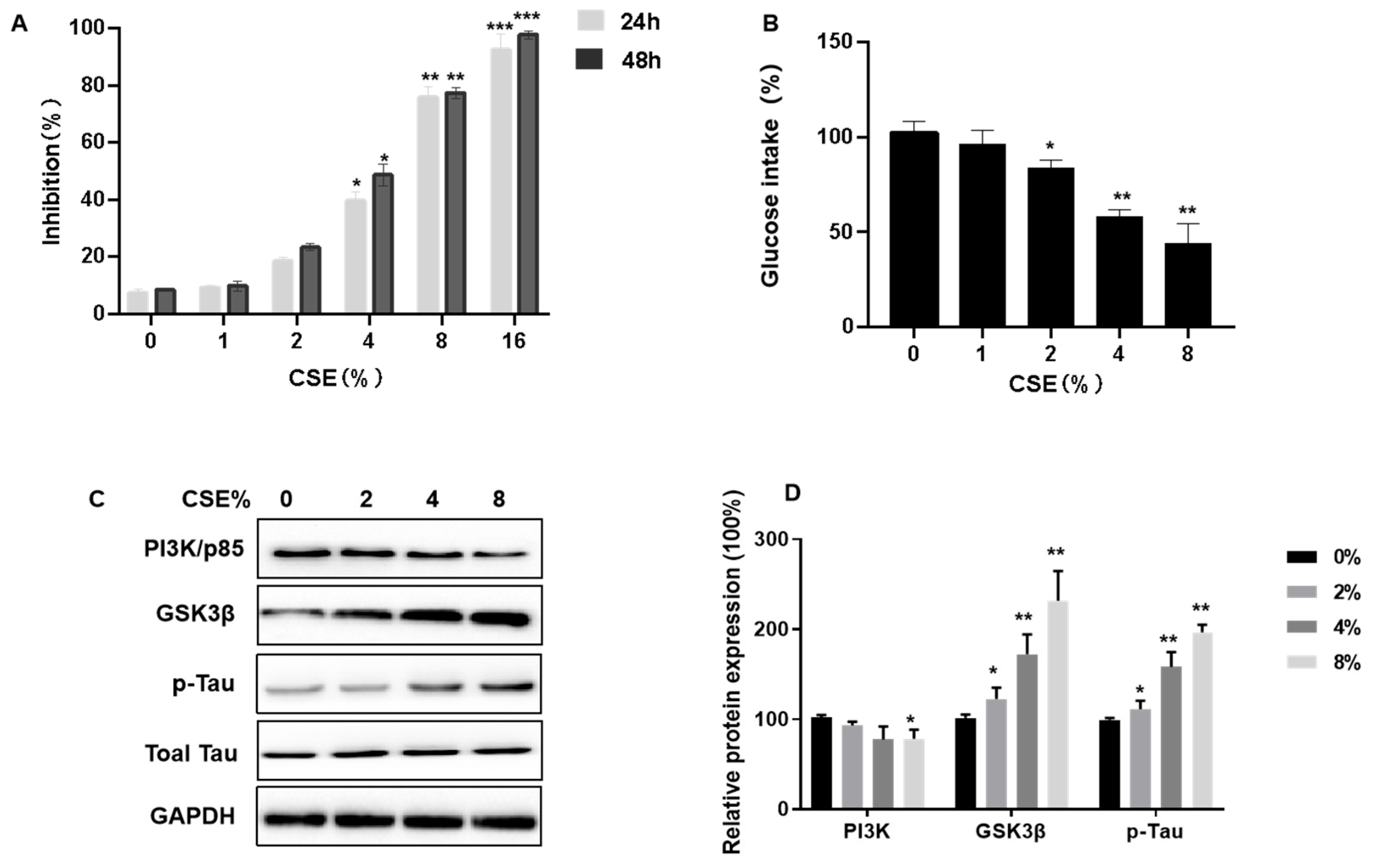
2.8. MTT (3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) Assay
2.9. Glucose Uptake Assay
2.10. RNA Extraction and Quantitative Real-Time Polymerase Chain Reaction (RT-PCR)
2.11. Cell Transfection
2.12. Protein Extraction and Western Blot Assays
2.13. Statistical Analysis
3. Results
3.1. Cigarette Smoke (CS) Impairs Learning Memory and Cognitive Abilities in Mice
3.2. CS Induces Pyramidal Cell Damage in the CA1 Region of the Hippocampus and Histopathological Changes in the Hippocampi of Mice
3.3. CS Causes Hippocampal Neuron Deficiency, Disruption of PI3K/GSK3β Insulin Signaling Pathways, and Tau Protein Hyperphosphorylation
3.4. CSE Exposure Inhibits Cell Viability, Decreases Insulin Sensitivity, Induces Tau Hyperphosphorylation, and Affects the PI3K/GSK3β Signaling Pathway in SH-SY5Y Cells
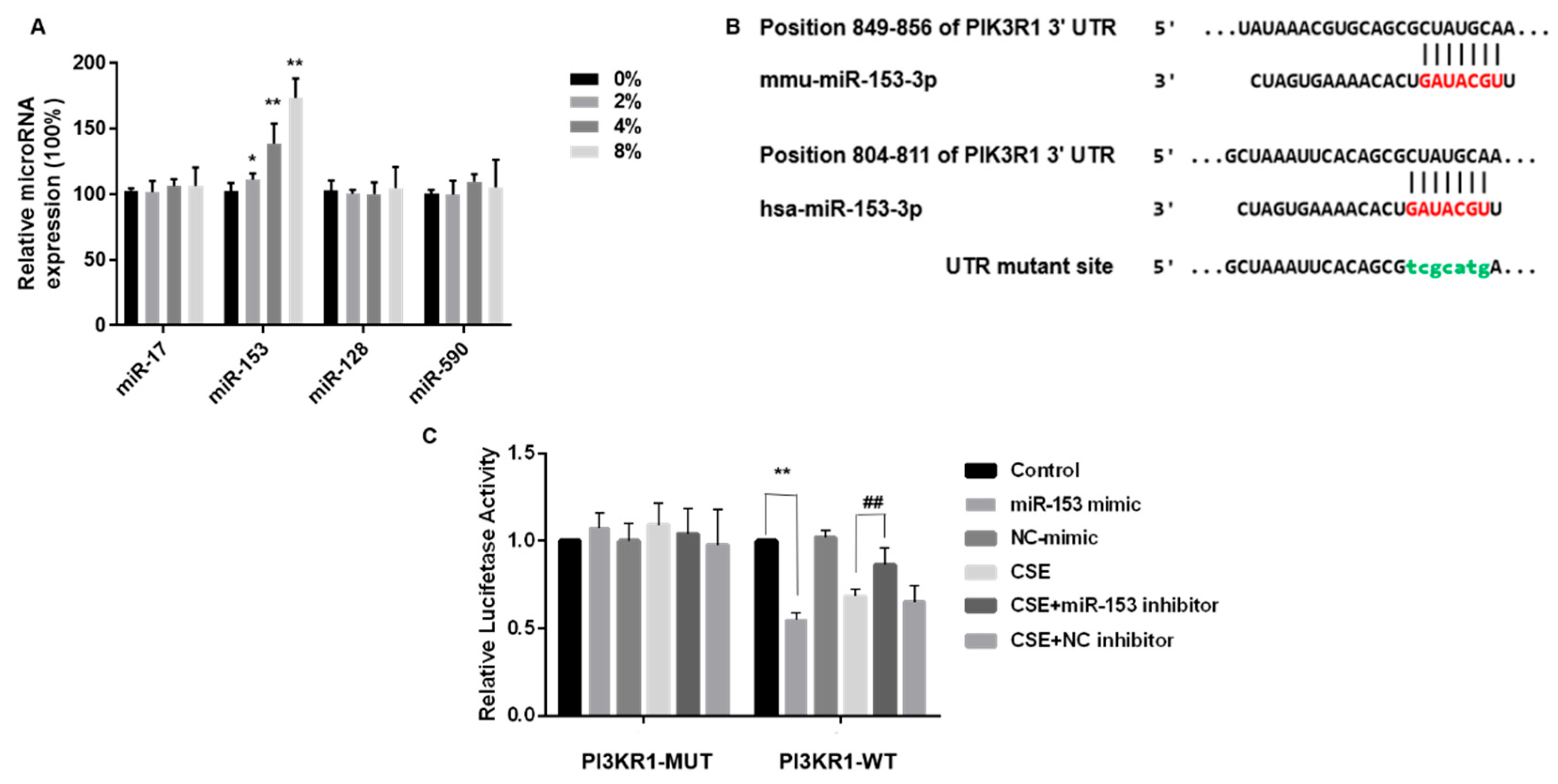
3.5. miR-153-5p Expression Is Elevated by CSE and Targets the 3′UTR Site of PIK3R1
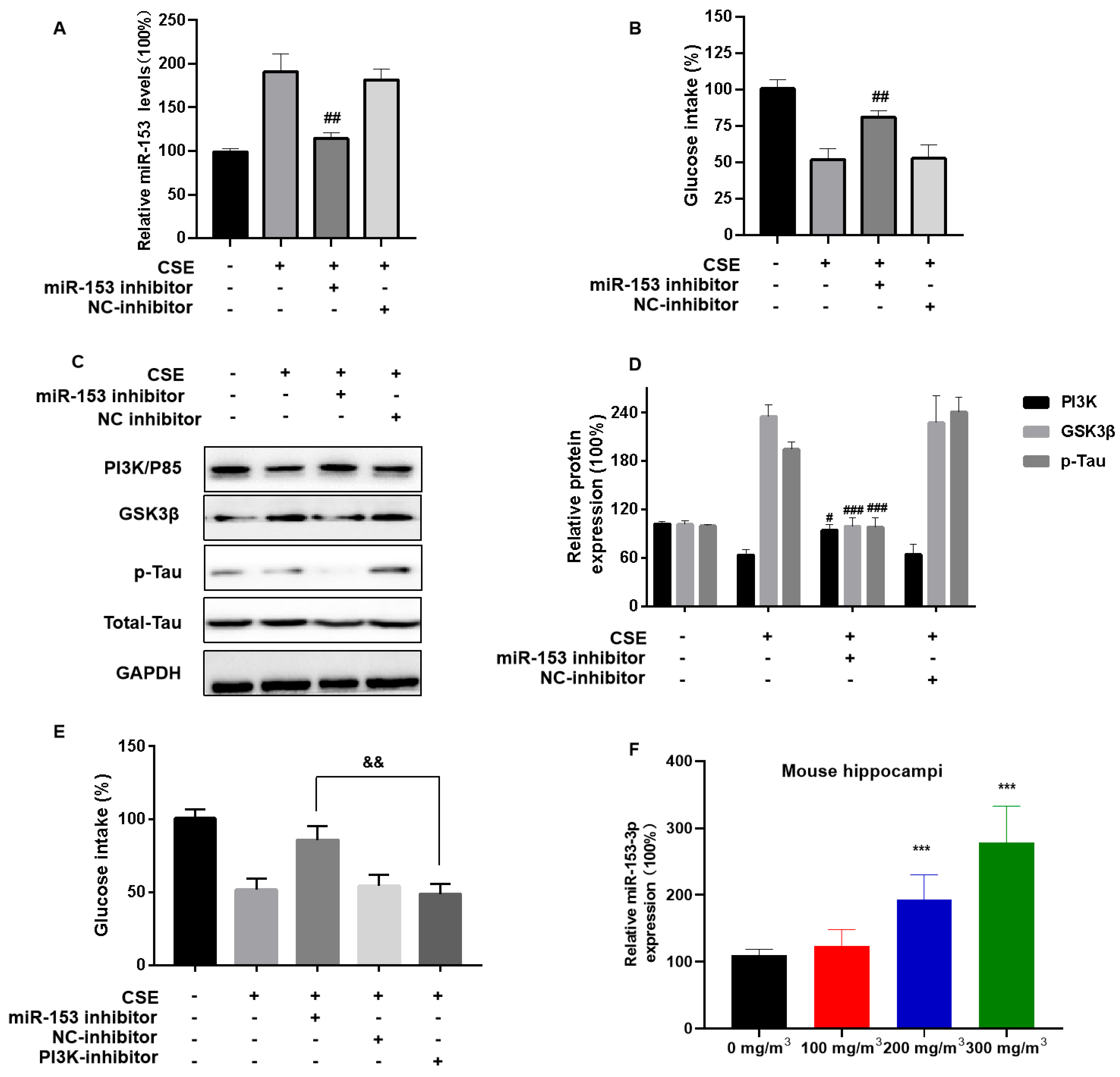
3.6. miR-153-5p via PI3K Is Involved in CSE-Induced Brain Insulin Resistance and Tau Protein Hyperphosphorylation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Lim, D.; Jeong, J.H.; Song, J. Lipocalin 2 regulates iron homeostasis, neuroinflammation, and insulin resistance in the brains of patients with dementia: Evidence from the current literature. CNS Neurosci. Ther. 2021, 27, 883–894. [Google Scholar] [CrossRef] [PubMed]
- Chandarana, C.V.; Roy, S. Comprehensive Review on Neuro-degenerative Type 3 DM. Curr. Diabetes Rev. 2022, 18, e1922787386. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Shu, X.O.; Li, H.; Cai, H.; Liu, Q.; Zheng, W.; Xiang, Y.B.; Villegas, R. Physical activity, smoking, and alcohol consumption in association with incidence of type 2 diabetes among middle-aged and elderly Chinese men. PLoS ONE 2013, 8, e77919. [Google Scholar] [CrossRef] [PubMed]
- Kellar, D.; Craft, S. Brain insulin resistance in Alzheimer’s disease and related disorders: Mechanisms and therapeutic approaches. Lancet Neurol. 2020, 19, 758–766. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.; Ta, Q.; Nguyen, T.; Nguyen, T.T.D.; Giau, V.V. Type 3 Diabetes and Its Role Implications in Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 3165. [Google Scholar] [CrossRef]
- Kroner, Z. The relationship between Alzheimer’s disease and diabetes: Type 3 diabetes? Altern. Med. Rev. 2009, 14, 373–379. [Google Scholar] [PubMed]
- Janoutova, J.; Machaczka, O.; Zatloukalova, A.; Janout, V. Is Alzheimer’s disease a type 3 diabetes? A review. Cent. Eur. J. Public Health 2022, 30, 139–143. [Google Scholar] [CrossRef]
- Kim, B.; Feldman, E.L. Insulin resistance in the nervous system. Trends Endocrinol. Metab. 2012, 23, 133–141. [Google Scholar] [CrossRef]
- Saltiel, A.R. Insulin signaling in health and disease. J. Clin. Investig. 2021, 131, e142241. [Google Scholar] [CrossRef]
- Flores-Opazo, M.; McGee, S.L.; Hargreaves, M. Exercise and GLUT4. Exerc. Sport Sci. Rev. 2020, 48, 110–118. [Google Scholar] [CrossRef]
- Huang, S.; Czech, M.P. The GLUT4 glucose transporter. Cell Metab. 2007, 5, 237–252. [Google Scholar] [CrossRef] [PubMed]
- Desale, S.E.; Chidambaram, H.; Chinnathambi, S. G-protein coupled receptor, PI3K and Rho signaling pathways regulate the cascades of Tau and amyloid-beta in Alzheimer’s disease. Mol. Biomed. 2021, 2, 17. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, F.; Lucas, J.J.; Avila, J. GSK3 and tau: Two convergence points in Alzheimer’s disease. J. Alzheimer Dis. 2013, 33 (Suppl. S1), S141–S144. [Google Scholar] [CrossRef] [PubMed]
- Pirscoveanu, D.; Pirici, I.; Tudorica, V.; Balşeanu, T.A.; Albu, V.C.; Bondari, S.; Bumbea, A.M.; Pirşcoveanu, M. Tau protein in neurodegenerative diseases—A review. Rom. J. Morphol. Embryol. 2017, 58, 1141–1150. [Google Scholar] [PubMed]
- Lucas, J.J.; Hernandez, F.; Gomez-Ramos, P.; Moran, M.A.; Hen, R.; Avila, J. Decreased nuclear beta-catenin, tau hyperphosphorylation and neurodegeneration in GSK-3beta conditional transgenic mice. EMBO J. 2001, 20, 27–39. [Google Scholar] [CrossRef]
- Chen, H.; Saad, S.; Sandow, S.L.; Bertrand, P.P. Cigarette smoking and brain regulation of energy homeostasis. Front. Pharmacol. 2012, 3, 147. [Google Scholar] [CrossRef]
- Tsermpini, E.E.; Kalogirou, C.I.; Kyriakopoulos, G.C.; Patrinos, G.P.; Stathopoulos, C. miRNAs as potential diagnostic biomarkers and pharmacogenomic indicators in psychiatric disorders. Pharmacogenomics J. 2022, 22, 211–222. [Google Scholar] [CrossRef]
- Klyucherev, T.O.; Olszewski, P.; Shalimova, A.A.; Chubarev, V.N.; Tarasov, V.V.; Attwood, M.M.; Syvanen, S.; Schioth, H.B. Advances in the development of new biomarkers for Alzheimer’s disease. Transl. Neurodegener. 2022, 11, 25. [Google Scholar] [CrossRef]
- Koh, H.S.; Lee, S.; Lee, H.J.; Min, J.W.; Iwatsubo, T.; Teunissen, C.E.; Cho, H.J.; Ryu, J.H. Targeting MicroRNA-485-3p Blocks Alzheimer’s Disease Progression. Int. J. Mol. Sci. 2021, 22, 13136. [Google Scholar] [CrossRef]
- Da, S.F.; Rode, M.P.; Vietta, G.G.; Iop, R.D.R.; Creczynski-Pasa, T.B.; Martin, A.S.; Da Silva, R. Expression levels of specific microRNAs are increased after exercise and are associated with cognitive improvement in Parkinson’s disease. Mol. Med. Rep. 2021, 24, 618. [Google Scholar]
- Kiko, T.; Nakagawa, K.; Tsuduki, T.; Furukawa, K.; Arai, H.; Miyazawa, T. MicroRNAs in plasma and cerebrospinal fluid as potential markers for Alzheimer’s disease. J. Alzheimers Dis. 2014, 39, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Cui, Y.; Luo, F.; Liu, X.; Wang, Q.; Bai, J.; Dong, F.; Sun, Q.; Lu, L.; Xu, H.; et al. MicroRNA-191, acting via the IRS-1/Akt signaling pathway, is involved in the hepatic insulin resistance induced by cigarette smoke extract. Environ. Sci. Pollut. Res. Int. 2018, 25, 22400–22407. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Xu, H.; Xue, J.; Yang, Q.; Chen, C.; Yang, P.; Han, A.; Tu, Q.; Lu, J.; Gao, X.; et al. MALAT1 via microRNA-17 regulation of insulin transcription is involved in the dysfunction of pancreatic beta-cells induced by cigarette smoke extract. J. Cell Physiol. 2018, 233, 8862–8873. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Wang, Q.; Sun, Q.; Qin, Y.; Han, A.; Cao, Y.; Yang, Q.; Yang, P.; Lu, J.; Liu, Q.; et al. In type 2 diabetes induced by cigarette smoking, activation of p38 MAPK is involved in pancreatic beta-cell apoptosis. Environ. Sci. Pollut. Res. Int. 2018, 25, 9817–9827. [Google Scholar] [CrossRef]
- Azevedo, P.S.; Polegato, B.F.; Paiva, S.; Costa, N.; Santos, P.; Bazan, S.; Fernandes, A.A.H.; Fabro, A.; Pires, V.; Tanni, S.E.; et al. The role of glucose metabolism and insulin resistance in cardiac remodelling induced by cigarette smoke exposure. J. Cell Mol. Med. 2021, 25, 1314–1318. [Google Scholar] [CrossRef] [PubMed]
- Hartog, M.; Zhang, Q.Y.; Ding, X. Role of Mouse Cytochrome P450 Enzymes of the Cyp2abfgs Subfamilies in the Induction of Lung Inflammation by Cigarette Smoke Exposure. Toxicol. Sci. 2019, 172, 123–131. [Google Scholar] [CrossRef]
- Jang, E.S.; Jeong, S.H.; Hwang, S.H.; Kim, H.Y.; Ahn, S.Y.; Lee, J.; Lee, S.H.; Park, Y.S.; Hwang, J.H.; Kim, J.W.; et al. Effects of coffee, smoking, and alcohol on liver function tests: A comprehensive cross-sectional study. BMC Gastroenterol. 2012, 12, 145. [Google Scholar] [CrossRef]
- He, K.; Nie, L.; Ali, T.; Wang, S.; Chen, X.; Liu, Z.; Li, W.; Zhang, K.; Xu, J.; Liu, J.; et al. Adiponectin alleviated Alzheimer-like pathologies via autophagy-lysosomal activation. Aging Cell 2021, 20, e13514. [Google Scholar] [CrossRef]
- Lueptow, L.M. Novel Object Recognition Test for the Investigation of Learning and Memory in Mice. J. Vis. Exp. 2017, 126, e55718. [Google Scholar]
- Liu, F.; Iqbal, K.; Grundke-Iqbal, I.; Gong, C.X. Involvement of aberrant glycosylation in phosphorylation of tau by cdk5 and GSK-3beta. FEBS Lett. 2002, 530, 209–214. [Google Scholar] [CrossRef]
- Wooten, K.G.; McGuire, L.C.; Olivari, B.S.; Jackson, E.M.J.; Croft, J.B. Racial and Ethnic Differences in Subjective Cognitive Decline—United States, 2015–2020. Morb. Mortal. Wkly. Rep. 2023, 72, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Nianogo, R.A.; Rosenwohl-Mack, A.; Yaffe, K.; Carrasco, A.; Hoffmann, C.M.; Barnes, D.E. Risk Factors Associated With Alzheimer Disease and Related Dementias by Sex and Race and Ethnicity in the US. JAMA Neurol. 2022, 79, 584–591. [Google Scholar] [CrossRef] [PubMed]
- Nichols, E.; Steinmetz, J.D.; Vollset, S.E.; Fukutaki, K.; Chalek, J.; Abd-Allah, F.; Abdoli, A.; Abualhasan, A.; Abu-Gharbieh, E.; Akram, T.T.; et al. Estimation of the global prevalence of dementia in 2019 and forecasted prevalence in 2050: An analysis for the Global Burden of Disease Study 2019. Lancet Public Health 2022, 7, e105–e125. [Google Scholar] [CrossRef]
- Aseervatham G, S.B.; Choi, S.; Krishnan, J.; Ruckmani, K. Cigarette smoke and related risk factors in neurological disorders: An update. Biomed. Pharmacother. 2017, 85, 79–86. [Google Scholar]
- Liu, D.; Zhao, Y.; Qi, Y.; Gao, Y.; Tu, D.; Wang, Y.; Gao, H.M.; Zhou, H. Benzo(a)pyrene exposure induced neuronal loss, plaque deposition, and cognitive decline in APP/PS1 mice. J. Neuroinflamm. 2020, 17, 258. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.Y.; Guo, L.L.; Wang, P.; Song, J.; Le, Y.Y.; Viollet, B.; Miao, C.Y. Chronic exposure to nicotine enhances insulin sensitivity through alpha7 nicotinic acetylcholine receptor-STAT3 pathway. PLoS ONE 2012, 7, e51217. [Google Scholar] [CrossRef] [PubMed]
- Facchini, F.S.; Hollenbeck, C.B.; Jeppesen, J.; Chen, Y.D.; Reaven, G.M. Insulin resistance and cigarette smoking. Lancet 1992, 339, 1128–1130. [Google Scholar] [CrossRef]
- Chen, Z.; Liu, X.A.; Kenny, P.J. Central and peripheral actions of nicotine that influence blood glucose homeostasis and the development of diabetes. Pharmacol. Res. 2023, 194, 106860. [Google Scholar] [CrossRef]
- Khanna, A.; Guo, M.; Mehra, M.; Royal, W. Inflammation and oxidative stress induced by cigarette smoke in Lewis rat brains. J. Neuroimmunol. 2013, 254, 69–75. [Google Scholar] [CrossRef]
- Wang, H.; Chen, H.; Han, S.; Fu, Y.; Tian, Y.; Liu, Y.; Wang, A.; Hou, H.; Hu, Q. Decreased mitochondrial DNA copy number in nerve cells and the hippocampus during nicotine exposure is mediated by autophagy. Ecotoxicol. Environ. Saf. 2021, 226, 112831. [Google Scholar] [CrossRef]
- La Maestra, S.; Kisby, G.E.; Micale, R.T.; Johnson, J.; Kow, Y.W.; Bao, G.; Sheppard, C.; Stanfield, S.; Tran, H.; Woltjer, R.L.; et al. Cigarette smoke induces DNA damage and alters base-excision repair and tau levels in the brain of neonatal mice. Toxicol. Sci. 2011, 123, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, P.; Horn, K.H.; Greene, R.M.; Michele, M. Prenatal exposure to environmental tobacco smoke alters gene expression in the developing murine hippocampus. Reprod. Toxicol. 2010, 29, 164–175. [Google Scholar] [CrossRef] [PubMed]
- Dobric, A.; De Luca, S.N.; Seow, H.J.; Wang, H.; Brassington, K.; Chan, S.M.H.; Mou, K.; Erlich, J.; Liong, S.; Selemidis, S.; et al. Cigarette Smoke Exposure Induces Neurocognitive Impairments and Neuropathological Changes in the Hippocampus. Front. Mol. Neurosci. 2022, 15, 893083. [Google Scholar] [CrossRef] [PubMed]
- James, D.E.; Stockli, J.; Birnbaum, M.J. The aetiology and molecular landscape of insulin resistance. Nat. Rev. Mol. Cell Biol. 2021, 22, 751–771. [Google Scholar] [CrossRef]
- Taniguchi, C.M.; Emanuelli, B.; Kahn, C.R. Critical nodes in signalling pathways: Insights into insulin action. Nat. Rev. Mol. Cell Biol. 2006, 7, 85–96. [Google Scholar] [CrossRef]
- Goda, S.; Kaneshita, Y.; Inoue, H.; Domae, E.; Ikeo, T.; Iida, J.; Domae, N. Enamel matrix derivative protein stimulated wound healing via phosphoinositide 3-kinase. J. Periodontol. 2009, 80, 1631–1637. [Google Scholar] [CrossRef]
- Prickaerts, J.; Moechars, D.; Cryns, K.; Lenaerts, I.; van Craenendonck, H.; Goris, I.; Daneels, G.; Bouwknecht, J.A.; Steckler, T. Transgenic mice overexpressing glycogen synthase kinase 3beta: A putative model of hyperactivity and mania. J. Neurosci. 2006, 26, 9022–9029. [Google Scholar] [CrossRef]
- Narasimhan, M.; Riar, A.K.; Rathinam, M.L.; Vedpathak, D.; Henderson, G.; Mahimainathan, L. Hydrogen peroxide responsive miR153 targets Nrf2/ARE cytoprotection in paraquat induced dopaminergic neurotoxicity. Toxicol. Lett. 2014, 228, 179–191. [Google Scholar] [CrossRef]
- Gui, Y.; Liu, H.; Zhang, L.; Lv, W.; Hu, X. Altered microRNA profiles in cerebrospinal fluid exosome in Parkinson disease and Alzheimer disease. Oncotarget 2015, 6, 37043–37053. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| miR-153 | 5′-GCUAAAUUCACAGCGCUAUGCAA-3′ |
| 3′-CUAGUGAAAACACUGAUACGUU-5′ | |
| miR-17 | 5′-GAGGCAAGGUUAUAUGCACUUUC-3′ |
| 3′-GAUGGACGUGACAUUCGUGAAAC-5′ | |
| miR-128 | 5′-GACCCAGACACAUCGCACUGUGG-3′ |
| 3′-UUUCUCUGGCCAA-GUGACACU-5′ | |
| miR-590 | 5′-AAAUGUACCUUCAGAAUAAGCUUC-3′ |
| 3′-GACGUGAAAAUACUUAUUCGAG-5′ | |
| U6 | 5′-CGCTTCGGCAGCACATATACTAAAATTGGAAC-3′ |
| 3′-CGTTCCTACTGTGCGTTTAAGCACTTCG-5′ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, Q.; Wang, H.; Yang, M.; Xia, H.; Wu, Y.; Liu, Q.; Tang, H. miR-153-3p via PIK3R1 Is Involved in Cigarette Smoke-Induced Neurotoxicity in the Brain. Toxics 2023, 11, 969. https://doi.org/10.3390/toxics11120969
Sun Q, Wang H, Yang M, Xia H, Wu Y, Liu Q, Tang H. miR-153-3p via PIK3R1 Is Involved in Cigarette Smoke-Induced Neurotoxicity in the Brain. Toxics. 2023; 11(12):969. https://doi.org/10.3390/toxics11120969
Chicago/Turabian StyleSun, Qian, Hailan Wang, Mingxue Yang, Haibo Xia, Yao Wu, Qizhan Liu, and Huanwen Tang. 2023. "miR-153-3p via PIK3R1 Is Involved in Cigarette Smoke-Induced Neurotoxicity in the Brain" Toxics 11, no. 12: 969. https://doi.org/10.3390/toxics11120969
APA StyleSun, Q., Wang, H., Yang, M., Xia, H., Wu, Y., Liu, Q., & Tang, H. (2023). miR-153-3p via PIK3R1 Is Involved in Cigarette Smoke-Induced Neurotoxicity in the Brain. Toxics, 11(12), 969. https://doi.org/10.3390/toxics11120969