
Effects of Chronic Hydrocodone Exposure and Ceftriaxone on the Expression of Astrocytic Glutamate Transporters in Mesocorticolimbic Brain Regions of C57/BL Mice



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal
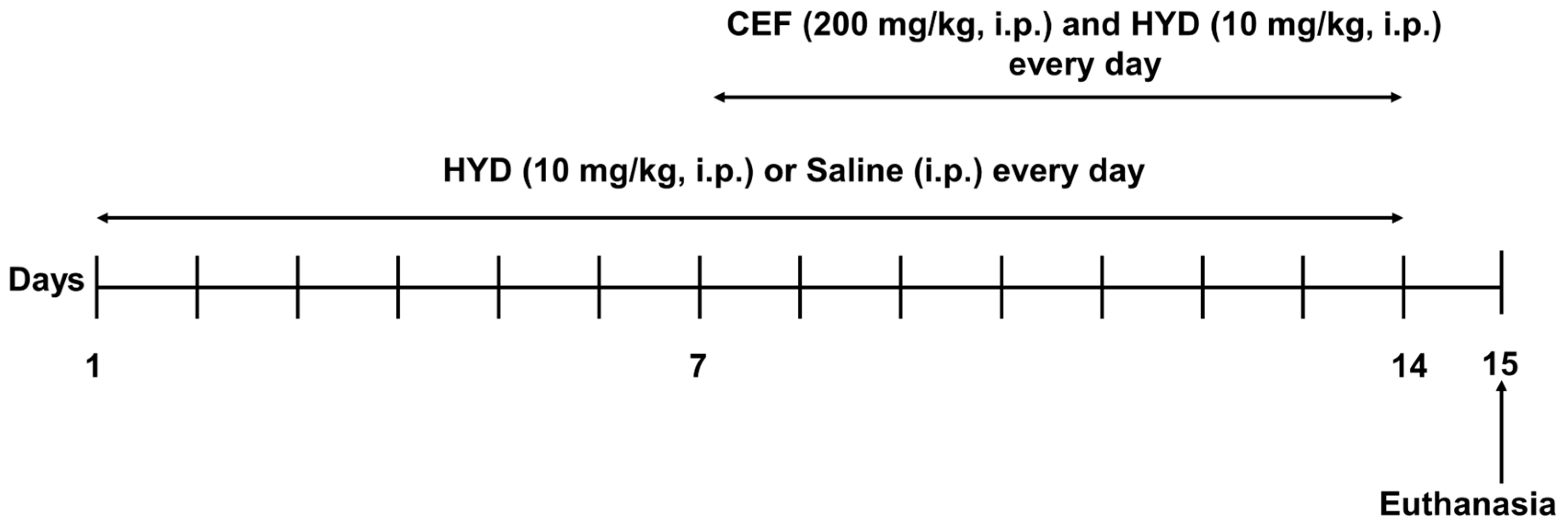
2.2. Drugs and Dosing
2.3. Brain Tissue Extraction
2.4. Western Blot Analysis
2.5. Statistical Analysis
3. Results
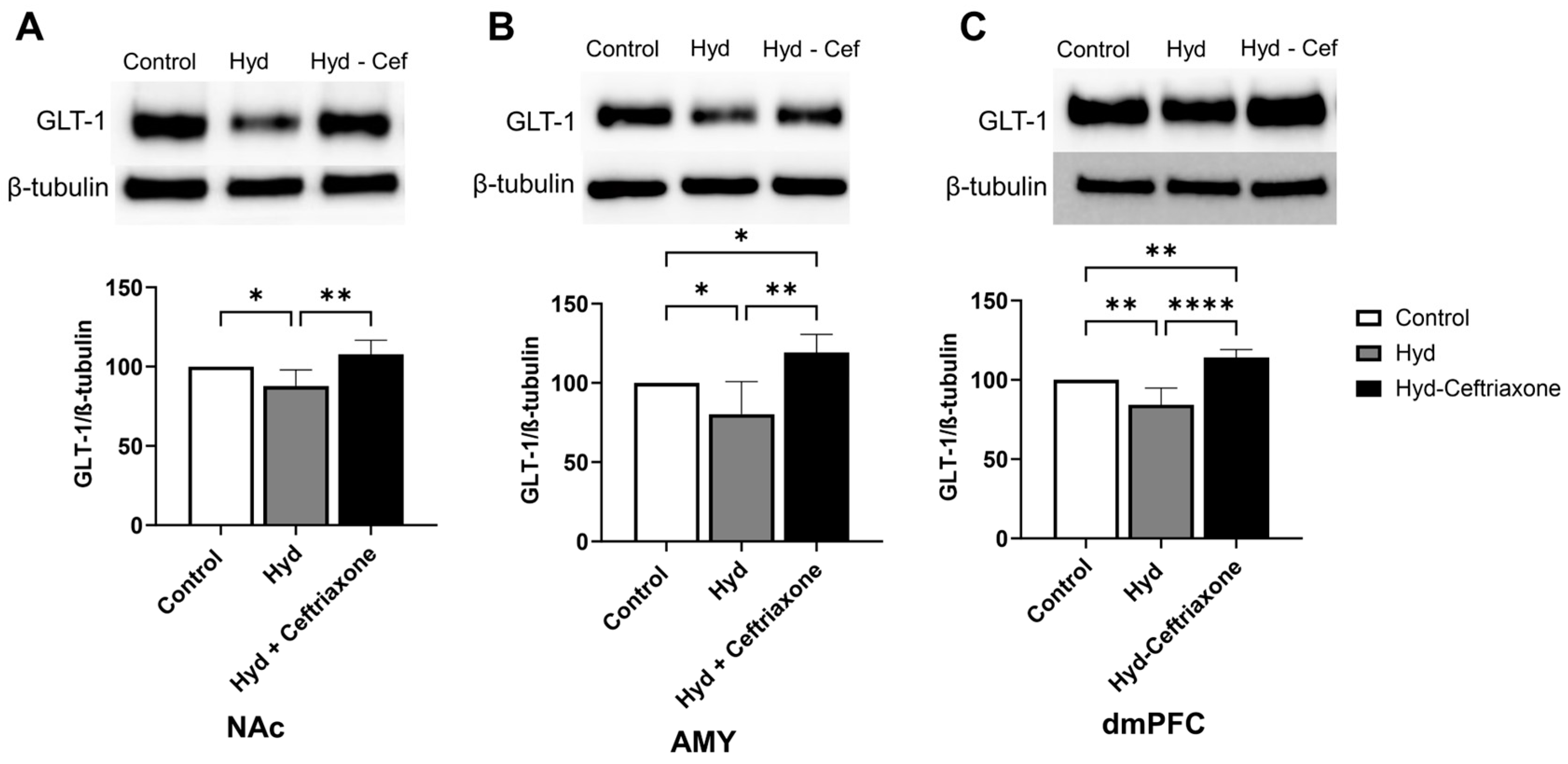
3.1. Effect of Chronic Hydrocodone Exposure and Ceftriaxone on GLT-1 Protein Expression in the NAC, AMY, and dmPFC
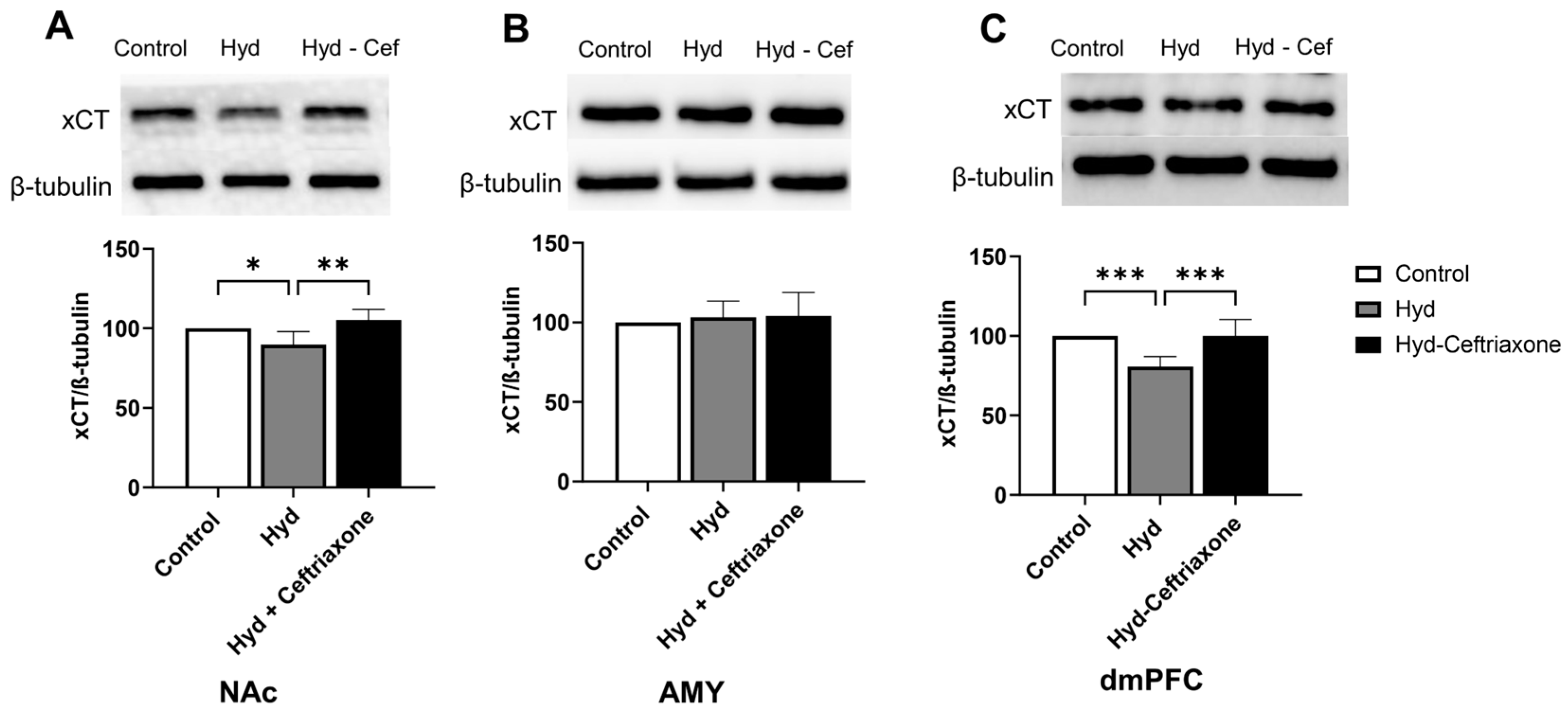
3.2. Effect of Chronic Hydrocodone Exposure and Ceftriaxone on xCT Protein Expression in the NAc, AMY, and dmPFC
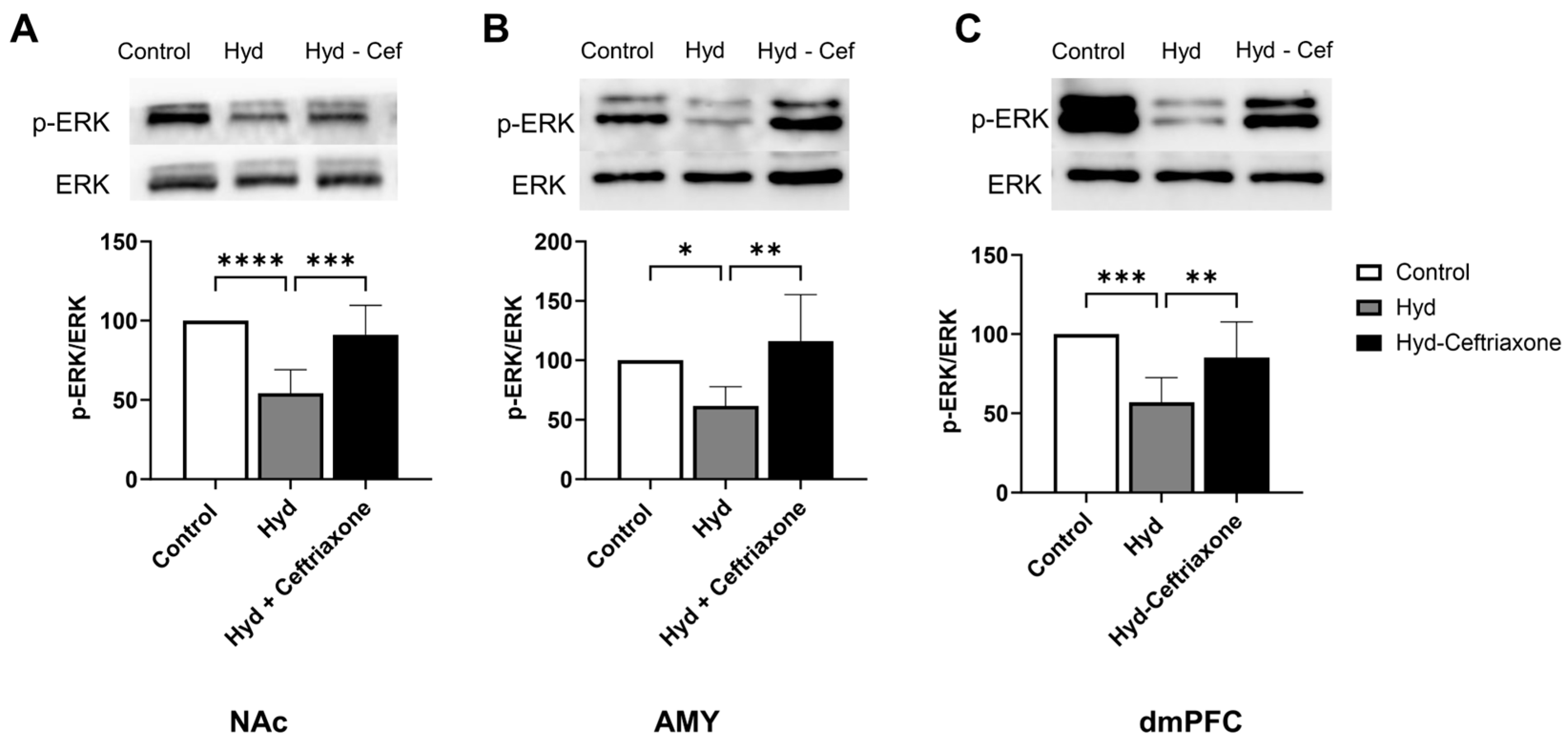
3.3. Effects of Chronic Hydrocodone Exposure and Ceftriaxone on p-ERK Protein Expression in the NAc, AMY, and dmPFC
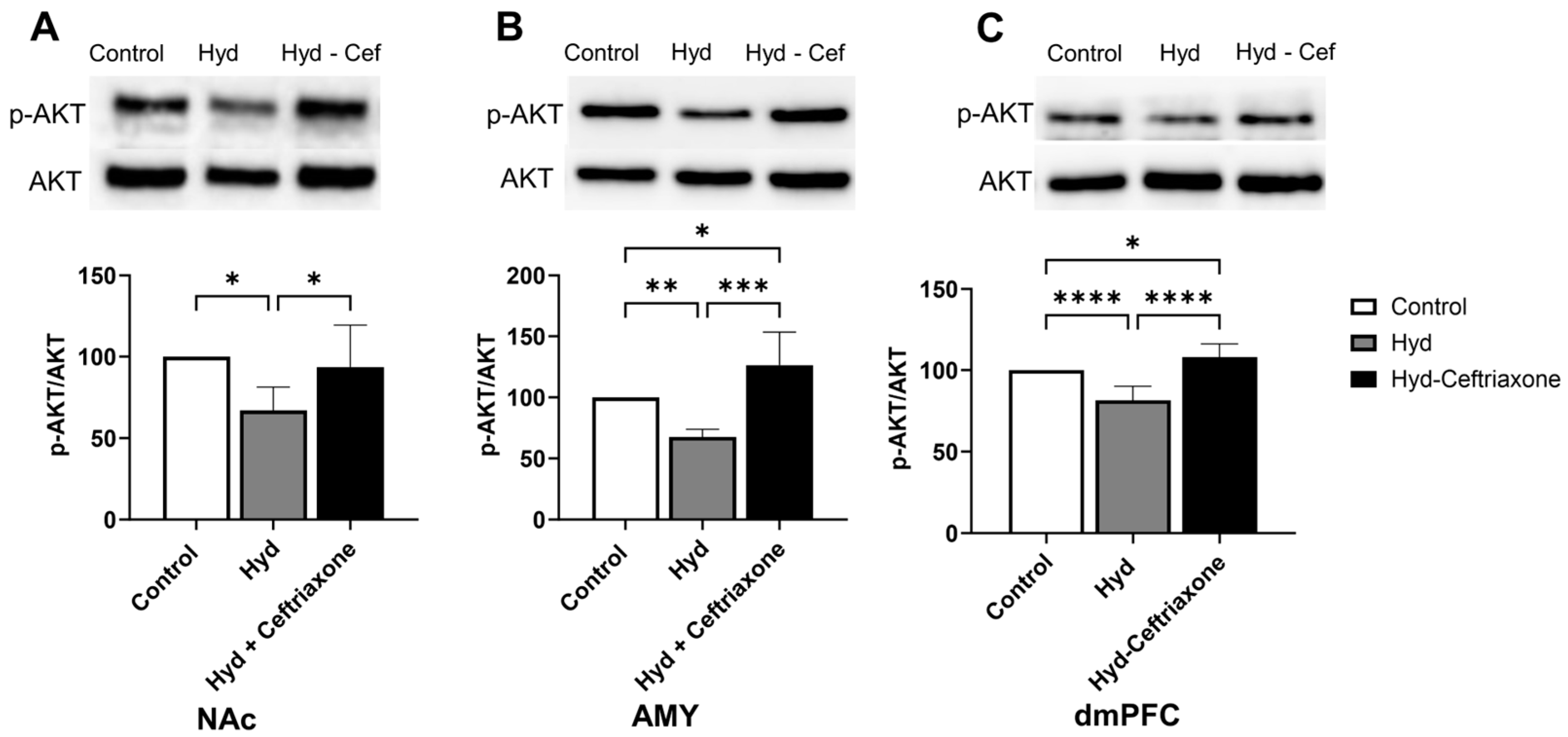
3.4. Effect of Chronic Hydrocodone Exposure and Ceftriaxone on p-AKT Protein Expression in the NAc, AMY, and dmPFC
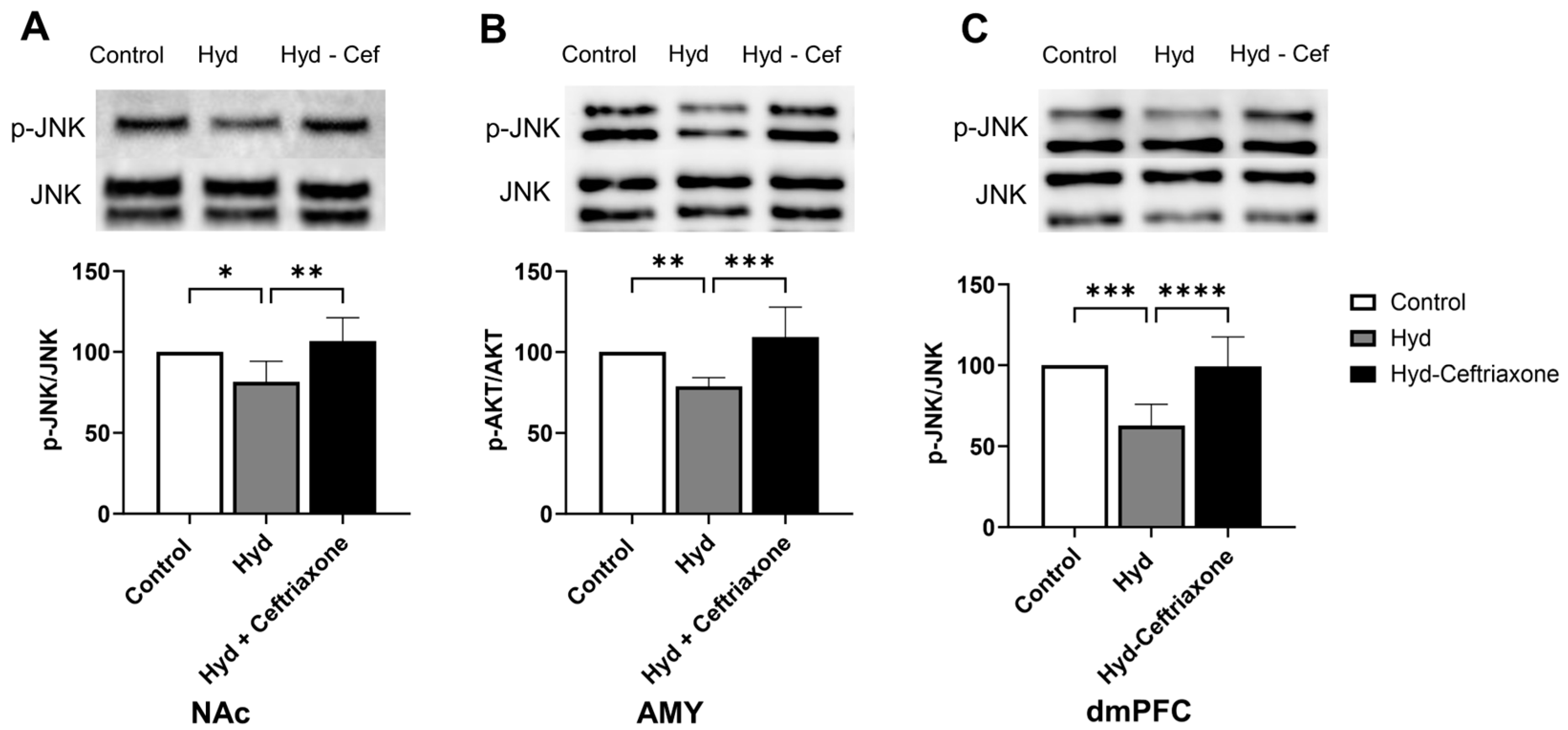
3.5. Effect of Chronic Hydrocodone Exposure and Ceftriaxone on p-JNK Protein Expression in the NAc, AMY, and dmPFC
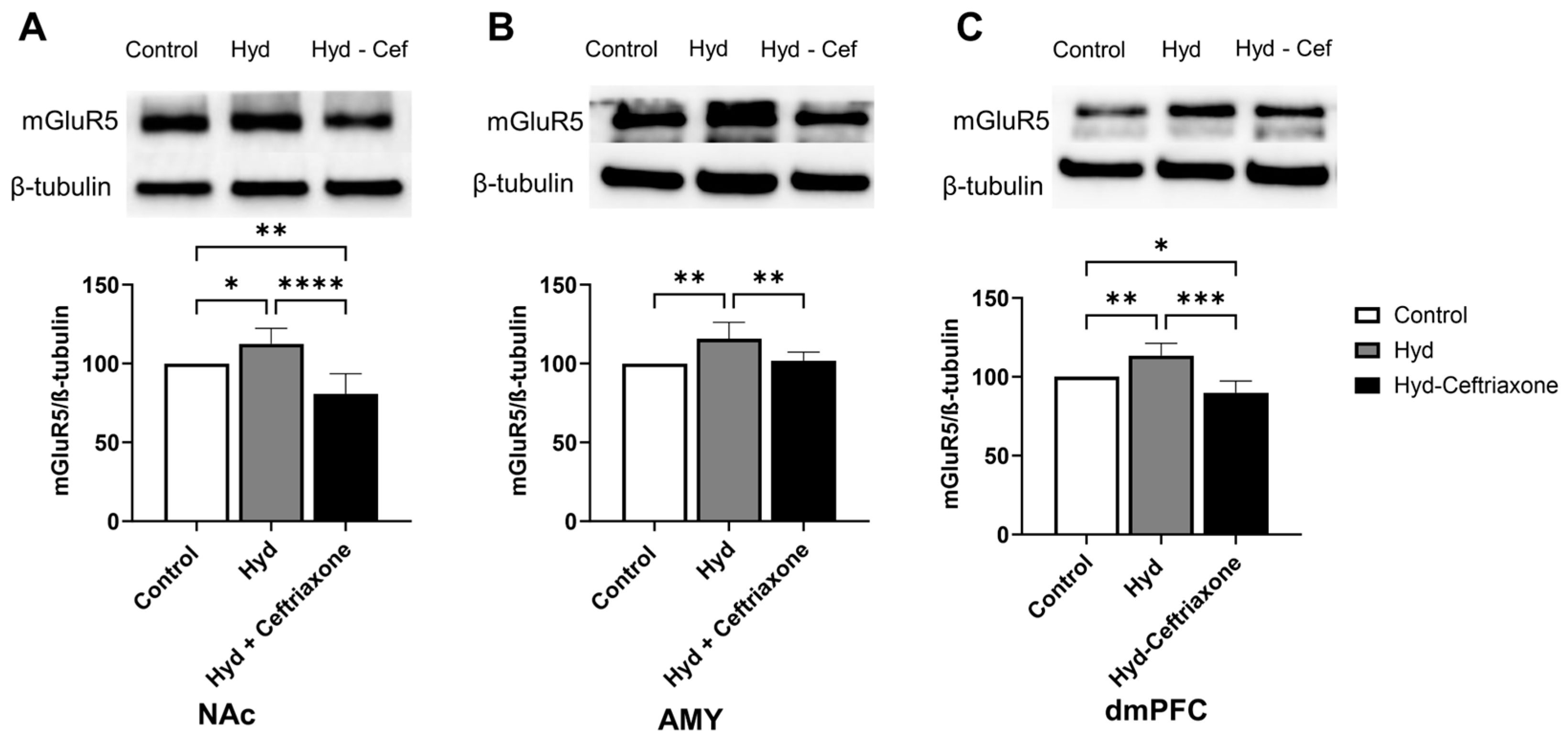
3.6. Effect of Chronic Hydrocodone Exposure and Ceftriaxone on mGluR5 Protein Expression in the NAc, AMY, and dmPFC
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rudd, R.A.; Seth, P.; David, F.; Scholl, L. Increases in Drug and Opioid-Involved Overdose Deaths—United States, 2010–2015. MMWR Morb. Mortal. Wkly. Rep. 2016, 65, 1445–1452. [Google Scholar] [CrossRef]
- Singla, A.; Sloan, P. Pharmacokinetic evaluation of hydrocodone/acetaminophen for pain management. J. Opioid Manag. 2013, 9, 71–80. [Google Scholar] [CrossRef]
- Gould, H.J., III; Paul, D. Hydrocodone extended-release: Pharmacodynamics, pharmacokinetics and behavioral pharmacology of a controversy. Pharmacol. Res. 2015, 91, 99–103. [Google Scholar] [CrossRef]
- Reeves, K.C.; Shah, N.; Munoz, B.; Atwood, B.K. Opioid Receptor-Mediated Regulation of Neurotransmission in the Brain. Front. Mol. Neurosci. 2022, 15, 919773. [Google Scholar] [CrossRef]
- Corkrum, M.; Rothwell, P.E.; Thomas, M.J.; Kofuji, P.; Araque, A. Opioid-Mediated Astrocyte-Neuron Signaling in the Nucleus Accumbens. Cells 2019, 8, 586. [Google Scholar] [CrossRef]
- Alasmari, F.; Goodwani, S.; McCullumsmith, R.E.; Sari, Y. Role of glutamatergic system and mesocorticolimbic circuits in alcohol dependence. Prog. Neurobiol. 2018, 171, 32–49. [Google Scholar] [CrossRef]
- Baker, D.A.; Xi, Z.X.; Shen, H.; Swanson, C.J.; Kalivas, P.W. The origin and neuronal function of in vivo nonsynaptic glutamate. J. Neurosci. 2002, 22, 9134–9141. [Google Scholar] [CrossRef]
- Danbolt, N.C. Glutamate uptake. Prog. Neurobiol. 2001, 65, 1–105. [Google Scholar] [CrossRef]
- Rao, P.S.; Sari, Y. Glutamate transporter 1: Target for the treatment of alcohol dependence. Curr. Med. Chem. 2012, 19, 5148–5156. [Google Scholar] [CrossRef]
- Alasmari, F.; Crotty Alexander, L.E.; Nelson, J.A.; Schiefer, I.T.; Breen, E.; Drummond, C.A.; Sari, Y. Effects of chronic inhalation of electronic cigarettes containing nicotine on glial glutamate transporters and alpha-7 nicotinic acetylcholine receptor in female CD-1 mice. Prog. Neuropsychopharmacol. Biol. Psychiatry 2017, 77, 1–8. [Google Scholar] [CrossRef]
- Alhaddad, H.; Alasmari, F.; Alhamadani, B.; Wong, W.; Bell, R.L.; Sari, Y. Effects of chronic ethanol consumption on the expression of GLT-1 and neuroplasticity-related proteins in the nucleus accumbens of alcohol-preferring rats. Brain Res. Bull. 2020, 165, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Alhaddad, H.; Wong, W.; Sari, A.T.; Crotty Alexander, L.E.; Sari, Y. Effects of 3-Month Exposure to E-Cigarette Aerosols on Glutamatergic Receptors and Transporters in Mesolimbic Brain Regions of Female C57BL/6 Mice. Toxics 2020, 8, 95. [Google Scholar] [CrossRef] [PubMed]
- Althobaiti, Y.S.; Alshehri, F.S.; Almalki, A.H.; Sari, Y. Effects of Ceftriaxone on Glial Glutamate Transporters in Wistar Rats Administered Sequential Ethanol and Methamphetamine. Front. Neurosci. 2016, 10, 427. [Google Scholar] [CrossRef]
- Das, S.C.; Yamamoto, B.K.; Hristov, A.M.; Sari, Y. Ceftriaxone attenuates ethanol drinking and restores extracellular glutamate concentration through normalization of GLT-1 in nucleus accumbens of male alcohol-preferring rats. Neuropharmacology 2015, 97, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Knackstedt, L.A.; Melendez, R.I.; Kalivas, P.W. Ceftriaxone restores glutamate homeostasis and prevents relapse to cocaine seeking. Biol. Psychiatry 2010, 67, 81–84. [Google Scholar] [CrossRef]
- Alhaddad, H.; Das, S.C.; Sari, Y. Effects of ceftriaxone on ethanol intake: A possible role for xCT and GLT-1 isoforms modulation of glutamate levels in P rats. Psychopharmacology 2014, 231, 4049–4057. [Google Scholar] [CrossRef]
- Das, S.C.; Althobaiti, Y.S.; Hammad, A.M.; Alasmari, F.; Sari, Y. Role of suppressing GLT-1 and xCT in ceftriaxone-induced attenuation of relapse-like alcohol drinking in alcohol-preferring rats. Addict. Biol. 2022, 27, e13178. [Google Scholar] [CrossRef]
- Knackstedt, L.A.; LaRowe, S.; Mardikian, P.; Malcolm, R.; Upadhyaya, H.; Hedden, S.; Markou, A.; Kalivas, P.W. The role of cystine-glutamate exchange in nicotine dependence in rats and humans. Biol. Psychiatry 2009, 65, 841–845. [Google Scholar] [CrossRef]
- Sondheimer, I.; Knackstedt, L.A. Ceftriaxone prevents the induction of cocaine sensitization and produces enduring attenuation of cue-and cocaine-primed reinstatement of cocaine-seeking. Behav. Brain Res. 2011, 225, 252–258. [Google Scholar] [CrossRef]
- Mao, J.; Sung, B.; Ji, R.R.; Lim, G. Chronic morphine induces downregulation of spinal glutamate transporters: Implications in morphine tolerance and abnormal pain sensitivity. J. Neurosci. 2002, 22, 8312–8323. [Google Scholar] [CrossRef]
- Ozawa, T.; Nakagawa, T.; Shige, K.; Minami, M.; Satoh, M. Changes in the expression of glial glutamate transporters in the rat brain accompanied with morphine dependence and naloxone-precipitated withdrawal. Brain Res. 2001, 905, 254–258. [Google Scholar] [CrossRef]
- Wang, X.F.; Zhao, T.Y.; Su, R.B.; Wu, N.; Li, J. Agmatine Prevents Adaptation of the Hippocampal Glutamate System in Chronic Morphine-Treated Rats. Neurosci. Bull. 2016, 32, 523–530. [Google Scholar] [CrossRef]
- Alshehri, F.S.; Hakami, A.Y.; Althobaiti, Y.S.; Sari, Y. Effects of ceftriaxone on hydrocodone seeking behavior and glial glutamate transporters in P rats. Behav. Brain Res. 2018, 347, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Abulseoud, O.A.; Alasmari, F.; Hussein, A.M.; Sari, Y. Ceftriaxone as a Novel Therapeutic Agent for Hyperglutamatergic States: Bridging the Gap Between Preclinical Results and Clinical Translation. Front. Neurosci. 2022, 16, 841036. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Yang, L.; Du, H.; Sun, Q.; Wang, X.; Cong, L.; Liu, X.; Yin, L.; Li, S.; Du, Y. Insulin Attenuates Beta-Amyloid-Associated Insulin/Akt/EAAT Signaling Perturbations in Human Astrocytes. Cell. Mol. Neurobiol. 2016, 36, 851–864. [Google Scholar] [CrossRef] [PubMed]
- Karki, P.; Hong, P.; Johnson, J., Jr.; Pajarillo, E.; Son, D.S.; Aschner, M.; Lee, E.Y. Arundic Acid Increases Expression and Function of Astrocytic Glutamate Transporter EAAT1 Via the ERK, Akt, and NF-kappaB Pathways. Mol. Neurobiol. 2018, 55, 5031–5046. [Google Scholar] [CrossRef]
- Li, L.B.; Toan, S.V.; Zelenaia, O.; Watson, D.J.; Wolfe, J.H.; Rothstein, J.D.; Robinson, M.B. Regulation of astrocytic glutamate transporter expression by Akt: Evidence for a selective transcriptional effect on the GLT-1/EAAT2 subtype. J. Neurochem. 2006, 97, 759–771. [Google Scholar] [CrossRef]
- Paxinos, G.; Halliday, G.; Watson, C.; Kassem, M.S. Atlas of the Developing Mouse Brain; Academic Press: Cambridge, MA, USA, 2020. [Google Scholar]
- Alshehri, F.S.; Althobaiti, Y.S.; Sari, Y. Effects of administered ethanol and methamphetamine on glial glutamate transporters in rat striatum and hippocampus. J. Mol. Neurosci. 2017, 61, 343–350. [Google Scholar] [CrossRef]
- Karki, P.; Lee, E.; Aschner, M. Manganese neurotoxicity: A focus on glutamate transporters. Ann. Occup. Environ. Med. 2013, 25, 4. [Google Scholar] [CrossRef]
- Pajarillo, E.; Rizor, A.; Lee, J.; Aschner, M.; Lee, E. The role of astrocytic glutamate transporters GLT-1 and GLAST in neurological disorders: Potential targets for neurotherapeutics. Neuropharmacology 2019, 161, 107559. [Google Scholar] [CrossRef]
- Sari, Y.; Sakai, M.; Weedman, J.M.; Rebec, G.V.; Bell, R.L. Ceftriaxone, a beta-lactam antibiotic, reduces ethanol consumption in alcohol-preferring rats. Alcohol Alcohol. 2011, 46, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.W.; Scofield, M.D.; Boger, H.; Hensley, M.; Kalivas, P.W. Synaptic Glutamate Spillover Due to Impaired Glutamate Uptake Mediates Heroin Relapse. J. Neurosci. 2014, 34, 5649–5657. [Google Scholar] [CrossRef] [PubMed]
- Althobaiti, Y.S.; Alshehri, F.S.; Hakami, A.Y.; Hammad, A.M.; Sari, Y. Effects of Clavulanic Acid Treatment on Reinstatement to Methamphetamine, Glial Glutamate Transporters, and mGluR 2/3 Expression in P Rats Exposed to Ethanol. J. Mol. Neurosci. 2019, 67, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Logan, C.N.; Bechard, A.R.; Hamor, P.U.; Wu, L.; Schwendt, M.; Knackstedt, L.A. Ceftriaxone and mGlu2/3 interactions in the nucleus accumbens core affect the reinstatement of cocaine-seeking in male and female rats. Psychopharmacology 2020, 237, 2007–2018. [Google Scholar] [CrossRef] [PubMed]
- Sari, Y.; Toalston, J.E.; Rao, P.S.S.; Bell, R.L. Effects of ceftriaxone on ethanol, nicotine or sucrose intake by alcohol-preferring (P) rats and its association with GLT-1 expression. Neuroscience 2016, 326, 117–125. [Google Scholar] [CrossRef]
- Sari, Y.; Smith, K.D.; Ali, P.K.; Rebec, G.V. Upregulation of GLT1 attenuates cue-induced reinstatement of cocaine-seeking behavior in rats. J. Neurosci. 2009, 29, 9239–9243. [Google Scholar] [CrossRef]
- Muller, D.L.; Unterwald, E.M. In vivo regulation of extracellular signal-regulated protein kinase (ERK) and protein kinase B (Akt) phosphorylation by acute and chronic morphine. J. Pharmacol. Exp. Ther. 2004, 310, 774–782. [Google Scholar] [CrossRef]
- Hudson, R.; Green, M.; Wright, D.J.; Renard, J.; Jobson, C.E.L.; Jung, T.; Rushlow, W.; Laviolette, S.R. Adolescent nicotine induces depressive and anxiogenic effects through ERK 1-2 and Akt-GSK-3 pathways and neuronal dysregulation in the nucleus accumbens. Addict. Biol. 2021, 26, e12891. [Google Scholar] [CrossRef]
- Zhu, H.; Zhuang, D.; Lou, Z.; Lai, M.; Fu, D.; Hong, Q.; Liu, H.; Zhou, W. Akt and its phosphorylation in nucleus accumbens mediate heroin-seeking behavior induced by cues in rats. Addict. Biol. 2021, 26, e13013. [Google Scholar] [CrossRef]
- Rao, P.S.; Saternos, H.; Goodwani, S.; Sari, Y. Effects of ceftriaxone on GLT1 isoforms, xCT and associated signaling pathways in P rats exposed to ethanol. Psychopharmacology 2015, 232, 2333–2342. [Google Scholar] [CrossRef]
- Zhang, X.; Shi, M.; Bjoras, M.; Wang, W.; Zhang, G.; Han, J.; Liu, Z.; Zhang, Y.; Wang, B.; Chen, J.; et al. Ginsenoside Rd promotes glutamate clearance by up-regulating glial glutamate transporter GLT-1 via PI3K/AKT and ERK1/2 pathways. Front. Pharmacol. 2013, 4, 152. [Google Scholar] [CrossRef] [PubMed]
- Goodwani, S.; Rao, P.S.S.; Bell, R.L.; Sari, Y. Amoxicillin and amoxicillin/clavulanate reduce ethanol intake and increase GLT-1 expression as well as AKT phosphorylation in mesocorticolimbic regions. Brain Res. 2015, 1622, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Kihara, T.; Akaike, A.; Niidome, T.; Sugimoto, H. PI3K/Akt/mTOR signaling regulates glutamate transporter 1 in astrocytes. Biochem. Biophys. Res. Commun. 2010, 393, 514–518. [Google Scholar] [CrossRef] [PubMed]
- Lein, E.S.; Hawrylycz, M.J.; Ao, N.; Ayres, M.; Bensinger, A.; Bernard, A.; Boe, A.F.; Boguski, M.S.; Brockway, K.S.; Byrnes, E.J.; et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature 2007, 445, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Rosas, M.; Porru, S.; Fenu, S.; Ruiu, S.; Peana, A.T.; Papale, A.; Brambilla, R.; Di Chiara, G.; Acquas, E. Role of nucleus accumbens mu opioid receptors in the effects of morphine on ERK1/2 phosphorylation. Psychopharmacology 2016, 233, 2943–2954. [Google Scholar] [CrossRef]
- Rosas, M.; Porru, S.; Sabariego, M.; Piludu, M.A.; Giorgi, O.; Corda, M.G.; Acquas, E. Effects of morphine on place conditioning and ERK1/2 phosphorylation in the nucleus accumbens of psychogenetically selected Roman low-and high-avoidance rats. Psychopharmacology 2018, 235, 59–69. [Google Scholar]
- Lee, E.; Sidoryk-Wegrzynowicz, M.; Wang, N.; Webb, A.; Son, D.S.; Lee, K.; Aschner, M. GPR30 regulates glutamate transporter GLT-1 expression in rat primary astrocytes. J. Biol. Chem. 2012, 287, 26817–26828. [Google Scholar] [CrossRef]
- Zhi, Y.; Lu, C.; Zhu, G.; Li, Z.; Zhu, P.; Liu, Y.; Shi, W.; Su, L.; Jiang, J.; Qu, J.; et al. Positive regulation of the CREB phosphorylation via JNK-dependent pathway prevents antimony-induced neuronal apoptosis in PC12 cell and mice brain. Neurotoxicology 2020, 81, 101–108. [Google Scholar] [CrossRef]
- Walton, M.R.; Dragunow, M. Is CREB a key to neuronal survival? Trends Neurosci. 2000, 23, 48–53. [Google Scholar] [CrossRef]
- Walters, C.L.; Blendy, J.A. Different requirements for cAMP response element binding protein in positive and negative reinforcing properties of drugs of abuse. J. Neurosci. 2001, 21, 9438–9444. [Google Scholar] [CrossRef]
- Valverde, O.; Mantamadiotis, T.; Torrecilla, M.; Ugedo, L.; Pineda, J.; Bleckmann, S.; Gass, P.; Kretz, O.; Mitchell, J.M.; Schutz, G.; et al. Modulation of anxiety-like behavior and morphine dependence in CREB-deficient mice. Neuropsychopharmacology 2004, 29, 1122–1133. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Miguel, A.; García-Fuster, M.; Callado, L.; La Harpe, R.; Meana, J.; García-Sevilla, J. Phosphorylation of FADD (Fas-associated death domain protein) at serine 194 is increased in the prefrontal cortex of opiate abusers: Relation to mitogen activated protein kinase, phosphoprotein enriched in astrocytes of 15 kDa, and Akt signaling pathways involved in neuroplasticity. Neuroscience 2009, 161, 23–38. [Google Scholar] [PubMed]
- Ferraguti, F.; Shigemoto, R. Metabotropic glutamate receptors. Cell Tissue Res. 2006, 326, 483–504. [Google Scholar]
- Ghasemzadeh, M.B.; Nelson, L.C.; Lu, X.Y.; Kalivas, P.W. Neuroadaptations in ionotropic and metabotropic glutamate receptor mRNA produced by cocaine treatment. J. Neurochem. 1999, 72, 157–165. [Google Scholar] [CrossRef]
- Qi, C.; Wang, X.; Ge, F.; Li, Y.; Shen, F.; Wang, J.; Cui, C. mGluR 5 in the nucleus accumbens shell regulates morphine-associated contextual memory through reactive oxygen species signaling. Addict. Biol. 2015, 20, 927–940. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wong, W.; Sari, Y. Effects of Chronic Hydrocodone Exposure and Ceftriaxone on the Expression of Astrocytic Glutamate Transporters in Mesocorticolimbic Brain Regions of C57/BL Mice. Toxics 2023, 11, 870. https://doi.org/10.3390/toxics11100870
Wong W, Sari Y. Effects of Chronic Hydrocodone Exposure and Ceftriaxone on the Expression of Astrocytic Glutamate Transporters in Mesocorticolimbic Brain Regions of C57/BL Mice. Toxics. 2023; 11(10):870. https://doi.org/10.3390/toxics11100870
Chicago/Turabian StyleWong, Woonyen, and Youssef Sari. 2023. "Effects of Chronic Hydrocodone Exposure and Ceftriaxone on the Expression of Astrocytic Glutamate Transporters in Mesocorticolimbic Brain Regions of C57/BL Mice" Toxics 11, no. 10: 870. https://doi.org/10.3390/toxics11100870
APA StyleWong, W., & Sari, Y. (2023). Effects of Chronic Hydrocodone Exposure and Ceftriaxone on the Expression of Astrocytic Glutamate Transporters in Mesocorticolimbic Brain Regions of C57/BL Mice. Toxics, 11(10), 870. https://doi.org/10.3390/toxics11100870